
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-NIR absorption spectroscopy study of the formation of ternary plutonyl(VI) carbonate complexes†
Yongheum
Jo
a,
Hye-Ryun
Cho
b and
Jong-Il
Yun
 *a
*a
aDepartment of Nuclear and Quantum Engineering, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: jiyun@kaist.ac.kr
bNuclear Chemistry Research Team, Korea Atomic Energy Research Institute, 111 Daedeok-daero 989 beon-gil, Yuseong-gu, Daejeon 34057, Republic of Korea
First published on 27th July 2020
Abstract
We present the first experimental evidence for the ternary complexation of calcium and magnesium ions with plutonyl(VI)tricarbonate species in carbonate-containing aqueous solutions using visible-NIR spectrophotometric titration. Prior to studying the ternary plutonyl(VI) carbonate complexation, visible-NIR absorption spectral information of PuO2(CO3)22− and PuO2(CO3)34− was successfully obtained. PuO2(CO3)22− has a prominent peak at 853 nm and its molar absorptivity was determined to be ε853, PuO2(CO3)22− = 49.0 ± 4.2 M−1·cm−1. The spectrophotometric titration results by adding calcium or magnesium to the plutonyl(VI) carbonate system consisting of PuO2(CO3)22− and PuO2(CO3)34− indicate the formation of CaPuO2(CO3)32− and MgPuO2(CO3)32− complexes and provide the formation constants at 0.1 M H/NaClO4 for MPuO2(CO3)32− from PuO2(CO3)34−, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 4.33 ± 0.50 and 2.58 ± 0.18 for M = Ca2+ and Mg2+, respectively. In addition, the formation constants of CaPuO2(CO3)32− and MgPuO2(CO3)32− from PuO2(CO3)34− at infinite dilution (logK°) were proposed to be 6.05 ± 0.50 and 4.29 ± 0.18, respectively, based on the correction of ionic strength using the Davies equation. The absorption spectrum of the ternary plutonyl(VI) complexes of CaPuO2(CO3)32− is similar to that of PuO2(CO3)34− with the exception of a characteristic absorption peak at 808 nm (ε808, CaPuO2(CO3)32− = 42.9 ± 1.6 M−1·cm−1). According to the calculated aqueous plutonyl(VI) speciation including the ternary plutonyl(VI) complexes, CaPuO2(CO3)32− is considered the dominant Pu(VI) species under environmental conditions, and plutonyl(VI) may be more mobile than expected in previous assessments.
K = 4.33 ± 0.50 and 2.58 ± 0.18 for M = Ca2+ and Mg2+, respectively. In addition, the formation constants of CaPuO2(CO3)32− and MgPuO2(CO3)32− from PuO2(CO3)34− at infinite dilution (logK°) were proposed to be 6.05 ± 0.50 and 4.29 ± 0.18, respectively, based on the correction of ionic strength using the Davies equation. The absorption spectrum of the ternary plutonyl(VI) complexes of CaPuO2(CO3)32− is similar to that of PuO2(CO3)34− with the exception of a characteristic absorption peak at 808 nm (ε808, CaPuO2(CO3)32− = 42.9 ± 1.6 M−1·cm−1). According to the calculated aqueous plutonyl(VI) speciation including the ternary plutonyl(VI) complexes, CaPuO2(CO3)32− is considered the dominant Pu(VI) species under environmental conditions, and plutonyl(VI) may be more mobile than expected in previous assessments.
1. Introduction
Plutonium is generally produced during nuclear power generation and by nuclear weapons programmes and is one of the most strictly regulated elements in the world because of national security, nuclear proliferation, and radiological hazard concerns. Anthropogenic activities such as nuclear weapons testing,1 severe accidents in nuclear power plants, improper waste management, and disposal of legacy waste2 can cause plutonium contamination in the environment. The fate and transport of plutonium in the environment must be assessed for the remediation of polluted sites and nuclear waste management.3 However, it is very difficult to predict the chemical behaviour of plutonium in the environment because plutonium can simultaneously exist in multiple oxidation states (+3, +4, +5, and +6) in a single aquatic system, and aquatic plutonium (bio)geochemistry is exclusively governed by the oxidation state.4 For instance, tetravalent plutonium, Pu(IV), is considered insoluble and readily adsorbed, but Pu(IV) transport can be facilitated by colloids. However, hexavalent plutonium, Pu(VI), is relatively soluble and behaves as an aqueous species due to its complexation with a variety of inorganic and organic ligands.5Natural waters such as rainwater, groundwater, and seawater can not only leach plutonium from plutonium-bearing pollutants but also transport plutonium. The oxidation state and aqueous speciation of plutonium are of utmost importance for the understanding of plutonium transport and migration via natural fluids. In contact with atmospheric air, natural waters are oxidizing, and the higher oxidation states of plutonyl ions, namely, Pu(V) and Pu(VI), are widely observed in oxic rainwater, surface groundwater, seawater, and drinking water.2,6–9 Radiolysis also plays an important role in the oxidation of plutonium and the stabilization of a high oxidation state.9,10 In the weakly acidic to weakly alkaline pH range (pH 5–9) of natural water, carbonate ion (CO32−) is one of the most influential ligands in aquatic chemistry due to its high concentration and strong affinity for cations. In oxidizing natural waters of neutral to weakly alkaline pH, plutonyl(V,VI)carbonato species such as PuO2CO3− and PuO2(CO3)22− are expected to be the dominant aqueous plutonium species5,11,12 based on a thermodynamic database13 and modelling.
Beyond the binary carbonato complexes, ternary complexes containing divalent alkaline earth ions were found for hexavalent uranium, U(VI),14 and are regarded as the major U(VI) complexes. Over the past two decades, the formation of ternary species (MxUO2(CO3)32x−4, M = Ca2+ and Mg2+) has been intensively studied and the MxUO2(CO3)32x−4 complexes are the dominant aqueous U(VI) species in the presence of naturally abundant Ca2+ and Mg2+ ions in groundwater and seawater.15–25 In addition to their overwhelming predominance in aqueous U(VI) species in natural waters, the ternary complexes are U(VI) species with significantly increased mobility resulting from decreased U(VI) sorption26–28 and hindered reduction to immobile tetravalent uranium.29–32 In a field study, the occurrence of Ca2UO2(CO3)3(aq) was observed even in deep, reducing, Fe(II)-containing groundwater (Eh – 140 mV, ∼500 metres below sea level).33 A study of uranium speciation in sea urchins identified the uptake of uranium in the form of ternary calcium carbonato species.34 Although ternary complexation is very striking with respect to the common fate of uranium under various environmental conditions, it has not been reported for hexavalent actinides other than U(VI).
Here, we identify the ternary complexation of plutonyl(VI) tricarbonate with calcium and magnesium ions for the first time. Visible-near infrared (vis-NIR) absorption spectroscopy is employed to elucidate the chemical interaction of Ca2+ and Mg2+ with plutonyl(VI) carbonate species. Among the various chemical models, a chemical model involving the formation of CaPuO2(CO3)32− confirms our spectrophotometric results. Finally, the noticeable impacts of the ternary complexation of Pu(VI) on its aqueous chemistry and mobility in the environment are discussed based on the obtained formation constants for CaPuO2(CO3)32− and MgPuO2(CO3)32−.
2. Experimental section
2.1. Cautions
Plutonium is radioactive and toxic. For experiments with plutonium, a restricted area with appropriate safety equipment and authorization is required. In this work, the plutonium solutions were carefully handled in a chemical fume hood and negative pressure glove box.2.2. Chemicals
Plutonium stock solution was prepared by the dissolution of PuO2 (ORNL, 242Pu: 99.932% with trace levels of 238Pu, 239Pu, 240Pu, 241Pu, and 244Pu) with concentrated HNO3 and a small amount of HF. 241Am generated from the β-decay of 241Pu was separated from the plutonium stock solution by anion exchange (AG 1-X8, Bio-Rad). The background electrolyte (HNO3 and HF) was converted to HClO4 by fuming with concentrated HClO4 solution before drying and re-dissolution. The absence of the oxidation states of plutonium other than +6 was confirmed by vis-NIR absorption spectroscopy. Ultrafiltration (10 kDa, Ultracel YM regenerated cellulose membrane, Centricon-10, Millipore) was employed to eliminate particulates and the breakdown probability obtained using laser-induced breakdown detection (LIBD) of the Pu(VI) stock solution was comparable to that of ultrapure water (Milli-Q, Merck Millipore), which indicates the absence of particulates in the solution. The concentration of the Pu(VI) stock solution was determined to be 6 mM by liquid scintillation counting (LSC). The detailed experimental setups for LIBD have been described elsewhere.35,36 Aliquots of plutonium stock solution were taken to obtain the desired plutonium concentration.Stock solutions of 0.5 M Na2CO3 and 1.5 M NaClO4 were prepared by the dissolution of anhydrous Na2CO3 (99.999%, Aldrich) and hydrate NaClO4 (99.99%, Aldrich), respectively, in Milli-Q water. Pu(VI) samples for vis-NIR spectrophotometric titration were prepared with a Pu concentration of 0.35–0.40 mM. The initial carbonate concentration was set to 18 mM. To minimize the reduction of Pu, NaOCl was spiked at a 0.2 mM concentration of hypochlorite in the samples. HClO4 (99.999%, Aldrich) was used to adjust the pH of the Pu samples. The pH was measured using a combination glass Ross-type electrode (Thermo Scientific Orion) filled with 3 M KCl calibrated with pH buffers (pH 4.01, 7.00, and 10.01, Thermo Scientific Orion). The pH measurement was carried with a separated and duplicated sample solution in each titration step. The pH values mentioned throughout this manuscript are the measured values. The magnesium and calcium titrants were prepared by the dissolution of Mg(ClO4)2·4H2O (99%, Aldrich) and Ca(ClO4)2·4H2O (99%, Aldrich) in 0.1 M NaClO4 to obtain 0.23 M Mg2+ and 0.14 M Ca2+. The ionic strength of the Pu(VI) sample was set to 0.1 M H/NaClO4. All solutions were prepared with ultrapure Milli-Q water (18.2 MΩ cm−1). The plutonium concentration was measured using LSC (Tri-Carb 3110TR, PerkinElmer) and the concentrations of magnesium and calcium were analysed by inductively coupled plasma-optical emission spectroscopy (ICP-OES).
2.3. Vis-NIR spectrophotometry and data processing
A 2.5 mL Pu sample was contained in a sealable quartz cell (Hellma Analytics) with an optical path length of 1 cm, and the cell was mounted in a temperature-controllable cuvette holder (Agilent Technologies) during the measurement. The vis-NIR absorption spectra were recorded by a spectrophotometer (Cary 5000, Agilent) at 25 °C. To increase the concentrations of H+, Mg2+, and Ca2+, aliquots of HClO4 and titrants, Mg(ClO4)2 and Ca(ClO4)2 in 0.1 M NaClO4, were added, and the sample was equilibrated for 10 min with magnetic stirring before the absorption measurements. The equilibrium constants and absorption spectra of each species were calculated from the recorded spectra using a minimization program (Hypspec2014).37 For the fitting of the spectrophotometric results and the calculation of formation constants and absorption spectra of CaPuO2(CO3)32−, the aqueous species H2CO3, HCO3−, NaHCO3, NaCO3−, CaHCO3+, CaCO3, PuO2(CO3)22−, PuO2(CO3)34−, H+, and OH− were included in the model as the existing species in the system. The equilibrium constants were obtained from ref. 13 and 38 and corrected to those at 0.1 M H/NaClO4 based on the specific ion interaction theory (SIT)39 and the corresponding coefficients in ref. 40 and 41. The absorption spectrum in the range of 873 nm to 899 nm was excluded from data processing due to the meaningless signals with noisy fluctuation generated while exchanging the detectors of the spectrometer. All experiments were performed in triplicate. Errors of the data obtained in this work represent ±1σ. The formation constant of CaPuO2(CO3)32− was calculated to inherently include the errors of the formation constants of PuO2(CO3)22− and PuO2(CO3)34− since the logK values of PuO2(CO3)22− and PuO2(CO3)34− were taken into account in the calculation with their errors (±0.50). On the other hand, the error for the logK of MgPuO2(CO3)32− was obtained independent of the errors for PuO2(CO3)22− and PuO2(CO3)34−.
The stepwise formation constants of MgPuO2(CO3)32− and CaPuO2(CO3)32− species (logKMPuO2(CO3)32−, M2+ + PuO2(CO3)34− ↔ MPuO2(CO3)2−, M = Mg and Ca) at infinite dilution were determined using the Davies equation.43
2.4. Geochemical modelling
PHREEQC (version 3)44 was used to calculate the aqueous Pourbaix diagram (Eh-pH diagram) and Pu(VI) speciation with pH at 25 °C and infinite dilution. The thermodynamic data for Pu(VI) species were basically obtained from NEA-TDB in PHREEQC format (updated in November, 2018).45 For the formation constants of MgPuO2(CO3)32− and CaPuO2(CO3)32−, the data obtained in this work were used. For the aqueous and solid species for calcium, magnesium, and carbonate, the thermodynamic data of WATEQ4F38 were used. In the calculations, calcite, magnesite, dolomite, PuO2CO3(cr), and PuO2(OH)2·H2O(cr) were taken into account as solubility limiting phases. The Pourbaix diagram was illustrated using PhreePlot software.463. Results and discussion
3.1. Vis-NIR absorption spectral properties of PuO2(CO3)22− and PuO2(CO3)34−
Prior to the investigation of Ca2+ complexation with plutonyl(VI)carbonate species, the vis-NIR absorption characteristics of PuO2(CO3)22− and PuO2(CO3)34− species were identified. Fig. 1a and b show the results of a vis-NIR spectrophotometric titration with decreasing pH. At the starting point of the spectrophotometric titration (pH 9.78, Fig. 1a and b), the aqueous Pu(VI) speciation is calculated to be 100% PuO2(CO3)34− by thermodynamic modelling based on the critically reviewed NEA-TDB (ESI, Fig. S1†), and the recorded spectrum at pH 9.78 is identical to the absorption spectrum of Pu(VI) observed under concentrated carbonate conditions at a strongly alkaline pH (2 M Na2CO3, pH 12.6) in a previous study.47 It is therefore assumed that the spectrophotometric titration starts at 100% PuO2(CO3)34−, and the observed spectrum of Pu(VI) at the initial point (pH 9.78) is ascribed to the characteristic absorption spectrum of PuO2(CO3)34−. The characteristic sharp absorption peak of PuO22+ ions at 830 nm completely disappears due to carbonato complexation, and instead, broad absorption from 500 nm to 650 nm (molar absorptivity ε = 55.7 ± 2.8 M−1 cm−1 at the peak position of 569 nm) is observed. In addition, the observed absorption bands in the ranges of 840–870 nm, 900–960 nm, and 1000–1120 nm are comparable to the reported absorption bands of Pu(VI) carbonate species in previous work.47,48 | ||
| Fig. 1 Representative results of the spectrophotometric pH titration of the plutonyl(VI) carbonate system ([Pu(VI)] = 0.36 mM, [CO32−]total = 18 mM, pH 9.78 (black line) to 7.73 (red line), and I = 0.1 M H/NaClO4) with decreasing pH in the wavelength ranges of (a) 500–870 nm and (b) 900–1120 nm. The arrows indicate a gradual increase (up) and decrease (down) in absorbance due to acidification. (c) Vis-NIR absorption spectra (500–1120 nm) of PuO2(CO3)34− (solid line, measured at pH 9.78) and PuO2(CO3)22− (dotted line, determined from the equilibrium constant of the transition reaction, PuO2(CO3)34− ↔ PuO2(CO3)22− + CO32−). | ||
As the pH decreased, the recorded absorption spectra gradually changed (Fig. 1a and b), indicating chemical changes in the aqueous Pu(VI) species. A decrease in absorbance with a continuous hypsochromic shift in the broad absorption between 500 nm and 650 nm and the appearance of isosbestic points at 540, 654, 823, 953, and 1054 nm representing a one-to-one transition of PuO2(CO3)34− to PuO2(CO3)22− are observed with decreasing pH. With acidification, absorption bands at 853 nm and 1010 nm appear and gradually grow, whereas the intensity of the double peaks in the 1000–1120 nm region decreases.
In addition to the characteristic changes in the absorption spectra, the calculated equilibrium constant also supports a change in aqueous Pu(VI) species from PuO2(CO3)34− to PuO2(CO3)22− after comparison with the reported thermodynamic data. The chemical reaction is as follows:
| PuO2(CO3)34− ↔ PuO2(CO3)22− + CO32− | (1) |
From the results of the spectrophotometric titration, the equilibrium constant (logK) of reaction (1) is calculated to be −4.02 ± 0.19 at 0.1 M H/NaClO4 by the data processing software37 and is corrected to −3.15 ± 0.20 at infinite dilution by SIT39 using ion interaction coefficients from previous work.41 The corrected formation constant of −3.15 ± 0.20 is very comparable to the value of −3.30 ± 1.02, which resulted from the difference in the Gibb's free energies of formation for PuO2(CO3)22− and PuO2(CO3)34− species.13 These critically reviewed formation constants of reactions (2) and (3) are 14.7 ± 0.50 and 18.0 ± 0.50, respectively.
| PuO22+ + 2CO32− ↔ PuO2(CO3)22− | (2) |
| PuO22+ + 3CO32− ↔ PuO2(CO3)34− | (3) |
The stability constants and interaction coefficients of Pu(VI) carbonate species in 0.202 m NaClO4 were recently revisited by coupling capillary electrophoresis and ICP-MS.41 The newly obtained stability constants at infinite dilution based on the reactions (2) and (3) were 14.99 ± 0.06 and 17.94 ± 0.30, respectively. This corresponds to −2.95 ± 0.31 for reaction (1), which is also comparable to our data (−3.15 ± 0.20).
The aqueous Pu(VI) speciation from the spectrophotometry in this study and from the thermodynamic modelling with NEA-TDB are in good agreement (ESI, Fig. S1†). Along with the equilibrium constant of reaction (1), the pure absorption spectrum of PuO2(CO3)22− is obtained accordingly based on the results of the potentiometric titration (Fig. 1c). The molar absorptivity of the most prominent peak of PuO2(CO3)22− at 853 nm is determined to be 49.0 ± 4.2 M−1 cm−1. Notably, a shoulder peak in the vicinity of the PuO2(CO3)22− band at 853 nm arises at approximately 843 nm below pH 8.2. Thermodynamic modelling reveals the formation of plutonyl(VI) monocarbonate species in very low abundance, <5%. To avoid interference from Pu(VI) species other than PuO2(CO3)22− and PuO2(CO3)34−, the equilibrium constant and the absorption spectrum of PuO2(CO3)22− species are determined using the result obtained above pH 8.2.
3.2. Interaction of Ca2+ with Pu(VI) carbonate species
On the basis of the absorption spectral information of PuO2(CO3)22− and PuO2(CO3)34−, the ternary complexation of plutonyl(VI)carbonate species with Ca2+ is investigated (Fig. 2). At 808 nm, a new sharp peak, which is not observed in the PuO2(CO3)22−/PuO2(CO3)34− system, appears after adding Ca2+ and is thus a strong indication of the interaction of Ca2+ with Pu(VI) carbonate species. Together with the growth of this peak at 808 nm characteristic of the interaction with Ca2+, the broad absorption band between 500 nm and 650 nm, representing the equilibrium of PuO2(CO3)22− and PuO2(CO3)34−, undergoes a bathochromic shift after addition of Ca2+ even at a constant pH of 7.9 during the titration. These spectral features are identical to other spectrophotometric Ca2+ titrations with varying starting pH values and different ratios of PuO2(CO3)22−/PuO2(CO3)34− (ESI, Fig. S2†). This unambiguously indicates that a simultaneous interaction of Ca2+ with PuO2(CO3)22− and PuO2(CO3)34− is unlikely. In addition, the clear isosbestic points at 540 and 651 nm are independent of the pH of the Ca2+ titration, indicating that only one Ca–Pu(VI) carbonate species is formed: either Ca2+–PuO2(CO3)22− or Ca2+–PuO2(CO3)34−. The redshift in the broad peak (500–650 nm) and the disappearance of absorbance at 853 nm correspond to the change in the aqueous Pu(VI) species from PuO2(CO3)22− to PuO2(CO3)34−. The absorption spectrum of the newly formed Ca2+–Pu(VI) carbonate species is similar to that of PuO2(CO3)34−, except for a distinguishable absorption band at 808 nm. In addition, the characteristic peak of PuO2(CO3)22− at 853 nm is attenuated by the addition of Ca2+ and nearly vanishes at the end of the Ca2+ titration ([Ca2+] = 0.72 mM), which reflects a significant decrease in the PuO2(CO3)22− concentration with Ca2+ addition. Thus, Ca2+ likely interacts with PuO2(CO3)34− rather than PuO2(CO3)22−. This interaction is also experimentally confirmed by the results of the spectrophotometric titration at pH 9.1, showing the appearance of the peak at 808 nm even in the absence of PuO2(CO3)22− (Fig. 2b). | ||
| Fig. 2 Results of the spectrophotometric Ca2+ titration. (a) Representative results of the spectrophotometric Ca2+ titration of the plutonyl(VI)carbonate system ([Pu(VI)] = 0.39 mM, [CO32−]total = 17 mM, [Ca2+] = 0 to 0.72 mM, pH 7.9, and I = 0.1 M H/NaClO4) with increasing Ca2+ in the wavelength range of 500–870 nm. The arrows indicate a gradual increase (up) and decrease (down) in absorbance due to Ca2+ complexation. (b) Increase in the peak at 808 nm under PuO2(CO3)34−-dominant conditions ([Pu(VI)] = 0.39 mM, [CO32−]total = 17 mM, [Ca2+] = 0 to 0.11 mM, pH 9.1, and I = 0.1 M H/NaClO4). (c) Results of a triplicate slope analysis (logR vs. log[Ca2+]total) with the slope (x) representing the stoichiometric number of complexed Ca2+ obtained from linear regression. (d) Deconvoluted vis-NIR absorption spectra of CaPuO2(CO3)32− from the results of the spectrophotometric Ca2+ titrations. | ||
Slope analysis (Fig. 2c) was conducted to determine the stoichiometric number (x) of Ca2+ complexing with PuO2(CO3)34−. The chemical reaction of Ca2+ with PuO2(CO3)34− and its equilibrium constant of CaxPuO2(CO3)32x−4 (logK) are described with the concentration ratio (R) of Ca2+-complexed and uncomplexed plutonyl(VI) species, i.e., [CaxPuO2(CO3)32x−4]/[PuO2(CO3)34−], as follows:
| xCa2+ + PuO2(CO3)34− ↔ CaxPuO2(CO3)32x−4 |
| logK = logR − x·log[Ca2+] | (4) |
At the first measurement point, the total Pu(VI) concentration ([Pu]total) is the sum of the concentrations of PuO2(CO3)22− and PuO2(CO3)34− in the absence of Ca2+. In each step of the titration, the concentration of PuO2(CO3)22− is determined from the absorbance (A853) and the molar absorptivity at 853 nm (ε853, PuO2(CO3)22−), where the characteristic peak of PuO2(CO3)22− is located. The concentration ratio of PuO2(CO3)34− to PuO2(CO3)22− can be assumed to be constant (C) in all steps of the Ca2+ titration at a consistent pH and CO32− concentration; thus, the concentration of CaxPuO2(CO3)32x−4 is calculated from the Pu(VI) concentration balance. Accordingly, the concentration ratio (R) of Ca2+-complexed and uncomplexed Pu species is rearranged in terms of the absorption properties as follows:
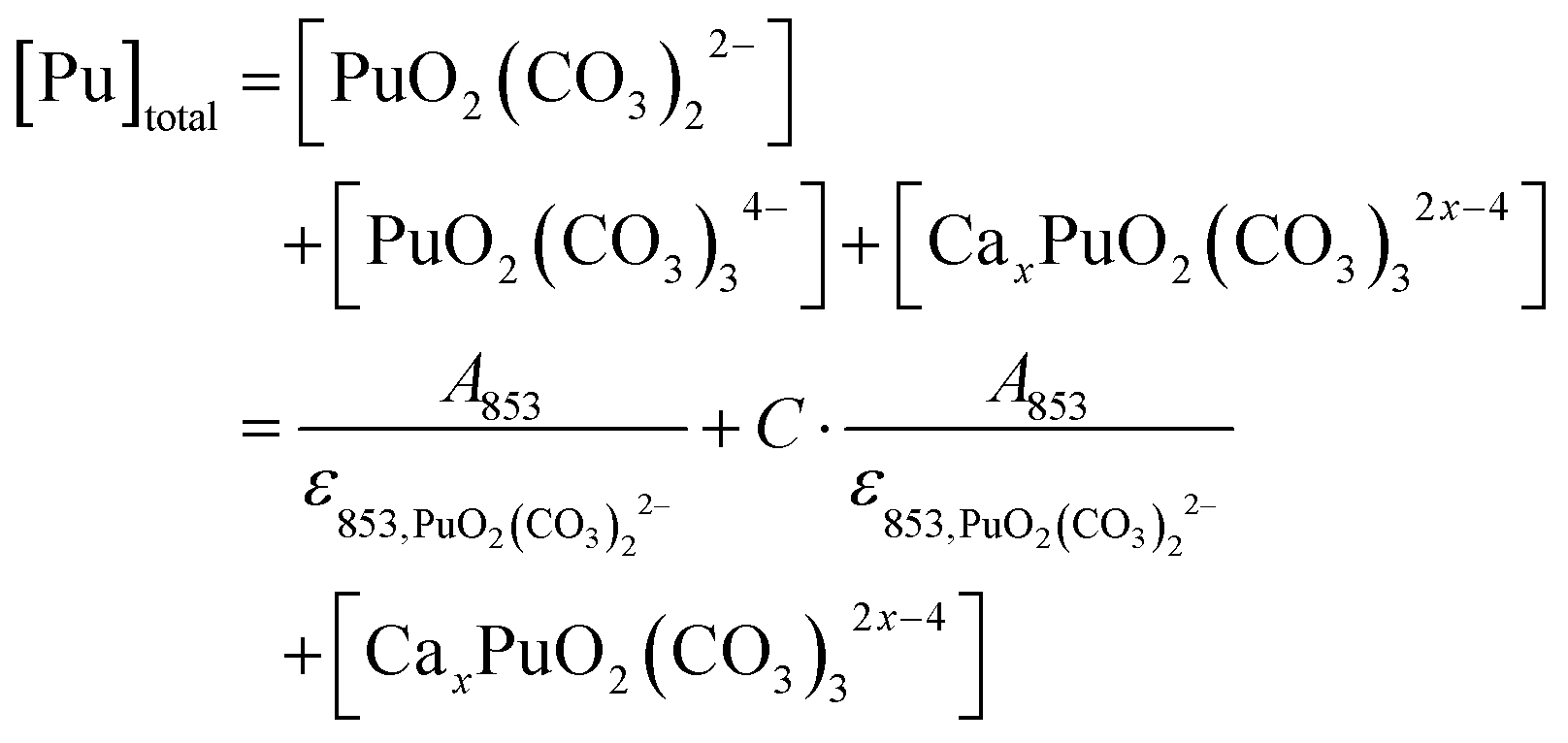
 | (5) |
As shown in Fig. 2c, the slope of logR vs. log[Ca2+]total is 1.14 ± 0.21, indicating that the stoichiometric number of Ca2+ ions complexing with PuO2(CO3)34− is close to 1 and CaPuO2(CO3)32− species are formed. The positive deviation of the obtained slope from the ideal value of 1 may be attributed to an overestimation of the free Ca2+ ion concentration because the amount of Ca2+ actually consumed by complexation is neglected. Taking into account the consumed Ca2+ concentration based on the assumption of x = 1, the slope is determined to be 0.94 ± 0.26 (ESI, Fig. S3†).
The formation of the monocalcium plutonyl(VI)tricarbonate, CaPuO2(CO3)32−, was also confirmed by the examination of possible chemical models. A chemical model based on the formation of CaPuO2(CO3)32− shows a very successful fitting result with the spectrophotometric titration, and the stepwise formation constant of CaPuO2(CO3)32− (logKCaPuO2(CO3)32−, Ca2+ + PuO2(CO3)34− ↔ CaPuO2(CO3)32−) corresponding to reaction (4) with x = 1 is calculated to be 4.33 ± 0.50 in 0.1 M H/NaClO4. The absorption spectrum of CaPuO2(CO3)32− calculated from the results of spectrophotometric titration is presented in Fig. 2d. The stepwise formation constant of the similar uranyl(VI) species, CaUO2(CO3)32−, in 0.1 M H/NaClO4 medium (logKCaUO2(CO3)32−, Ca2+ + UO2(CO3)34− ↔ CaUO2(CO3)32−) has been reported to be 3.13 ± 0.2220 and 3.00 ± 0.2523 in previous work. Other chemical models presuming the formation of neutral Ca2PuO2(CO3)3(aq) species show unsatisfactory spectrophotometric results with failures in the spectral fitting procedure (ESI, Fig. S4†) and implausible formation constants. Even though the ternary Ca2+ complexation of PuO2(CO3)34− is more favourable than that of UO2(CO3)34−, the di-calcium species, which is frequently observed for U(VI) as Ca2UO2(CO3)3(aq), appears to be difficult to form for PuO2(CO3)34−. UO2(CO3)34− can be dominant even at a low pH and carbonate concentration, where a millimolar concentration of Ca2+ is sufficient to form di-calcium species that can be dissolved. However, in the case of Pu(VI), the formation of a considerable amount of PuO2(CO3)34− requires a relatively high pH and carbonate concentration, but the amount of Ca2+ ions needed to form di-calcium species is limited due to the solubility limit imposed by the formation of CaCO3(s) under these conditions.
3.3. Mg2+–PuO2(CO3)34− system
Similar to Ca2+ complexation, an identical trend in spectrophotometric changes is also observed for the titration with Mg2+ ions (ESI, Fig. S5†). However, the evolution of the absorbance is more insensitive to the Mg2+ concentration than to the Ca2+ system, which represents a stronger affinity of Ca2+ than that of Mg2+ for PuO2(CO3)34−. Because of this small spectral change in absorbance, even with a large variation in the Mg2+ concentrations compared to the Ca2+ system, data processing by the minimization software37 and the convolution of the characteristic absorption spectrum of MgPuO2(CO3)32− are problematic. Instead, the concentration balance used in the slope analysis (eqn (4) and (5)) is utilized to quantitatively determine the concentration of Mg2+-complexed PuO2(CO3)34− in each step of the titration based on the assumed formation of MgPuO2(CO3)32−. The stepwise formation constant of MgPuO2(CO3)32− at 0.1 M H/NaClO4 (logKMgPuO2(CO3)32−) is calculated to be 2.58 ± 0.18, which is smaller than logKCaPuO2(CO3)32−. The favourability of Ca2+ over Mg2+ is widely observed in Mg2+/Ca2+–UO2(CO3)34− complexations as well.18,22,24
To make the formation constants applicable to thermodynamic modelling, the Davies equation43 is applied to correct the ionic strength to infinite dilution. The stepwise formations of MgPuO2(CO3)32− and CaPuO2(CO3)32− at infinite dilution are determined to be logK°MgPuO2(CO3)32− = 4.29 ± 0.18 and logK°CaPuO2(CO3)32− = 6.05 ± 0.50. To the best of our knowledge, the formations of MgPuO2(CO3)32− and CaPuO2(CO3)32− are reported for the first time in this work. For that reason, the ion interaction coefficients of the ternary plutonyl(VI) species are still unknown. Nonetheless, it is worth noting that the use of the SIT approach39 with the ion interaction coefficient of CaUO2(CO3)32− with Na+ suggested from PSI/Nagra-TDB,42ε(CaUO2(CO3)32−, Na+) = –(0.1 ± 0.1) kg·mol−1, as a surrogate coefficient for MPuO2(CO3)32− shows insignificant differences in logK° (4.27 ± 0.19 and 6.02 ± 0.50 for M = Mg2+ and Ca2+, respectively) with the values obtained using the Davies equation.43Table 1 summarizes the formation constants of ternary plutonyl(VI) complexes in 0.1 M H/NaClO4 and at infinite dilution (Davies equation43).
K) of Mg2+/Ca2+ complexation with PuO2(CO3)34−
| logK |
|||
|---|---|---|---|
| Reactions | I = 0.1 M H/NaClO4 | I = 0 M infinite dilution | Ref. |
| a Present work. b Corrected by using the Davies equation.43 | |||
| PuO22+ + 3CO32− ↔ PuO2(CO3)34− | — | 18.00 ± 0.50 | 13 |
| Mg2+ + PuO2(CO3)34− ↔ MgPuO2(CO3)32− | 2.58 ± 0.18 | 4.29 ± 0.18b | p.w.a |
| Mg2+ + PuO22+ + 3CO32− ↔ MgPuO2(CO3)32− | — | 22.29 ± 0.53 | p.w.a |
| Ca2+ + PuO2(CO3)34− ↔ CaPuO2(CO3)32− | 4.33 ± 0.50 | 6.05 ± 0.50b | p.w.a |
| Ca2+ + PuO22+ + 3CO32− ↔ CaPuO2(CO3)32− | — | 24.05 ± 0.50 | p.w.a |
3.4. Aqueous Pu speciation
Fig. 3 presents the Pourbaix diagram (Eh–pH diagram) and the aqueous Pu(VI) species distribution based on the obtained thermodynamic data of CaPuO2(CO3)32− and MgPuO2(CO3)32− and the data from NEA-TDB.13 It is worth noting that the modelling was performed at considerably reduced Pu concentrations (10−9 M) and carbonate amount (atmospheric CO2(g) equilibrium) as compared to those used in the experiments ([Pu(VI)] = 0.35–0.40 mM and [CO32−]initial = 18 mM). At weakly alkaline pH under oxidizing conditions, CaPuO2(CO3)32− is the dominant aqueous Pu(VI) species and the apparent Pu(VI/V) Nernstian potential is shifted toward a less positive value, which suggests a slight but further redox stabilization of Pu(VI). As a consequence of the formation of CaPuO2(CO3)32−, the effect of PuO2(CO3)22−, which was expected to be the major Pu(VI) species in previous work,5,11,12 would be weakened in the presence of calcium at environmental (millimolar) concentrations. Based on the aqueous Pu(VI) speciation (Fig. 3b), the predominance of CaPuO2(CO3)32− and a correspondingly decreased amount of PuO2(CO3)22− are expected in natural waters such as rainwater, groundwater, and seawater. In addition, the strong ternary complexation could increase Pu(VI) solubility up to approximately six times at pH 7.5–9.0 (ESI, Fig. S7†).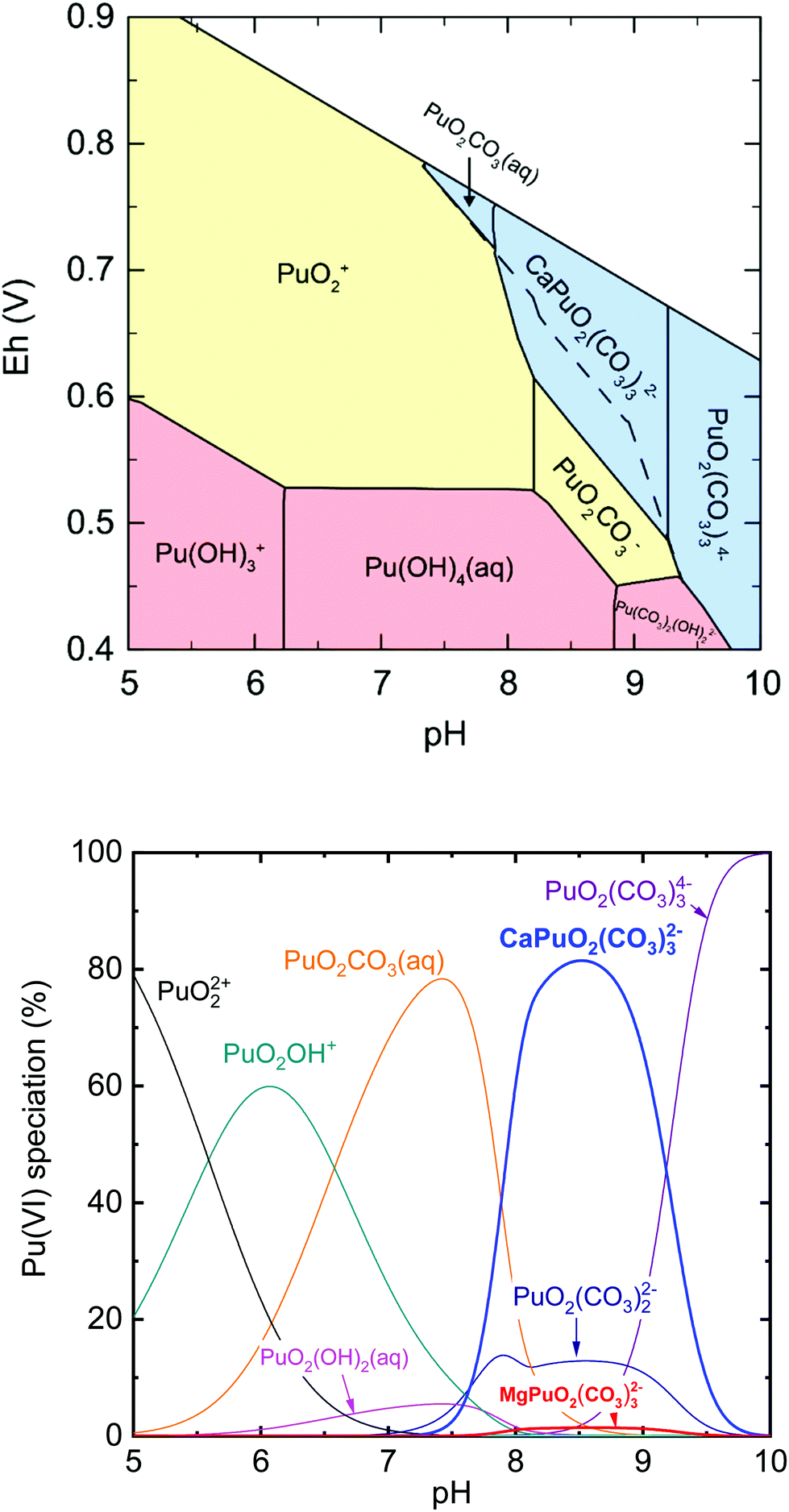 | ||
| Fig. 3 Environmental implications of the formation of CaPuO2(CO3)32− and MgPuO2(CO3)32− ([Pu] = 1 × 10−9 M, [Ca2+] = [Mg2+] = 1 mM, in equilibrium with pCO2(g) = 10−3.4 atm, at 25 °C and infinite dilution). (a) Calculated Pourbaix (Eh-pH) diagram of aqueous plutonium species. The dashed line represents the boundary of apparent Pu(VI/V) Nernstian potential in the absence of Ca2+ and Mg2+. (b) Calculated aqueous Pu(VI) species distribution. The abrupt change at pH 8.1 occurs due to the solubility of Ca2+ in the carbonate system. For the details of calculation, see section 2.4. | ||
4. Conclusions
The complexations of aqueous plutonyl(VI) carbonate species with Ca2+ and Mg2+ were identified for the first time using vis-NIR absorption spectroscopy. In Ca2+ or Mg2+ titrations, a characteristic absorption signal at 808 nm, which was not observed in the plutonyl carbonate system, i.e. PuO2(CO3)22− and PuO2(CO3)34−, appears and it is a clear indication of the ternary complexation of plutonyl(VI)carbonate with alkaline earth metals. The results of vis-NIR absorption spectroscopy evidently show that Ca2+ interacts with PuO2(CO3)34− rather than with PuO2(CO3)22−, and the results of slope analysis provide the stoichiometric number of Ca2+ complexing with PuO2(CO3)34− as 1, indicating the formation of MPuO2(CO3)32− (M = Ca2+ and Mg2+). Based on the results of spectrophotometric titration and the correction of ionic strength using the Davies equation, the formation constants of CaPuO2(CO3)32− and MgPuO2(CO3)32− from PuO2(CO3)34− are determined to be logK° = 6.05 ± 0.50 and 4.29 ± 0.18, respectively.
The prediction of the environmental behaviour of plutonium based on the geochemical calculation considering MPuO2(CO3)32− suggests that CaPuO2(CO3)32− would be the most predominant Pu(VI) species and increase Pu(VI) solubility in the natural waters. It may also affect the redox property of plutonium and stabilize Pu in the hexavalent state by strong complexation. Ternary Mg2+/Ca2+–PuO2(CO3)34− complexes can enhance Pu migration in aqueous systems under oxidizing conditions such as groundwater and seawater containing abundant calcium, magnesium, and carbonate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Nuclear R&D Program of the National Research Foundation of Korea funded by the Korean Ministry of Science and ICT (grant codes: 2016M2B2B1945252 and 2017M2A8A5014719).Notes and references
- E. P. Hardy, P. W. Krey and H. L. Volchok, Nature, 1973, 241, 444–445 CrossRef CAS.
- D. M. Nelson and M. B. Lovett, Nature, 1978, 276, 599–601 CrossRef CAS.
- K. J. Cantrell and A. R. Felmy, Plutonium and americium geochemistry at Hanford: a site wide review, Report PNNL-21651, Pacific Northwest National Lab. (PNNL), Richland, WA (United States), 2012 Search PubMed.
- D. L. Clark, The chemical complexities of plutonium, NM, 2000 Search PubMed.
- A. E. Hixon and B. A. Powell, Environ. Sci.: Processes Impacts, 2018, 20, 1306–1322 RSC.
- R. Fukai, A. Yamato, M. Thein and H. Bilinski, Geochem. J., 1987, 21, 51–57 CrossRef CAS.
- G. R. Choppin and A. Morgenstern, in Radioactivity in the Environment, ed. A. Kudo, Elsevier, Amsterdam, 2001, vol. 1, pp. 91–105 Search PubMed.
- G. R. Choppin and P. J. Wong, Aquat. Geochem., 1998, 4, 77–101 CrossRef CAS.
- R. P. Larsen and R. D. Oldham, Science, 1978, 201, 1008–1009 CrossRef CAS PubMed.
- I. Pashalidis, J. I. Kim, C. Lierse and J. C. Sullivan, Radiochim. Acta, 1993, 60, 99 CAS.
- K. Maher, J. R. Bargar and G. E. Brown, Inorg. Chem., 2013, 52, 3510–3532 CrossRef CAS PubMed.
- S. D. Reilly, W. Runde and M. P. Neu, Geochim. Cosmochim. Acta, 2007, 71, 2672–2679 CrossRef CAS.
- R. Guillaumont, T. Fanghänel, J. Fuger, I. Grenthe, V. Neck, D. A. Palmer and M. H. Rand, Chemical thermodynamics, in Update on the chemical thermodynamics of uranium, neptunium, plutonium, americium and technetium, Elsevier, Amsterdam, 2003, vol. 5 Search PubMed.
- G. Bernhard, G. Geipel, V. Brendler and H. Nitsche, Radiochim. Acta, 1996, 74, 87–91 CAS.
- S. N. Kalmykov and G. R. Choppin, Radiochim. Acta, 2000, 88, 603–606 CAS.
- G. Bernhard, G. Geipel, T. Reich, V. Brendler, S. Amayri and H. Nitsche, Radiochim. Acta, 2001, 89, 511–518 CAS.
- W. Dong and S. C. Brooks, Environ. Sci. Technol., 2006, 40, 4689–4695 CrossRef CAS PubMed.
- W. Dong and S. C. Brooks, Environ. Sci. Technol., 2008, 42, 1979–1983 CrossRef CAS PubMed.
- G. Geipel, S. Amayri and G. Bernhard, Spectrochim. Acta, Part A, 2008, 71, 53–58 CrossRef CAS PubMed.
- J.-Y. Lee and J.-I. Yun, Dalton Trans., 2013, 42, 9862–9869 RSC.
- F. Endrizzi and L. Rao, Chem. – Eur. J., 2014, 20, 14499–14506 CrossRef CAS PubMed.
- J.-Y. Lee, M. Vespa, X. Gaona, K. Dardenne, J. Rothe, T. Rabung, M. Altmaier and J.-I. Yun, Radiochim. Acta, 2017, 105, 171–185 CAS.
- Y. Jo, A. Kirishima, S. Kimuro, H.-K. Kim and J.-I. Yun, Dalton Trans., 2019, 48, 6942–6950 RSC.
- Y. Jo, H.-K. Kim and J.-I. Yun, Dalton Trans., 2019, 48, 14769–14776 RSC.
- C. Shang and P. E. Reiller, Dalton Trans., 2020, 49, 466–481 RSC.
- Z. Zheng, T. K. Tokunaga and J. Wan, Environ. Sci. Technol., 2003, 37, 5603–5608 CrossRef CAS PubMed.
- P. M. Fox, J. A. Davis and J. M. Zachara, Geochim. Cosmochim. Acta, 2006, 70, 1379–1387 CrossRef CAS.
- B. D. Stewart, M. A. Mayes and S. Fendorf, Environ. Sci. Technol., 2010, 44, 928–934 CrossRef CAS PubMed.
- S. C. Brooks, J. K. Fredrickson, S. L. Carroll, D. W. Kennedy, J. M. Zachara, A. E. Plymale, S. D. Kelly, K. M. Kemner and S. Fendorf, Environ. Sci. Technol., 2003, 37, 1850–1858 CrossRef CAS PubMed.
- J. Wan, T. K. Tokunaga, E. Brodie, Z. Wang, Z. Zheng, D. Herman, T. C. Hazen, M. K. Firestone and S. R. Sutton, Environ. Sci. Technol., 2005, 39, 6162–6169 CrossRef CAS PubMed.
- B. D. Stewart, P. S. Nico and S. Fendorf, Environ. Sci. Technol., 2009, 43, 4922–4927 CrossRef CAS PubMed.
- C. Liu, J. M. Zachara, L. Zhong, S. M. Heald, Z. Wang, B.-H. Jeon and J. K. Fredrickson, Environ. Sci. Technol., 2009, 43, 4928–4933 CrossRef CAS PubMed.
- E. L. Tullborg, J. Suksi, G. Geipel, L. Krall, L. Auqué, M. Gimeno and I. Puigdomenech, Procedia Earth Planet. Sci., 2017, 17, 440–443 CrossRef.
- B. Reeves, M. R. Beccia, P. L. Solari, D. E. Smiles, D. K. Shuh, C. Berthomieu, D. Marcellin, N. Bremond, L. Mangialajo, S. Pagnotta, M. Monfort, C. Moulin and C. Den Auwer, Environ. Sci. Technol., 2019, 53, 7974–7983 CrossRef CAS PubMed.
- H.-R. Cho, Y.-S. Youn, E. C. Jung and W. Cha, Dalton Trans., 2016, 45, 19449–19457 RSC.
- E. C. Jung and H.-R. Cho, in The delivery of nanoparticles, ed. A. A. Hashim, InTech, Rijeka, 2012 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS.
- J. W. Ball and D. K. Nordstrom, WATEQ4F – User's manual with revised thermodynamic data base and test cases for calculating speciation of major, trace and redox elements in natural waters, U.S. Geological Survey, 1991 Search PubMed.
- L. Ciavatta, Ann. Chim., 1980, 70, 551–567 CAS.
- I. Grenthe, F. Mompean, K. Spahiu and H. Wanner, TDB-2: guidelines for the extrapolation to zero ionic strength, OECD Nuclear Energy Agency, Issy-les-Moulineaux, 2013 Search PubMed.
- J.-C. Alexandre, N. Dacheux and J. Aupiais, Radiochim. Acta, 2018, 106, 801 CAS.
- T. Thoenen, W. Hummel, U. Berner and E. Curti, The PSI/Nagra chemical thermodynamic database 12/07, Paul Scherrer Institut, Villigen PSI, Switzerland, 2014 Search PubMed.
- C. W. Davies, Ion association, Butterworths, Washington D.C., 1962 Search PubMed.
- D. L. Parkhurst and C. Appelo, Description of input and examples for PHREEQC version 3: a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, U.S. Geological Survey, 2013 Search PubMed.
- J. S. Martinez, E.-F. Santillan, M. Bossant, D. Costa and M.-E. Ragoussi, Appl. Geochem., 2019, 107, 159–170 CrossRef CAS.
- D. G. Kinniburgh and D. M. Cooper, PhreePlot-Creating graphical output with PHREEQC Search PubMed, http://www.phreeplot.org/, version updated 4 May 2020.
- P. G. Varlashkin, G. M. Begun and J. R. Peterson, Radiochim. Acta, 1984, 35, 211 CAS.
- D. T. Reed, Radiochim. Acta, 1994, 66–67, 95 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/D0DT01982H |
| This journal is © The Royal Society of Chemistry 2020 |