
Allylic C–H functionalization via group 9 π-allyl intermediates

Abstract
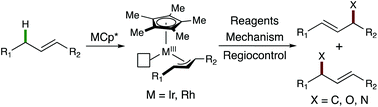
Allylic C–H functionalization catalysed by group 9 Cp* transition-metal complexes has recently gained significant attention. These reactions have expanded allylic C–H functionalization to include di- and trisubstituted olefins, and a broad range of coupling partners. More specifically, several catalytic C–N, C–O, and C–C bond forming allylic C–H functionalization reactions have been reported, proceeding via MCp*-π-allyl intermediates. Herein we present an overview of these reactions by mechanistic paradigm. We also place this information in context of recent advances, as well as, limitations that remain for this class of reactions.
- This article is part of the themed collection: 2020 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

