
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono and dinuclear iridium(III) complexes featuring bis-tridentate coordination and Schiff-base bridging ligands: the beneficial effect of a second metal ion on luminescence†
Emma V.
Puttock
 ,
Amit
Sil
,
Dmitry S.
Yufit
and
J. A. Gareth
Williams
*
,
Amit
Sil
,
Dmitry S.
Yufit
and
J. A. Gareth
Williams
*
Department of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 20th July 2020
Abstract
The synthesis and photophysical properties of a set of iridium(III) complexes featuring tridentate N^N^O-coordinating ligands are described, of generic structure [Ir(N^C^N-dpyx)(N^N^O-Ln)]+ (n = 1 to 4) (dpyx = 1,3-dipyridyl-4,6-dimethylbenzene). The proligands HLn are Schiff bases synthesised by condensation of salicylaldehydes with N-methyl-hydrazinopyridines: they are able to coordinate to the Ir(III) via lateral pyridine-N and phenolate-O− atoms and a central hydrazone-N atom; the four examples differ in the substitution pattern within the phenolate ring. The bis-tridentate coordination is confirmed by X-ray diffraction. The complexes are phosphorescent in solution at ambient temperature, with higher quantum yields and longer lifetimes than those of structurally related bis-cyclometallated complexes with an N^N^C-coordinating ligand. Related proligands H2L5 and H2L6 have been prepared from 4,6-bis(1-methyl-hydrazino)pyrimidine. They feature a central pyrimidine and two N^N^O units. They are shown to bind as ditopic, bis-tridentate ligands with two iridium(III) ions, leading to unprecedented dinuclear complexes of the form [{Ir(N^C^N)}2(O^N^N–N^N^O-Ln)]2+ (n = 5, 6; N^C^N = dpyx or 1,3-dipyridyl-4,6-difluoro-benzene), with an intramolecular Ir⋯Ir distance of around 6 Å determined crystallographically. Mononuclear analogues [Ir(N^C^N-dpyx)(N^N^O-HLn)]+ have also been isolated. The dinuclear complexes display a well-defined and unusually intense lowest-energy absorption band in the visible region, around 480 nm. They emit much more efficiently than their mononuclear counterparts, even though the emission wavelengths are comparable. Their superior performance appears to be due to an enhancement in the radiative rate constant, affirming conclusions drawn from recent related studies of dinuclear Ir(III) and Pt(II) complexes with ditopic, pyrimidine-based cyclometallating ligands.
Introduction
Interest in the light-emitting properties of iridium(III) complexes continues to be intense. Their large-scale use as triplet-emitting materials in organic light-emitting diodes (OLEDs)1 has been a catalyst for activity in other applications, for example, in light-emitting electrochemical cells (LEECs),2 for light-to-chemical/electrical energy conversion,3 photocatalysis,4 and as emitters in bio-imaging.5 Though the bulk of research has focused on mononuclear complexes, it is increasingly recognised that multinuclear complexes have much to offer.6,7 In terms of photophysical properties, multinuclear iridium structures tend to fall into one of two categories. In one case, the constituent units retain, to a large extent, properties that are similar to those of the isolated units: the bridging unit plays a relatively minor role other than that of bringing the units together. A “supramolecular” description is appropriate: the absorption profile of the assembly may be quite similar to the summation of the individual components. Examples include several compounds that make use of para-phenylene-bridged bis-bipyridines to link two Ir(ppy)2 units,8,9 together with related systems that feature tridentate ligands.10 On the other hand, the photophysical properties of the assembly may be fundamentally different from those of related mononuclear complexes: there may be very large effects on the singlet and triplet excited state energies. For example, we have shown how 4,6-diphenylpyrimidine can act as a bis-N^C-bridging ligand to form multinuclear complexes that are characterised by much lower absorption and emission energies than their mononuclear counterparts.11 Moreover, the radiative rate constants of the formally forbidden T1 → S0 process are greatly increased through more efficient spin–orbit coupling (SOC), leading to impressive quantum yields, even in the deep red region of the spectrum. Recent review articles collate other examples,6,7 and the beneficial effect on SOC extends to related dinuclear complexes of Pt(II)12,13 and to heterometallic assemblies featuring, for example, a combination of Ir(III) and Pt(II) centres.14Most multinuclear systems comprise metal complex units in which the metal ions are bound to bidentate ligands, e.g., of the form Ir(L^L)3. Since such units are normally racemic mixtures of Λ and Δ isomers, the resulting compounds are necessarily formed as mixtures of diastereoisomers that can be very difficult to separate (e.g., ΛΛ/ΔΔ + ΛΔ in the case of two metal centres). Tridentate ligands may be advantageous in this respect, since metal complexes of meridionally-coordinating tridentate ligands are normally achiral – e.g., complexes of the form Ir(L^L^L)2 – and hence give only a single product when incorporated into a multinuclear assembly.10,15 We have previously made use of N^C^N-coordinating ligands based on 1,3-dipyridylbenzene (dpyb) to access a diverse range of mononuclear iridium complexes containing one such ligand in conjunction with other bidentate or tridentate ligands, e.g., of the form Ir(N^C^N)(C^N^C), [Ir(N^C^N)(N^N)X]+, etc. (Fig. 1a, compounds 1, 3–6).16,17 Related ligands based on 1,3-bis(benzimidazolyl)benzene have been used in a similar way by Haga and colleagues, readily binding to Ir(III) in an N^C^N manner.18 Chi and co-workers have used the dpyb ligand platform, in conjunction with a second pyrazolyl-based tridentate ligand, to generate brightly emissive complexes (e.g., 2 in Fig. 1a)19 and have achieved even more impressive results with related carbene analogues (monoanionic :C^C–^C: coordination, with carbene units in place of the two pyridine rings).20N^C^N-coordinating units have been incorporated into multinuclear assemblies such as 7, 8 and 9 in Fig. 1. Whilst the behaviour of 7 can be rationalised using the supramolecular description (showing energy transfer from the Ir(III) units through to the lowest-energy Ru(II) unit),10b pyrimidine-bridged systems 8 and 9 and related derivatives display unusually efficient red emission, with luminescence quantum yields approaching unity in some cases.11
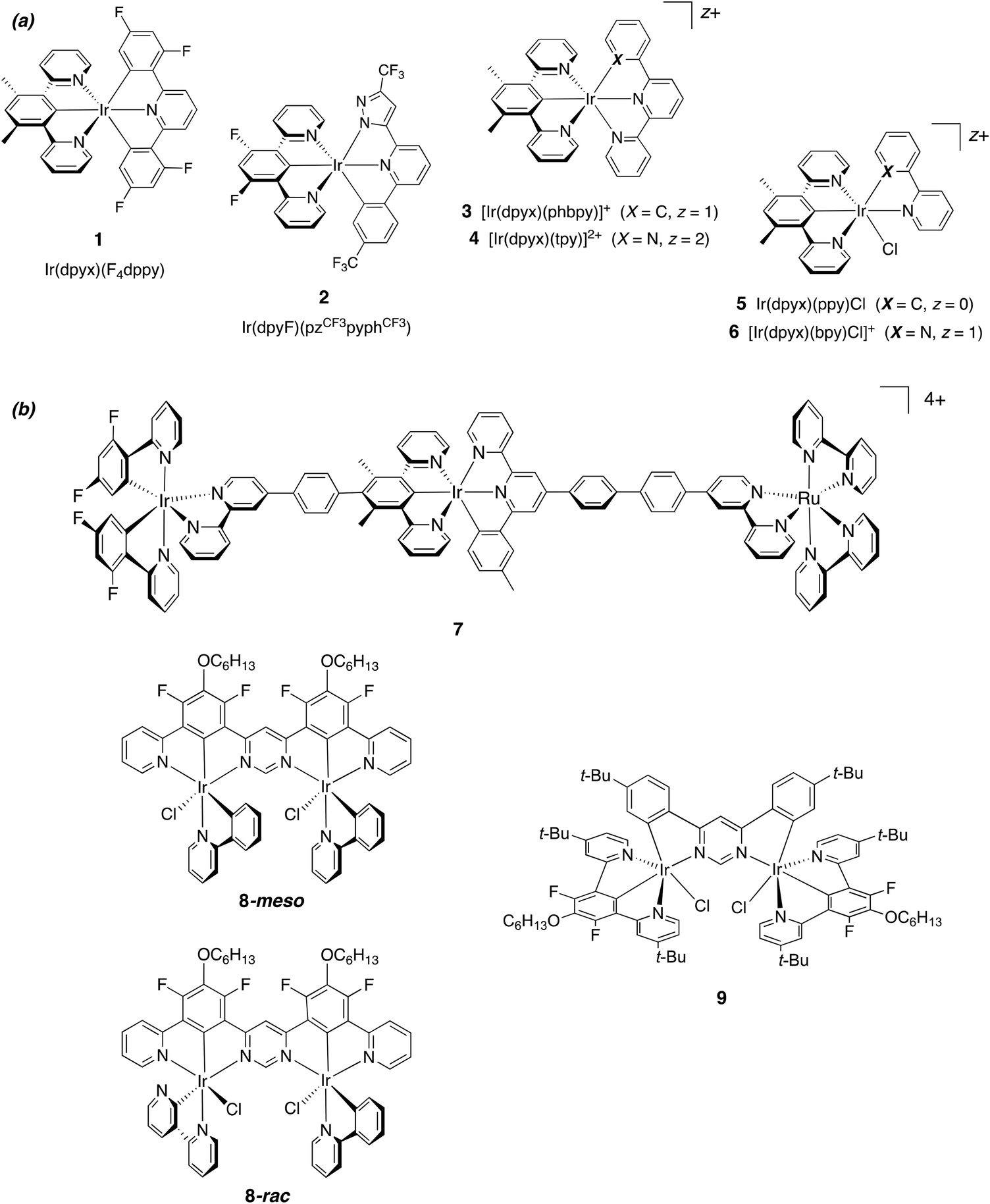 | ||
| Fig. 1 (a) Representative mononuclear Ir(III) complexes (charge-neutral and cationic) containing N^C^N-coordinating 1,3-dipyridylbenzene derivatives.16–20 (b) Examples of multinuclear complexes reported to date that feature N^C^N-coordinated Ir(III) centres. Trinuclear complex 7 behaves as a supramolecular system with energy transfer occurring between the constituent units.10b Dinuclear complexes 8 (which exists as separable meso and rac isomers) and 9 (achiral) are efficient deep-red emitters.11 | ||
The work described here had two objectives. The first was to attempt to prepare a new class of dpyb-based iridium(III) complexes, of the form [Ir(N^C^N)(N^N^O)]+, incorporating hydrazone-based N^N^O-coordinating ligands. Recently, we showed that proligands based on N-methyl-N-(2-pyridyl)-N′-(salicylidene)hydrazone – readily prepared from low-cost salicylaldehydes and 2-hydrazino-pyridines by simple Schiff-base condensation reactions – can be coordinated to Pt(II) to generate compounds of the form [Pt(N^N^O)Cl].21 These complexes are phosphorescent in solution under ambient conditions, and emission is further enhanced by metathesis of the monodentate halide to an acetylide, [Pt(N^N^O)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ar)]. It was of interest to examine whether such ligands could be used successfully with iridium(III), in combination with N^C^N-coordinating ligands, to generate new emissive materials. Secondly, we reasoned that the pyridylhydrazone structure of these ligands would lend itself well to the facile synthesis of ditopic, O^N^N–N^N^O-coordinating ligands, through the use of a pyrimidine unit in place of pyridine.22 Such ligands should be well set-up for bis-tridentate coordination to two Ir(N^C^N) units, by analogy with related cyclometallating pyrimidine and pyrazine-based systems.11,12 We report here the synthesis of several mononuclear and binuclear iridium(III) complexes with N^N^O and O^N^N–N^N^O-coordinating ligands respectively, together with the crystal structures of representative examples, and an evaluation of their photophysical properties. Interestingly, we find that the dinuclear complexes are much more strongly luminescent than their direct mononuclear counterparts, due to higher radiative rate constants and reinforcing an emerging picture as to the potential influence of a second metal ion on spin–orbit coupling.
C–Ar)]. It was of interest to examine whether such ligands could be used successfully with iridium(III), in combination with N^C^N-coordinating ligands, to generate new emissive materials. Secondly, we reasoned that the pyridylhydrazone structure of these ligands would lend itself well to the facile synthesis of ditopic, O^N^N–N^N^O-coordinating ligands, through the use of a pyrimidine unit in place of pyridine.22 Such ligands should be well set-up for bis-tridentate coordination to two Ir(N^C^N) units, by analogy with related cyclometallating pyrimidine and pyrazine-based systems.11,12 We report here the synthesis of several mononuclear and binuclear iridium(III) complexes with N^N^O and O^N^N–N^N^O-coordinating ligands respectively, together with the crystal structures of representative examples, and an evaluation of their photophysical properties. Interestingly, we find that the dinuclear complexes are much more strongly luminescent than their direct mononuclear counterparts, due to higher radiative rate constants and reinforcing an emerging picture as to the potential influence of a second metal ion on spin–orbit coupling.
Results and discussion
Proligand design and synthesis
A number of metal complexes of N-(2-pyridyl)-N′-(salicylidene)hydrazone have been reported with 1st row transition metals, e.g. with Co2+, Co3+, [V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]2+, Zn2+ and Cd2+.23 In our previous work investigating Pt(II) complexes of this ligand, and derivatives with substituents in the aromatic rings, we found that the resulting Pt(N^N^O)Cl complexes had poor stability, displaying quite rapid light-induced decomposition in solution, compromising the emission properties and rendering them difficult to assess.21 The instability was apparently associated with the facile deprotonation of the hydrazone unit. In contrast, complexes of the corresponding N-methylated ligands were robust, with no evidence of decomposition. In the present study, we therefore confined our attention to such N-methylated ligands. The parent proligand HL1 and three derivatives HL2–4 incorporating substituents in the phenolic ring were prepared from N-methyl-hydrazinopyridine and the corresponding salicylaldehyde as shown in Scheme 1a. Meanwhile, the synthesis of the ditopic, O^N^N–N^N^O proligands H2L5–7 was achieved by condensation of salicylaldehydes with 4,6-bis(1-methyl-hydrazino)pyrimidine, in turn prepared from 4,6-dichloropyrimidine and N-methyl-hydrazine (Scheme 2a). The proligands were obtained as fine white powders in high yield over the two steps, and characterised by 1H and 13C NMR spectroscopy, mass spectrometry and, for H2L5 and H2L7, by X-ray diffraction.
O]2+, Zn2+ and Cd2+.23 In our previous work investigating Pt(II) complexes of this ligand, and derivatives with substituents in the aromatic rings, we found that the resulting Pt(N^N^O)Cl complexes had poor stability, displaying quite rapid light-induced decomposition in solution, compromising the emission properties and rendering them difficult to assess.21 The instability was apparently associated with the facile deprotonation of the hydrazone unit. In contrast, complexes of the corresponding N-methylated ligands were robust, with no evidence of decomposition. In the present study, we therefore confined our attention to such N-methylated ligands. The parent proligand HL1 and three derivatives HL2–4 incorporating substituents in the phenolic ring were prepared from N-methyl-hydrazinopyridine and the corresponding salicylaldehyde as shown in Scheme 1a. Meanwhile, the synthesis of the ditopic, O^N^N–N^N^O proligands H2L5–7 was achieved by condensation of salicylaldehydes with 4,6-bis(1-methyl-hydrazino)pyrimidine, in turn prepared from 4,6-dichloropyrimidine and N-methyl-hydrazine (Scheme 2a). The proligands were obtained as fine white powders in high yield over the two steps, and characterised by 1H and 13C NMR spectroscopy, mass spectrometry and, for H2L5 and H2L7, by X-ray diffraction.
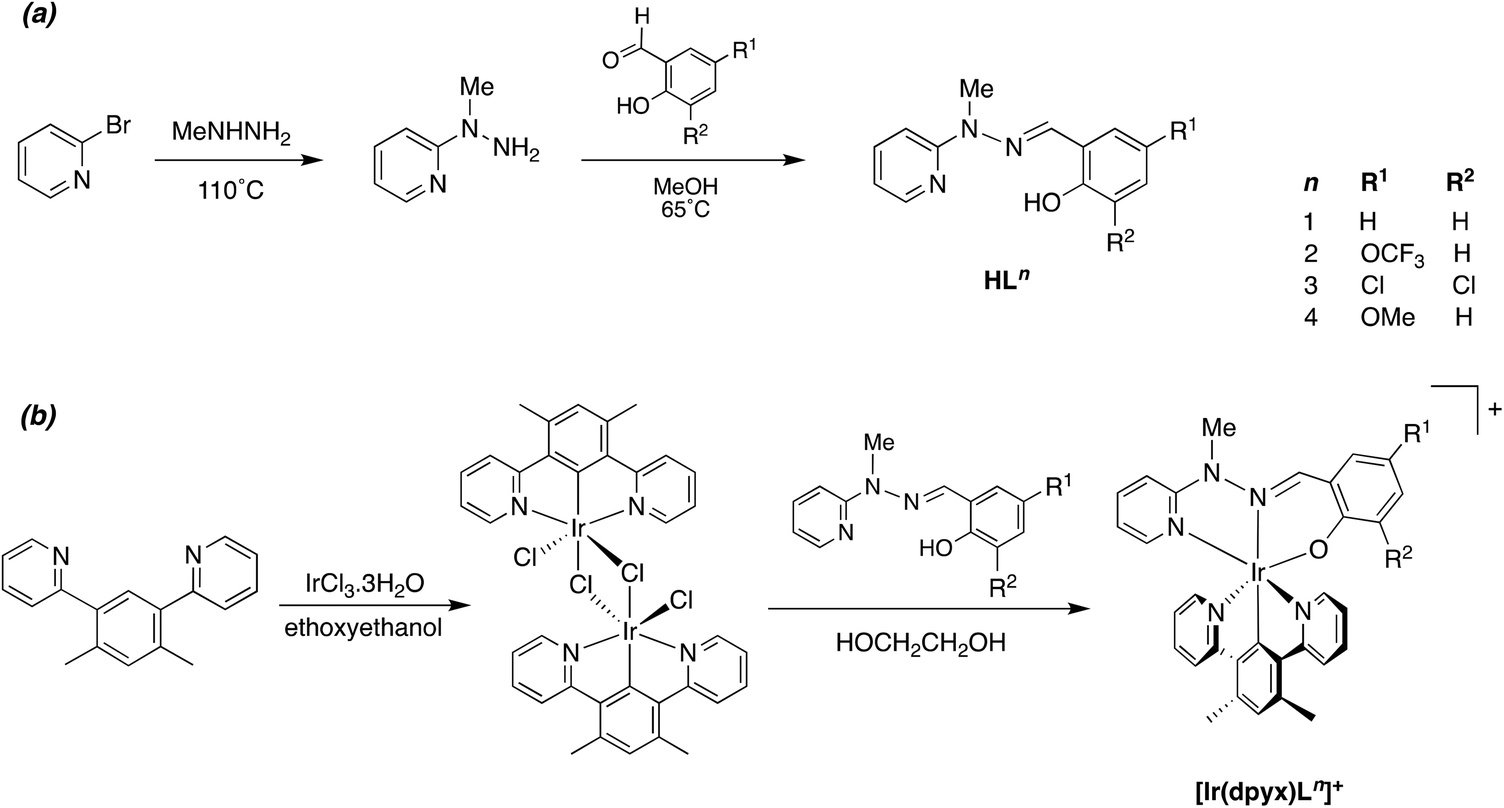 | ||
| Scheme 1 Synthesis of (a) the N^N^O proligands and (b) their mononuclear iridium(III) complexes described in this work. | ||
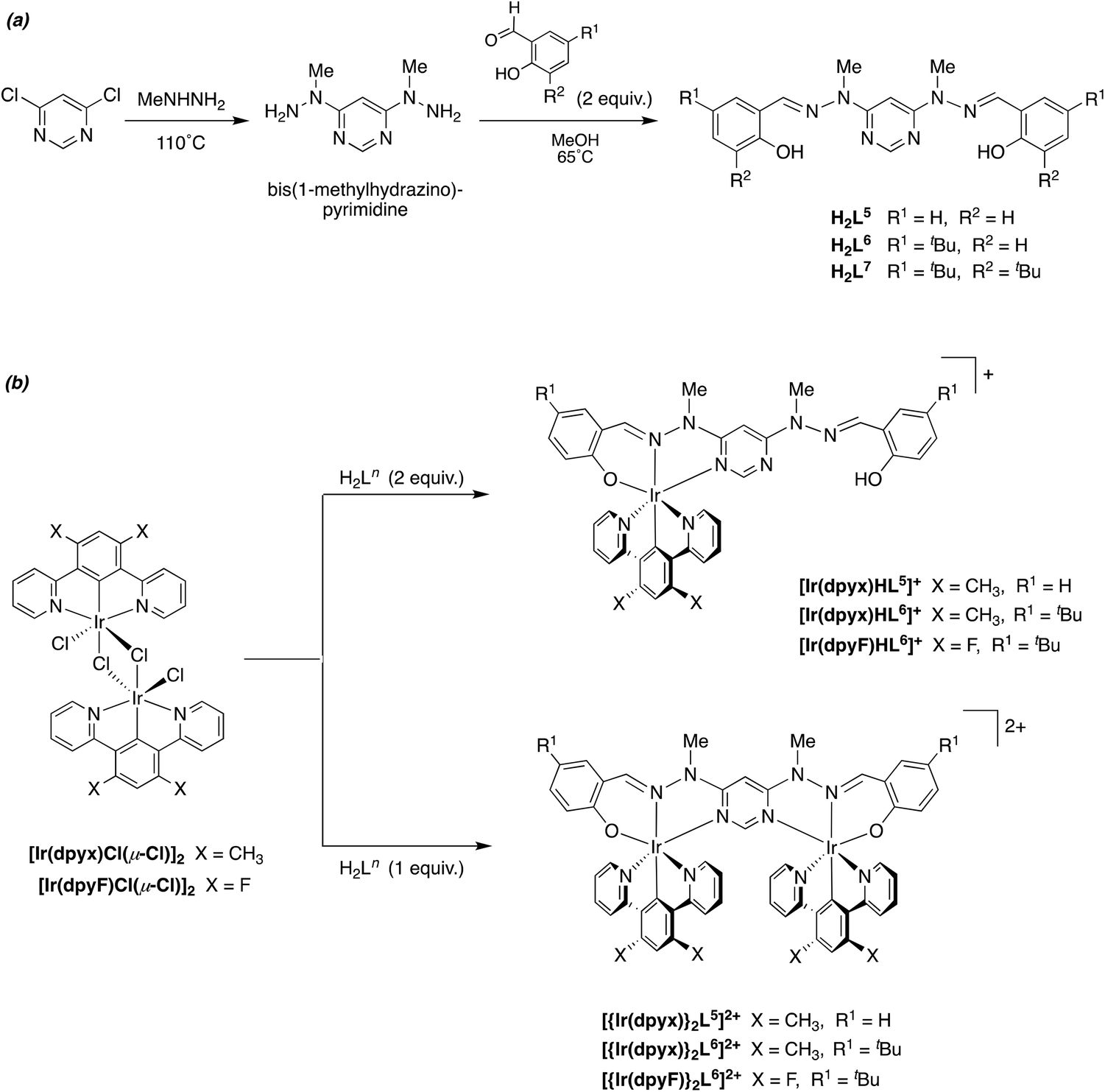 | ||
| Scheme 2 Synthesis of (a) the ditopic, O^N^N–N^N^O proligands and (b) their mono- and dinuclear iridium(III) complexes. | ||
Complexation to iridium
It has previously been shown that 1,3-dipyridylbenzene itself binds to iridium(III) primarily through bidentate N^C4 as opposed to tridentate N^C2^N coordination, but the latter binding mode can be attained by incorporating non-hydrogenic substituents at the 4,6-positions of the central ring (e.g., CH3, F, CF3).16–18 In the present instance, we focused on the xylene derivative incorporating methyl groups, 1,3-di(pyridyl)-4,6-dimethylbenzene (dpyxH), which reacts with hydrated iridium(III) chloride to generate the chloro-bridged dimer [Ir(N^C^N-dpyx)Cl(μ-Cl)]2. Reaction of this compound with proligands HL1–4 in ethylene glycol at 190 °C for 90 min gave the desired mononuclear complexes [Ir(N^C^N-dpyx)(N^N^O-L1–4)]+ as their chloride salts, which were subsequently metathesised to the corresponding hexafluorophosphate salts by precipitation from KPF6 (aq), and purified by recrystallisation from acetonitrile/diethyl ether (Scheme 1b). The identity of the complexes was confirmed by 1H and 13C NMR spectroscopy, mass spectrometry and, for [Ir(dpyx)L4]PF6, by X-ray diffraction (see below).The ditopic, bis-tridentate proligands H2L5–6 were reacted with [Ir(dpyx)Cl(μ-Cl)]2 under similar conditions (ethylene glycol at 190 °C for 90 minutes), and the identity of the main products was determined by the stoichiometry used. The use of two equivalents of the proligand relative to the iridium dimer gave the mononuclear complexes [Ir(dpyx)HL5–6]+ as the predominant product, whilst a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of materials gave primarily the dinuclear complexes [{Ir(dpyx)}2L5–6]2+. In each case, the initially formed chloride salts were metathesised to the hexafluorophosphates, and the products were purified by column chromatography, followed by recrystallisation from acetonitrile/ether. In the case of the bis(t-butyl) derivative H2L7, the mononuclear complex [Ir(dpyx)HL7]+ was successfully obtained (see ESI†), but we were unable to isolate its dinuclear complex in sufficient purity for photophysical study. Mono- and binuclear complexes incorporating 1,3-dipyridyl-4,6-difluorobenzene (dpyF, the bis-fluoro analogue of dpyx) were also synthesised for the tert-butyl bridging ligand; i.e., [Ir(dpyF)HL5–6]PF6 and [{Ir(dpyF)}2L5–6](PF6)2 (Scheme 2b). The identity of all six complexes was confirmed by 1H and 13C NMR spectroscopy, mass spectrometry and, for the three dinuclear complexes, by X-ray diffraction (see below).
:1 ratio of materials gave primarily the dinuclear complexes [{Ir(dpyx)}2L5–6]2+. In each case, the initially formed chloride salts were metathesised to the hexafluorophosphates, and the products were purified by column chromatography, followed by recrystallisation from acetonitrile/ether. In the case of the bis(t-butyl) derivative H2L7, the mononuclear complex [Ir(dpyx)HL7]+ was successfully obtained (see ESI†), but we were unable to isolate its dinuclear complex in sufficient purity for photophysical study. Mono- and binuclear complexes incorporating 1,3-dipyridyl-4,6-difluorobenzene (dpyF, the bis-fluoro analogue of dpyx) were also synthesised for the tert-butyl bridging ligand; i.e., [Ir(dpyF)HL5–6]PF6 and [{Ir(dpyF)}2L5–6](PF6)2 (Scheme 2b). The identity of all six complexes was confirmed by 1H and 13C NMR spectroscopy, mass spectrometry and, for the three dinuclear complexes, by X-ray diffraction (see below).
Structural characterisation
Crystals of H2L5 and H2L7 suitable for X-ray diffraction analysis were obtained by slow evaporation of chloroform solutions. The structures confirm the identities of the compounds and show the O–H⋯N intramolecular hydrogen bonding typical of ortho-iminophenols (Fig. 2 and Fig. S1 in the ESI†). The H2L5 molecule is located on a 2-fold axis. The molecules adopt a spiral conformation with the hydrazone units on opposite sides of the pyrimidine ring, reminiscent of structures reported by Lehn and co-workers for oligomeric hydrazone-pyrimidine.24 The dihedral angle between pyrimidine and phenyl cycles is larger in H2L7 than H2L5 {30.7° (av.) versus 13.29(5)°}, evidently as a result of the steric effect of the t-butyl substituents in the former. | ||
| Fig. 2 The molecular structures of H2L5 (left) and H2L7 (right), viewed from the face of the molecules (top) and from the side (bottom). The packing of the molecules in the two crystals is shown in Fig. S1 in the ESI.† | ||
Small crystals, suitable for X-ray diffraction, of the mononuclear iridium complex [Ir(dpyx)L4]PF6 were obtained by slow evaporation of the solvent from an acetonitrile solution; the crystal contains one molecule of MeCN per molecule of the complex. The molecular structure in the crystal is shown in Fig. 3, with key bond lengths and angles listed in the caption. The desired, bis-tridentate Ir(N^C^N)(N^N^O) coordination is evident: the Ir(III) centre exhibits a distorted pseudo-octahedral geometry. The dpyx ligand has an N–Ir–N bite angle of 161.42(6)°, quite typical of tridentate ligands like terpyridine that form two 5-membered chelate rings. The corresponding trans bite angle of 171.71(5)° in the N^N^O-coordinating ligand is significantly larger, reflecting the fact that one of the two chelate rings formed is six-membered in that case.25 The Ir–C and Ir–Npy bond lengths are similar to those of bis-terdentate complexes incorporating dpyx and terpyridine-based ligands.16 On the other hand, for the N^N^O ligand, the central Ir–Nhydrazone bond is significantly longer than the Ir–Npy bond {2.078(1) and 2.019(1) Å respectively}; this contrasts with terpyridine complexes where the M–N bond to the central pyridine is normally shorter than those to the lateral pyridines, owing to the constraints of the bite angle.26 The shortest intermolecular Ir⋯Ir contact in the crystal is 8.8037(3) Å.
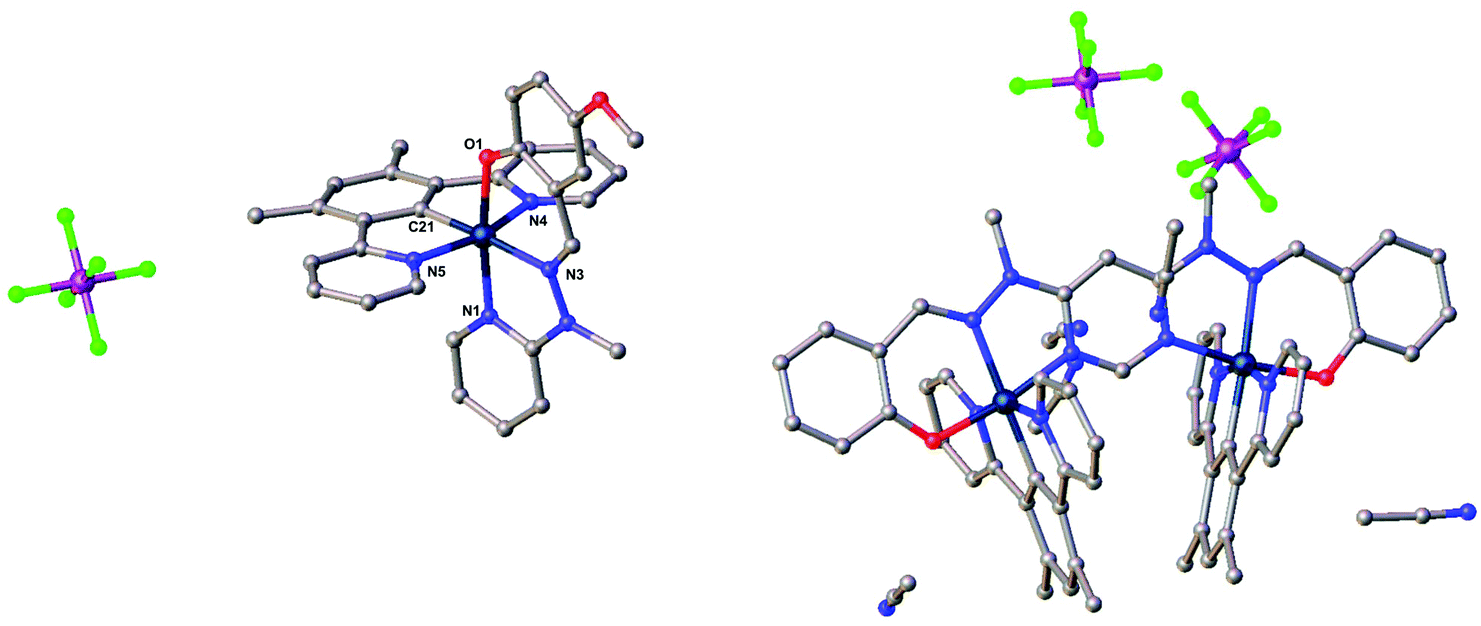 | ||
| Fig. 3 Left: The molecular structure of the mononuclear complex [Ir(dpyx)L4]PF6. Key bond lengths (Å) and angles (°): Ir–C21 1.948(2); Ir–N1 2.019(1); Ir–N3 2.078(1); Ir–N4 2.035(1); Ir–N5 2.053(1); Ir–O1 2.033(1); N4–Ir1–N5 161.42(6); N3–Ir1–O1 171.70(5); N1–Ir1–C19 175.57(6). Right: The molecular structure of the dinuclear complex [{Ir(dpyx)}2L5](PF6)2. For both structures, acetonitrile from the solvent is also present; hydrogen atoms are omitted for clarity; crystal packing diagrams are given in Fig. S2 and S3.† The structures of [{Ir(dpyx)}2L6](PF6)2 and [{Ir(dpyF)}2L6](PF6)2 are similar to [{Ir(dpyx)}2L5](PF6)2 and are shown in Fig. S4.† | ||
Crystals of the dinuclear complexes [{Ir(dpyx)}2L5](PF6)2 and [{Ir(dpyx)}2L6](PF6)2 were similarly obtained by slow evaporation of the solvent from acetonitrile solutions, whilst crystals of [{Ir(dpyF)}2L6](PF6)2 formed upon diffusion of diethyl ether into an acetonitrile solution. The crystals also contain solvent molecules (see Experimental section and ESI†). Key bond lengths and angles are given in Table S1 in the ESI.† In all three structures, both of the Ir atoms are hexacoordinated in a distorted pseudo-octahedral geometry, similar to that found in the mononuclear complex [Ir(dpyx)L4]PF6 discussed above and with a similar set of bond lengths and angles. The intermetallic Ir⋯Ir distance within the molecules is 5.9837(7), 6.0208(7) and 5.9426(7) Å respectively. The N^C^N ligands are tilted relative to one another, with dihedral angles of 33.8(1), 32.4(1) and 35.0(1)° between their average planes, respectively. It should be noted that the tilt is asymmetric: the intramolecular distances between centroids of juxtaposed pyridine rings on opposite sides of the L-ligand plane are 5.344 and 6.203 Å; 4.961 and 6.591 Å; and 4.481 and 6.494 Å respectively. In all cases a solvent acetonitrile molecule is wedged into the larger gap between the pyridine rings.
Photophysical properties of the mononuclear complexes [Ir(dpyx)L1–4]PF6
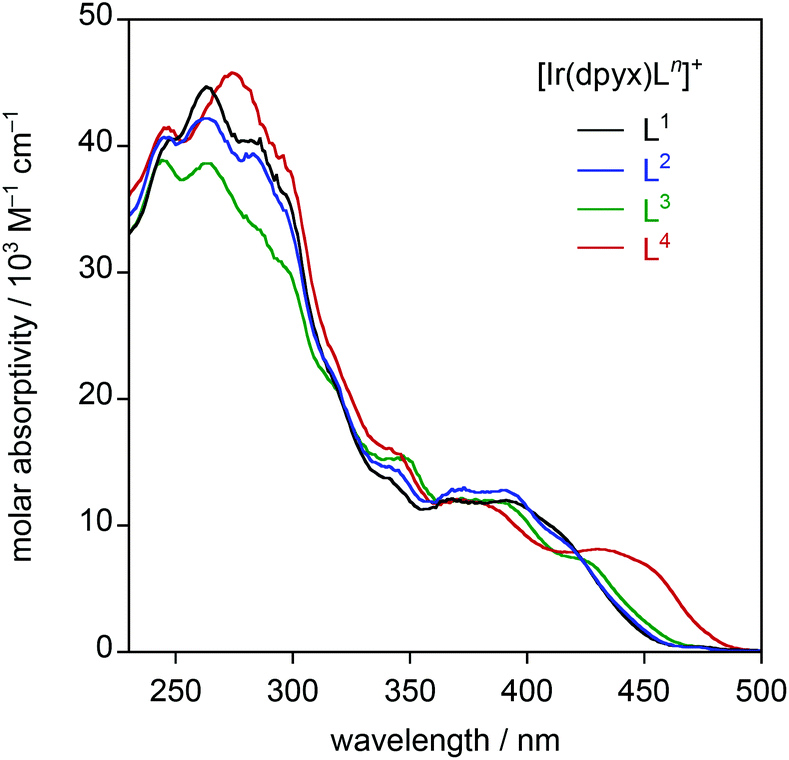 | ||
| Fig. 4 UV-visible absorption spectra of the series of mononuclear iridium(III) complexes [Ir(dpyx)Ln]PF6 in CH2Cl2 at 295 K. | ||
| Complex | Absorptiona at 295 K | Emission at 295 K | Emissionb at 77 K | ||||||
|---|---|---|---|---|---|---|---|---|---|
| λ max/nm (ε/M−1 cm−1) | λ max/nm | τ /ns | Φ lum × 102 | k r /103 s−1 | ∑knre/103 s−1 |
|
λ max/nm | τ/ns | |
|
a Maxima at λ > 230 nm are listed.
b In diethyl ether/isopentane/ethanol (2:2:1 v/v).
c Luminescence lifetimes in deoxygenated solution; values in parenthesis refer to air-equilibrated solutions.
d Luminescence quantum yield in deoxygenated solution, measured using [Ru(bpy)3]Cl2(aq) as the standard.
e
k
r and ∑knr are the radiative and non-radiative rate constants, estimated from the lifetime and quantum yield, assuming that the emitting state is formed with unitary efficiency: kr = Φlum/τ; ∑knr = (1 − Φlum)/τ.
f Bimolecular rate constant for quenching by molecular oxygen estimated from the lifetimes in deoxygenated and air-equilibrated solution, and taking [O2] = 2.1 mmol dm−3 in CH2Cl2 at atmospheric pressure of air at 295 K.
g Data from ref. 10a, with Φlum amended using Φlum = 0.040 for the standard.
|
|||||||||
| [Ir(dpyx)L1]PF6 | 246 (40200), 264 (44600), 283 (40400), 367 (12100), 391 (12000), 415sh (9240) |
560, 599 | 5400 [300] | 13 | 24 | 160 | 1.4 | 529, 576, 632 | 24000 |
| [Ir(dpyx)L2]PF6 | 246 (40600), 264 (42200), 283 (39400), 340 (14600), 372 (12800), 391 (12800), 418sh (8360) |
573, 611 | 4200 [300] | 11 | 16 | 210 | 1.4 | 537, 586, 643 | 19000 |
| [Ir(dpyx)L3]PF6 | 245 (38800), 264 (38600), 297 (30400), 317sh (21200), 344 (15300), 380 (11900), 422 (7360) |
578, 618 | 3000 [400] | 7.2 | 10 | 310 | 0.98 | 546, 596, 657 | 15000 |
| [Ir(dpyx)L4]PF6 | 246 (41400), 273 (45600), 296sh (39300), 342 (16000), 376 (11900), 430 (8140) |
614, 644 | 6200 [200] | 11 | 18 | 140 | 2.2 | 568, 620, 683 | 21000 |
| [Ir(dpyx)(phbpy)]PF6g | 240 (24500), 265 (22100), 294sh (16600), 367 (5360), 411 (5490), 479 (640) |
632 | 120 [50] | 3.3 | 270 | 8100 | 6.4 | 544, 576 | 3600 |
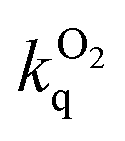
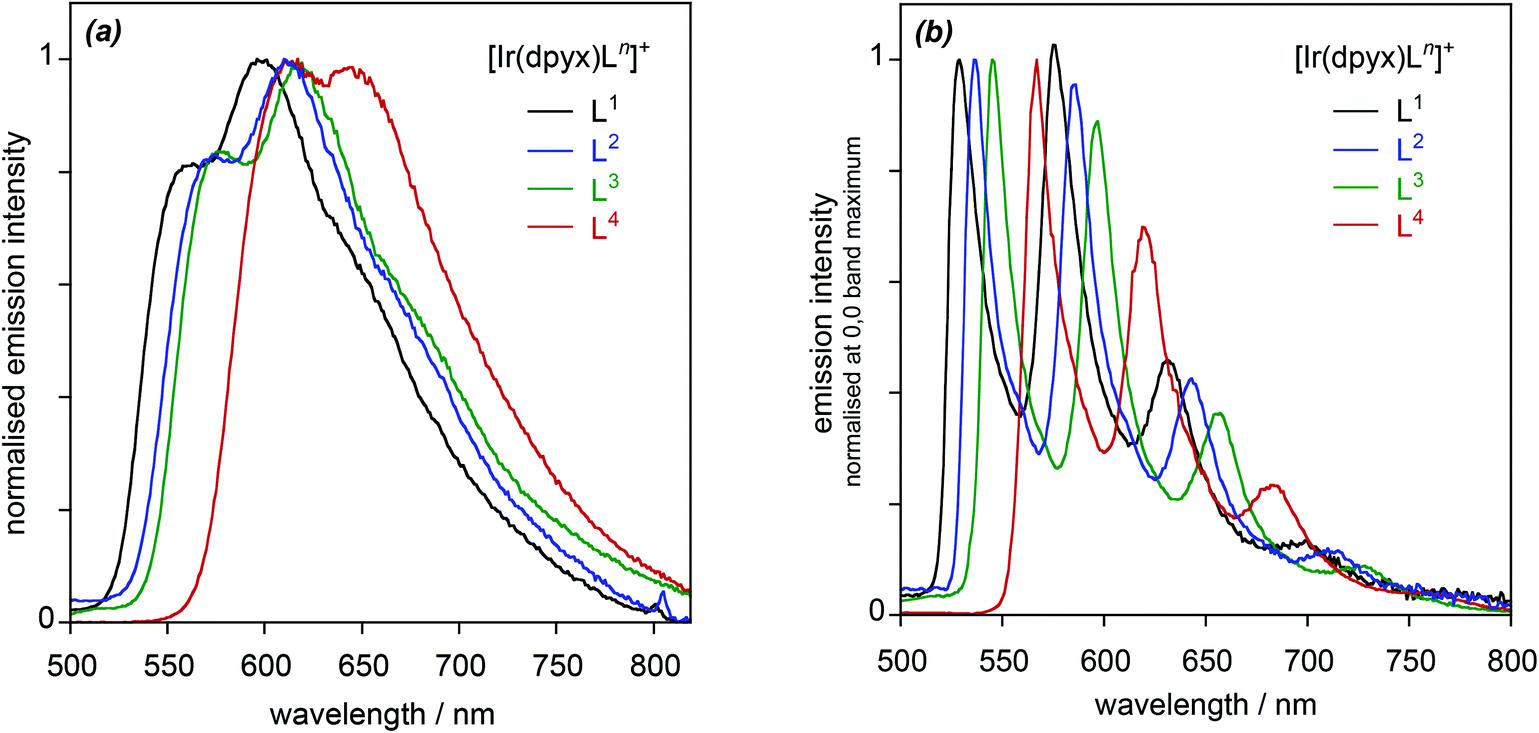 | ||
| Fig. 5 Normalised photoluminescence spectra of the series of mononuclear iridium(III) complexes [Ir(dpyx)Ln]PF6: (a) in CH2Cl2 at 295 K; (b) in EPA at 77 K. | ||
It is of interest to determine how the performance of these new complexes compares relative to previously reported monocationic systems incorporating a dipyridylbenzene ligand. A logical comparison is with [Ir(dpyx)(phbpy)]+ (phbpyH = 6-phenyl-2,2′-bipyridine; complex 3 in Fig. 1).10a It features an N^N^C-coordinating, cyclometallated ligand, as opposed to the N^N^O coordination of the new complexes. Its emission properties are listed in the final row of Table 1 for comparison. It can be seen that the new N^N^O complexes have somewhat superior quantum yields but substantially longer lifetimes. Some insight into the origins of the differences can be obtained through the estimation of the radiative kr and non-radiative ∑knr decay rate constants from the quantum yields and lifetimes {see Table 2, footnote (e)}. From the resulting data in Table 1, it is evident that kr is an order of magnitude smaller in the N^N^O complexes. This conclusion is perhaps not unexpected, given that one of the attractions of cyclometallation is the strong σ donation associated with the metallated aryl ring which favours the mixing of metal and ligand orbitals and thus enhanced spin–orbit coupling to promote the formally forbidden T1 → S0 phosphorescence process.28 Nevertheless, ∑knr is reduced by a larger factor of up to around 50, which leads to the net enhancement in quantum yields for the set of new complexes.
| Complex | Absorptiona at 298 K | Emission at 298 K | Emissionb at 77 K | ||||||
|---|---|---|---|---|---|---|---|---|---|
| λ max/nm (ε/M−1 cm−1) | λ max/nm | τ /ns | Φ lum × 102 | k r /103 s−1 | ∑knre/103 s−1 |
|
λ max/nm | τ/ns | |
|
a Maxima at λ > 230 nm are listed.
b In diethyl ether/isopentane/ethanol (2:2:1 v/v).
c Luminescence lifetimes in deoxygenated solution; values in parenthesis refer to air-equilibrated solutions.
d Luminescence quantum yield in deoxygenated solution, measured using [Ru(bpy)3]Cl2(aq) as the standard.
e
k
r and ∑knr are the radiative and non-radiative rate constants, estimated from the lifetime and quantum yield, assuming that the emitting state is formed with unitary efficiency: kr = Φlum/τ; ∑knr = (1 − Φlum)/τ.
f Bimolecular rate constant for quenching by molecular oxygen estimated from the lifetimes in deoxygenated and air-equilibrated solution, and taking [O2] = 2.1 mmol dm−3 in CH2Cl2 at atmospheric pressure of air at 295 K.
|
|||||||||
| [Ir(dpyx)HL5]PF6 | 270sh (33000), 302 (36500), 314 (36000), 335 (31900), 366 (25000), 424 (13200) |
563, 600 | 4600 [300] | 0.86 | 1.9 | 220 | 1.4 | 535, 579, 633, 696 | 18000 |
| [Ir(dpyx)HL6]PF6 | 274 (37300), 303 (39800), 320 (42000), 366 (25500), 424 (13400), 450sh (11300) |
569, 610 | 6400 [300] | 2.3 | 3.6 | 150 | 1.5 | 536, 584, 640, 704 | 15000 |
| [Ir(dpyF)HL6]PF6 | 268 (39700), 288 (34300), 318 (41400), 350 (31600), 368 (28700), 427 (14300), 444sh (13300) |
566, 603 | 4400 [180] | 0.31 | 0.72 | 230 | 2.4 | 536, 582, 637, 701 | 24000 |
| [{Ir(dpyx)}2L5](PF6)2 | 273 (36500), 290 (37700), 337 (36200), 378 (18700), 448 (11700), 476 (18400) |
564, 605 | 2100 [200] | 10 | 49 | 430 | 2.1 | 536, 586, 642, 706 | 15000 |
| [{Ir(dpyx)}2L6](PF6)2 | 290 (43100), 341 (43700), 378 (21600), 450 (12500), 483 (22900) |
574, 613 | 4000 [200] | 16 | 40 | 210 | 2.2 | 545, 596, 655, 721 | 18000 |
| [{Ir(dpyF)}2L6](PF6)2 | 275 (38400), 342 (41800), 372sh (18200), 447 (11200), 478 (22400) |
569, 611 | 6700 [220] | 17 | 24 | 120 | 2.0 | 538, 588, 646, 712 | 23000 |
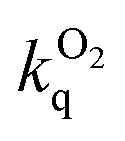
Photophysical properties of the mono- and dinuclear complexes featuring the pyrimidine-based bridging ligand
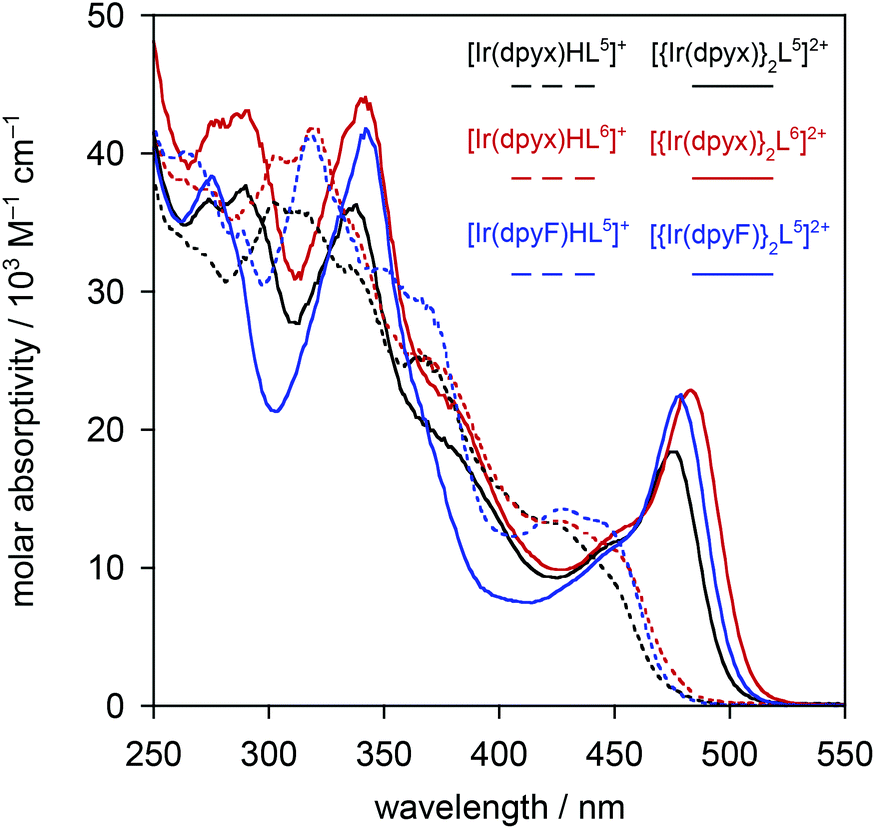 | ||
| Fig. 6 UV-visible absorption spectra of the dinuclear iridium(III) complexes [{Ir(dpyx)}2L5]+ (black), [{Ir(dpyx)}2L6]+ (red) and [{Ir(dpyF)}2L5]+ (blue) (solid lines), and of the corresponding complexes with only one iridium(III) bound (dashed lines); PF6− salts in CH2Cl2 at 295 K. | ||
The absorption spectra of the three dinuclear complexes [{Ir(dpyx)}2L5]2+, [{Ir(dpyx)}2L6]2+ and [{Ir(dpyF)}2L5]2+, are strikingly different from those of the mononuclear analogues (Fig. 6). They each show a very strong, well-defined, lowest-energy band, around 480 nm, significantly red-shifted compared to the lowest-energy absorption of the mononuclear complexes. These bands, with ε around 20000 M−1 cm−1, are unusually intense compared with those of the lowest-energy bands of most cyclometallated Ir(III) complexes (usually around 5000 M−1 cm−1). Similar observations have, however, been made recently for dinuclear Ir(III) and Pt(II) complexes with pyrimidine-based cyclometallated ligands, including 8 and 9 in Fig. 1, and we return to this point below.
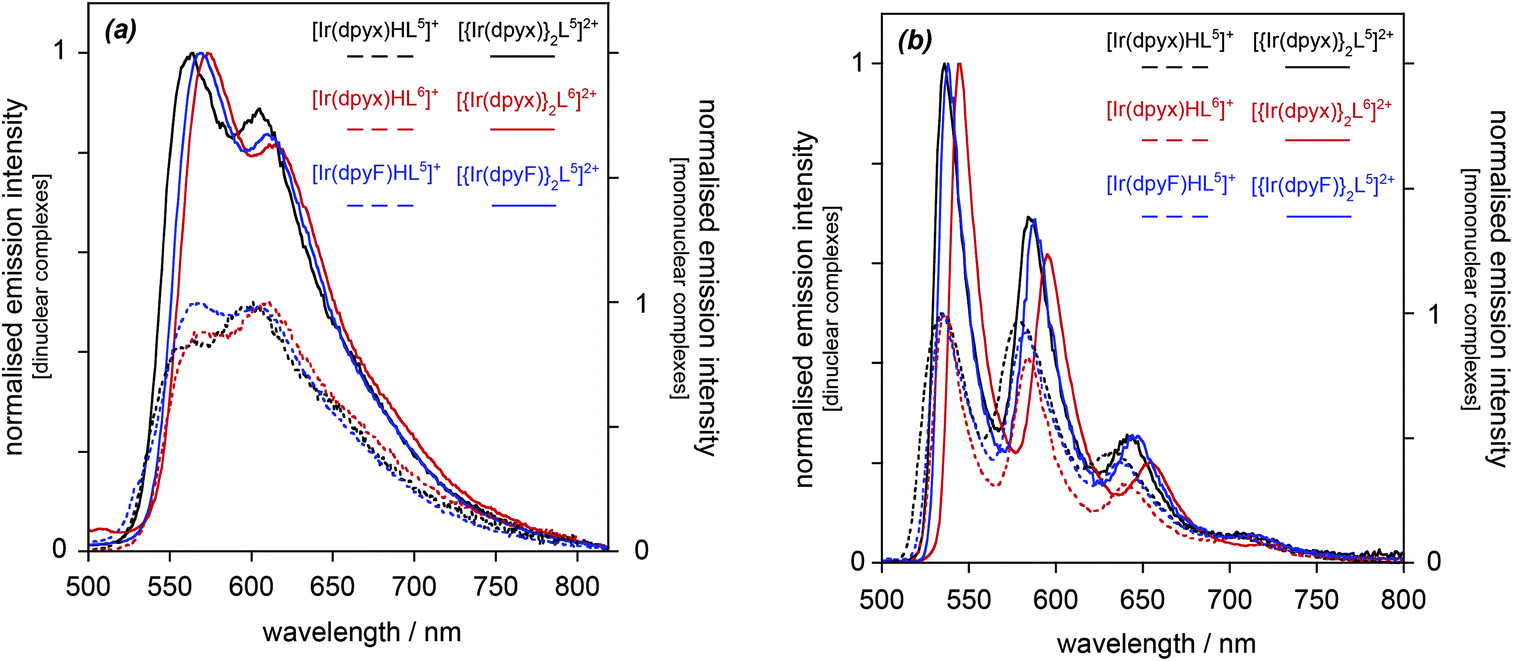 | ||
| Fig. 7 Normalised photoluminescence spectra of the dinuclear iridium(III) complexes [{Ir(dpyx)}2L5]+ (black), [{Ir(dpyx)}2L6]+ (red) and [{Ir(dpyF)}2L5]+ (blue) (solid lines), and of the corresponding complexes with one Ir(III) bound (dashed lines), PF6− salts in each case: (a) in CH2Cl2 at 295 K; (b) in EPA at 77 K. The mono- and dinuclear series are normalised separately for clarity. | ||
The order-of-magnitude higher radiative rate constants kr in the dinuclear complexes [{Ir(N^C^N)}2Ln]2+ compared to their mononuclear counterparts [Ir(N^C^N)HLn]+ is intriguing, but it mirrors the results of an increasing number of studies with cyclometallating, bridging ligands based on pyrimidines and pyrazines, such as 8 and 9 in Fig. 1. Though it may be tempting to attribute the increase to the additional spin–orbit coupling associated with a second metal ion, it is notable that the oscillator strength of the lowest-energy spin-allowed transition is also enhanced significantly, as noted above (Fig. 6), and as it is, too, in systems with bridging cyclometallating ligands like 8 and 9. It is important to note that the SOC process involves coupling of the triplet state to energetically accessible spin-allowed singlet states. The more allowed the latter, the higher the triplet radiative rate constant is expected to be.28 Thus, the good performance of the dinuclear complexes in terms of kr and hence Φlum is likely to be a consequence, at least in part, of the high oscillator strength of the lowest spin-allowed transition.30
Concluding remarks
In summary, simple Schiff base chemistry allows pyridyl and phenolate donors to be linked via a central hydrazone unit, generating tridentate N^N^O-coordinating ligands that are used for the first time in this study to prepare bis-tridentate complexes of Ir(III). They offer a readily-synthesised alternative to cyclometallating N^N^C ligands such as 6-phenylbipyridines and, indeed, the simple mononuclear complexes of the form [Ir(dpyx)(N^N^O)]+ reported here display somewhat superior emission properties to those of [Ir(dpyx)(phbpy)]+. More significantly, the use of 4,6-bis(N-methyl-hydrazino)pyrimidine – as opposed to N-methyl-hydrazinopyridine – provides easy access to ditopic, bis-N^N^O-coordinating ligands that are able to bridge two Ir(III) centres, leading to unprecedented dinuclear complexes in which each metal centre is coordinated in a bis-tridentate manner. The presence of a second metal ion appears to increase the allowedness of both spin-allowed and spin-forbidden transitions, such that the dinuclear complexes absorb unusually strongly in the visible region as well as showing an enhanced rate of phosphorescence. Clearly, these types of Schiff-base ligands may offer considerable scope as alternatives to polypyridyl and cyclometallating ligands such as those based on terpyridines, aryl-bipyridines and related pyrimidine and pyrazine systems.Experimental details
General
Reagents were obtained from commercial sources and used without further purification unless stated otherwise. All solvents used in preparative work were at least Analar grade and water was purified using the PuriteSTILL plus™ system. 1H and 13C NMR spectra were recorded on a Bruker Avance-400 spectrometer. Two-dimensional NMR (COSY, NOESY, HSQC and HMBC) spectra were acquired on Varian VNMRS-600 (600 MHz) or VNMRS-700 (700 MHz) instruments. Chemical shifts (δ) are in ppm, referenced to residual protio-solvent resonances, and coupling constants are given in hertz. Mass spectra were obtained by electrospray ionisation (positive and negative ionisation modes) on a Waters TQD mass spectrometer interfaced with an Acquity UPLC system with acetonitrile as the carrier solvent. Measurements requiring the use of an atmospheric solids analysis probe (ASAP) for ionisation were performed on Waters Xevo QToF mass spectrometer.X-ray crystallography
The X-ray single crystal data have been collected using λMoKα radiation (λ = 0.71073 Å) on an Agilent XCalibur (Sapphire-3 CCD detector, fine-focus sealed tube, graphite monochromator; complex H2L5) and a Bruker D8Venture (Photon100 CMOS detector, IμS-microsource, focusing mirrors; all other complexes) diffractometers equipped with Cryostream (Oxford Cryosystems) open-flow nitrogen cryostats at the temperature 120.0(2) K. All structures were solved by direct methods and refined by full-matrix least squares on F2 for all data using Olex231 and SHELXTL32 software. All non-disordered non-hydrogen atoms were refined anisotropically, hydrogen atoms were placed in the calculated positions and refined in riding mode. It is not always possible in such heavy-atom structures to see electron-density peaks in difference maps which would correspond to acceptable locations for the various solvent H atoms, so optimisation of the orientation of methyl groups of the solvent molecules was deemed to be unnecessary. Disordered atoms were refined in isotropic approximation with various fixed SOF's. Crystal [{Ir(dpyx)2}L6](PF6)2 contains severely disordered 0.5 PF6 anion and some solvent molecules the exact number and chemical identity of which could not be found and satisfactory modeled. Their contribution (total 140e per unit cell) has been taken into account by application of the MASK procedure of Olex2 package. Crystal data and parameters of refinement are listed in Table 3. Crystallographic data for the six structures have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 2006844–2006849.†
| Compound | H2L5 | H2L7 | [Ir(dpyx)L4]PF6 | [{Ir(dpyx)}2L5](PF6)2 | [{Ir(dpyx)}2L6](PF6)2 | [{Ir(dpyF)}2L6](PF6)2 |
| Empirical formula | C20H20N6O2 | C36H52N6O2 | C34H32F6IrN6O2P | C64H60F12Ir2N14O2P2 | C141H150F18Ir4N26O5P3 | C70H71F16Ir2N13O3P2 |
| Formula weight | 376.42 | 600.83 | 893.83 | 1731.60 | 3492.57 | 1892.73 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group | I2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P |
P |
| a/Å | 15.5025(12) | 12.1186(12) | 11.4532(5) | 12.7200(11) | 12.2674(11) | 12.4261(12) |
| b/Å | 10.4734(8) | 12.2241(10) | 15.8034(7) | 14.3155(12) | 16.3422(15) | 15.8044(15) |
| c/Å | 11.3532(9) | 12.9851(11) | 18.1543(8) | 19.9170(17) | 21.227(2) | 19.8347(19) |
| α/° | 90 | 72.172(3) | 90.00 | 86.983(3) | 75.235(3) | 76.196(3) |
| β/° | 93.231(7) | 77.714(3) | 95.0909(16) | 79.538(3) | 76.582(3) | 74.210(3) |
| γ/° | 90 | 88.934(3) | 90.00 | 78.218(2) | 87.493(3) | 81.438(3) |
| Volume/Å3 | 1840.4(3) | 1787.0(3) | 3273.0(2) | 3490.9(5) | 4002.2(6) | 3625.3(6) |
| Z | 4 | 2 | 4 | 2 | 1 | 2 |
| ρ calc, g cm−3 | 1.359 | 1.117 | 1.814 | 1.647 | 1.449 | 1.734 |
| μ/mm−1 | 0.092 | 0.070 | 4.206 | 3.939 | 3.422 | 3.809 |
| F(000) | 792.0 | 652.0 | 1760.0 | 1700.0 | 1733.0 | 1868.0 |
| Reflections collected | 15142 |
28016 |
71211 |
74344 |
83046 |
64406 |
| Independent refl., Rint | 2446, 0.1019 | 9488, 0.0442 | 9548, 0.0310 | 16016, 0.1680 |
21245, 0.0351 |
15791, 0.1711 |
| Data/restraints/parameters | 2446/0/133 | 9488/2/417 | 9548/0/456 | 16016/914/872 |
21245/64/891 |
15791/198/966 |
| Goodness-of-fit on F2 | 1.020 | 1.021 | 1.054 | 0.958 | 1.053 | 0.989 |
| Final R1 [I ≥ 2σ(I)] | 0.0565 | 0.0556 | 0.0161 | 0.0735 | 0.0484 | 0.0640 |
| Final wR2 [all data] | 0.1299 | 0.1369 | 0.0367 | 0.1700 | 0.1166 | 0.1398 |
Solution-state photophysics
UV-visible absorption spectra were recorded on a Biotek Instruments UVIKON XS spectrometer operating with LabPower software. Emission spectra were acquired on a Jobin Yvon Fluoromax-2 spectrometer equipped with a Hamamatsu R928 photomultiplier tube. All samples were contained within 1 cm pathlength quartz cuvettes modified for connection to a vacuum line. Degassing was achieved by at least three freeze–pump–thaw cycles whilst connected to the vacuum manifold: final vapour pressure at 77 K was <5 × 10−2 mbar. Emission was recorded at 90° to the excitation source, and spectra were corrected after acquisition for dark count and for the spectral response of the detector. The quantum yields were determined relative to an aqueous solution of [Ru(bpy)3]Cl2, for which Φlum = 0.040.33 Emission spectra at 77 K were recorded in 4 mm diameter tubes held within a liquid-nitrogen-cooled quartz dewar, using the same spectrometer.The luminescence lifetimes in solution at 295 K were measured by time-correlated single-photon counting, using an EPL405 pulsed-diode laser as excitation source (405 nm excitation, pulse length of 60 ps, repetition rate 20 kHz). The emission was detected at 90° to the excitation source, after passage through a monochromator, using an R928 PMT thermoelectrically cooled to −20 °C. The luminescence lifetimes at 77 K were recorded using the same detector operating in multichannel scaling mode, following excitation with a pulsed xenon lamp.
Synthetic and characterisation details for representative proligands and complexes
Proligands HL1–HL4 were prepared by condensation of N-methylhydrazinopyridine with the appropriate salicylaldehyde, as described in our earlier work on Pt(II) complexes.21 Starting materials for the preparation of the ditopic ligands were obtained from commercial suppliers and used as supplied. Details are given below, and the numbering system for the assignment of NMR resonances is provided in Fig. 8. The preparation of H2L7 and its mononuclear Ir(III) complex are described in the ESI.† | ||
| Fig. 8 Numbering system for NMR assignments. | ||
1H NMR (DMSO-d6, 700 MHz): 9.01 (1H, s), 8.21 (2H, d, J = 2.5, H6′′), 7.87 (2H, t, J = 8, H5′′), 7.79–7.78 (3H, m, H3′′ or H3), 7.64 (1H, ddd, J = 9, 7 and 1.5, H4′), 7.48 (1H, d, J = 9, H3′), 7.15 (1H, s, H4′′′), 7.11 (2H, t, J = 6.5, H4′′), 7.00 (1H, ddd, J = 8, 7 and 2, H5), 6.86 (1H, d, J = 6, H6′), 6.62 (1H, t, J = 7, H4), 6.57 (1H, t, J = 7), 6.43 (1H, d, J = 8.5), 4.20 (3H, s, HNMe), 2.85 (6H, s, HMe). 13C NMR (DMSO-d6, 176 MHz): 178.7, 168.9, 160.8, 154.5, 150.4, 149.6, 139.8, 139.2, 138.0, 137.9, 137.6, 135.0, 131.5, 131.4, 123.8, 123.2, 121.8, 119.5, 116.6, 115.1, 109.3, 33.7, 21.9. MS (ES+): m/z 678 [M]+; HRMS (ES+): m/z 676.1840 [M]+; calculated for [C31H27N5OIr]+ 676.1822.
:1 mixture of H2L5 (36 mg, 0.096 mmol) and [Ir(dpyx)Cl(μ-Cl)]2 (50 mg, 0.048 mmol) in ethylene glycol (1.5 mL) was heated to 195 °C for 90 min under argon. Upon cooling to ambient temperature, water (5 mL) was added and the resulting orange-brown solid separated by filtration. The crude material was dissolved in the minimum volume of hot DMSO and the solution added dropwise into saturated aqueous KPF6 (5 mL). The resulting yellow solid was separated by centrifugation and washed with water (3 × mL) to yield the product as a yellow solid (22 mg, 0.023 mmol, 24% yield). 1H NMR (CD3CN, 700 MHz): 10.41 (1H, s), 8.90 (1H, s), 8.25 (2H, d, J = 9), 8.16 (1H, s), 7.85–7.82 (4H, m), 7.72 (1H, dd, J = 8 and 1.5), 7.51 (1H, d, J = 8), 7.34 (1H, t, J = 8), 7.18 (1H, s), 7.11–7.06 (4H, m), 7.00–6.98 (2H, m), 6.69 (1H, t, J = 7), 6.62 (1H, s), 6.42 (1H, d, J = 8), 4.10 (3H, s), 3.48 (3H, s), 2.91 (6H, s). 13C NMR (CD3CN, 176 MHz): 179.7, 178.0, 175.0, 170.7, 162.8, 159.1, 158.1, 151.6, 141.1, 140.6, 139.8, 139.0, 136.2, 133.5, 132.5, 131.9, 125.1, 123.8, 123.0, 120.9, 120.5, 119.8, 118.3, 117.4, 116.8, 85.1, 41.4, 33.9, 30.6, 22.5. MS (ES+): m/z 827 [M + H]+; HRMS (E+): m/z 825.2443 [M]+; calculated for [C30H34N8O2Ir]+ 825.2411.
:1 mixture of H2L5 (22 mg, 0.058 mmol) and [Ir(dpyx)Cl(μ-Cl]2 (61 mg, 0.058 mmol) was heated to 195 °C in ethylene glycol (1.5 mL) for 90 min under argon. Upon cooling to ambient temperature, water (2 mL) was added. The precipitated material was dissolved in the minimum volume of hot DMSO, and added dropwise into saturated aqueous KPF6 solution (5 mL). The resulting precipitate was collected by centrifugation, washed with water (3 × mL), and recystallised from MeCN/Et2O to yield the desired product as a yellow solid (4 mg, 0.003 mmol, 5% yield). 1H NMR (CD3CN, 700 MHz): 8.83 (2H, s, Himine), 8.01 (4H, d, J = 8, H3′′), 7.70 (4H, ddd, J = 8, 7.5 and 1.5, H4′′), 7.64 (2H, dd, J = 8 and 2, H3′), 7.53 (4H, dd, J = 6 and 1, H6′′), 7.15 (2H, s, H4′′′), 7.04 (2H, ddd, J = 9, 7 and 2, H5′), 6.87 (4H, ddd, J = 7.5, 5.5 and 1), 6.63 (2H, ddd, J = 8, 7 and 1), 6.46 (1H, s, H6), 6.28 (2H, d, J = 9, H6′), 5.97 (1H, s, H3), 4.10 (6H, s, HNMe), 2.88 (12H, s, HMe). 13C NMR (CD3CN, 176 MHz): 175.7 (C1′′′), 170.0 (C2′′), 163.0 (C1′), 162.1 (C2′′′), 156.2 (C2), 151.6 (C6′′), 142.4 (Cimine), 140.6 (C4′′), 139.0 (C3′′′), 138.50 (C2′′′), 136.3 (C3′), 134.0 (C5′), 133.5 (C4′′′), 124.5 (C3′′), 123.5 (C5′′), 122.9 (C6′), 119.9 (C2′), 117.1 (C4′), 84.65 (C6), 34.70 (CNMe), 23.0 (CMe). MS (ES+): m/z 639 [M]2+.
:1 mixture of H2L6 (24 mg, 0.048 mmol) and [Ir(dpyx)Cl(μ-Cl]2 (50 mg, 0.048 mmol). The crude material was purified by column chromatography (5% MeOH in DCM), followed by recrystallisation from MeCN/Et2O, to yield the final product as a yellow solid (8 mg, 0.006 mmol, 12% yield). 1H NMR (CD3CN, 700 MHz): 8.87 (2H, s, Himine), 8.02 (4H, d, J = 8, H3′′), 7.71 (4H, ddd, J = 8, 7.5 and 1.5, H4′′), 7.64 (2H, d, J = 2.5, H3′), 7.53 (4H, ddd, J = 6, 2 and 0.5, H6′′), 7.16–7.14 (4H, m, H5′ and H4′′′), 6.89 (4H, ddd, J = 7, 6 and 1, H5′′), 6.45 (1H, s, H6), 6.25 (2H, d, J = 9, H6′), 5.97 (1H, s, H3), 4.12 (6H, s, HNMe), 2.89 (12H, s, HMe), 1.26 (18H, s, Hbutyl). 13C NMR (CD3CN, 176 MHz): 175.9 (C1′′′), 170.0 (C2′′′), 162.1 (C3), 161.2 (C1′), 156.0 (C2), 151.5 (C6′′), 142.7 (Cimine), 140.6 (C4′′), 139.5 (C4′), 139.0 (C3′′′), 138.5 (C2′′), 133.4 (C5′ or C4′′′), 132.2 (C5′ or C4′′′), 131.9 (C3′), 124.5 (C3′′), 123.5 (C5′′), 122.4 (C6′), 118.9 (C2′), 84.5 (C6), 34.7 (CNMe), 34.3 (C7′), 31.6 (Cbutyl), 23.0 (CMe). MS (ES+): m/z 695 [M]2+; HRMS (ES+): m/z 694.2226 [M]2+; calculated for [C64H62N10O2Ir2]2+ 694.2188.
:1 mixture of H2L6 (46 mg, 0.094 mmol) and [Ir(dpyF)Cl(μ-Cl)]2 (100 mg, 0.094 mmol). The crude material was purified by column chromatography (2% MeOH in DCM), followed by recrystallisation from MeCN/Et2O, to yield the final product as a yellow solid (18 mg, 0.013 mmol, 12% yield). 1H NMR (CD3CN, 600 MHz) δ 8.90 (2H, s, Himine), 8.09 (4H, d, J = 8.2, H3′′), 7.88–7.73 (4H, m, H4′′), 7.67 (2H, d, J = 2.2, H3′), 7.54 (4H, d, J = 5.7, H6′′), 7.24 (2H, t, J = 10.2, H4′′′), 7.19 (2H, dt, J = 9.0, 2.0, H5′), 6.97 (4H, td, J = 6.6, 5.7, 1.5, H5′′), 6.48 (1H, s, H6), 6.33 (2H, dd, J = 9.1, 1.4, H6′), 5.91 (1H, s, H3), 4.13 (6H, s, HMe), 1.26 (18H, s, Hbutyl). 19F NMR (CD3CN, 376 MHz) δF = −107.1 (4F, d, J = 14), −72.9 (12F, d, J = 710, PF6−). HRMS (ES+): m/z 703.1696 [M]2+; calculated for [C60H50F4Ir2N10O2]2+ 703.1740.
Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
We thank EPSRC and Durham University for support.Notes and references
- (a) Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) C. Cebrian and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459 CrossRef PubMed; (c) J. Kalinwoski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2009, 253, 1709 CrossRef; (d) P. T. Chou, Y. Chi, M. W. Chung and C. C. Lin, Coord. Chem. Rev., 2011, 255, 2653 CrossRef.
- (a) R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef PubMed; (b) S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985 CrossRef; (c) B. Pashaei, S. Karmi, H. Shahroosvand and M. Pilkington, Adv. Funct. Mater., 2020, 1908103 CrossRef; (d) Q. Y. Zheng, F. Li, Z. Y. Chen, K. Yang, Y. Liu, T. L. Guo, G. G. Shan and Z. M. Su, ACS Appl. Mater. Interfaces, 2020, 12, 4649 CrossRef PubMed.
- (a) A. Genoni, D. N. Chirdon, M. Boniolo, A. Sartorel, S. Bernhard and M. Bonchio, ACS Catal., 2017, 7, 154 CrossRef; (b) I. N. Mills, J. A. Porras and S. Bernhard, Acc. Chem. Res., 2018, 51, 352 CrossRef PubMed; (c) R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Surinach and A. Llobet, Nat. Rev. Chem., 2019, 3, 331 CrossRef.
- (a) A. Trowbridge, D. Reich and M. J. Gaunt, Nature, 2018, 561, 522 CrossRef; (b) J. Zhang, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M. H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 13625 CrossRef PubMed; (c) P. S. Kim, S. Choi, S. Y. Kim, J. H. Jo, Y. S. Lee, B. Kim, W. Kim, W. Choi, C. H. Kim, H. J. Son, C. Pac and S. O. Kang, Chem. – Eur. J., 2019, 25, 13609 CrossRef PubMed; (d) M. Yang, J. E. Yarnell, K. El Roz and F. N. Castellano, ACS Appl. Energy Mater., 2020, 3, 1842 CrossRef.
- (a) K. K. W. Lo, A. W. T. Choi and W. H. T. Law, Dalton Trans., 2012, 41, 6021 RSC; (b) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef; (c) M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144 RSC; (d) Y. Chen, R. Guan, C. Zhang, J. Huang, L. Ji and H. Chao, Coord. Chem. Rev., 2016, 310, 16 CrossRef; (e) A. M. H. Yip and K. K. W. Lo, Coord. Chem. Rev., 2018, 361, 138 CrossRef; (f) A. M. Ranieri, C. Caporale, V. Fiorini, A. Hubbard, P. Rigby, S. Stagni, W. Watkin, M. I. Ogden, M. J. Hackett and M. Massi, Chem. – Eur. J., 2019, 25, 10566 CrossRef.
- J. A. G. Williams, Multinuclear iridium complexes in Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, Wiley, Chichester, 2017 Search PubMed.
- (a) G. Li, D. G. Congrave, D. Zhu, Z. Su and M. R. Bryce, Polyhedron, 2018, 140, 146 CrossRef CAS; (b) G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Chem. Soc. Rev., 2020, 49, 765 RSC.
- (a) S. Welter, F. Lafolet, E. Cecchetto, F. Verrgeer and L. De Cola, ChemPhysChem, 2005, 6, 2417 CrossRef PubMed; (b) S. Welter, N. Salluce, A. Benetti, N. Rot, P. Belser, P. Sonar, A. C. Grimsdale, K. Müllen, M. Lutz, A. L. Spek and L. De Cola, Inorg. Chem., 2005, 44, 4706 CrossRef PubMed; (c) M. Cavazzini, S. Quici, C. Scalera, F. Puntoriero, G. La Ganga and S. Campagna, Inorg. Chem., 2009, 48, 8578 CrossRef.
- (a) K. J. Arm and J. A. G. Williams, Chem. Commun., 2005, 230 RSC; (b) K. J. Arm and J. A. G. Williams, Dalton Trans., 2006, 2172 RSC; (c) W. Leslie, A. S. Batsanov, J. A. K. Howard and J. A. G. Williams, Dalton Trans., 2004, 623 Search PubMed; (d) C. Sabatini, A. Barbieri, F. Barigelletti, K. J. Arm and J. A. G. Williams, Photochem. Photobiol. Sci., 2007, 6, 397 RSC; (e) K. J. Knuckey and J. A. G. Williams, Eur. J. Inorg. Chem., 2017, 5205 CrossRef.
- (a) V. L. Whittle and J. A. G. Williams, Inorg. Chem., 2008, 47, 6596 CrossRef PubMed; (b) V. L. Whittle and J. A. G. Williams, Dalton Trans., 2009, 3929 RSC.
- (a) P. H. Lanoë, C. M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6381 RSC; (b) R. E. Daniels, S. Culham, M. Hunter, M. C. Durrant, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Dalton Trans., 2016, 45, 6949 RSC.
- (a) S. Culham, P. H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992 CrossRef; (b) M. Z. Shafikov, R. Daniels, P. Pander, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, ACS Appl. Mater. Interfaces, 2019, 11, 8182 CrossRef.
- E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127 CrossRef.
- (a) V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304 CrossRef; (b) G. Turnbull, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2017, 53, 2729 RSC.
- J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993 CrossRef.
- (a) A. J. Wilkinson, A. E. Goeta, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2004, 43, 6513 CrossRef; (b) A. J. Wilkinson, A. E. Goeta, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2006, 45, 8685 CrossRef; (c) P. Brulatti, R. J. Gildea, J. A. K. Howard, V. Fattori, M. Cocchi and J. A. G. Williams, Inorg. Chem., 2012, 51, 3813 CrossRef; (d) L. F. Gildea, A. S. Batsanov and J. A. G. Williams, Dalton Trans., 2013, 42, 10388 RSC.
- (a) J. A. G. Williams, A. J. Wilkinson and V. L. Whittle, Dalton Trans., 2008, 2081 RSC; (b) J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783 RSC.
- (a) S. Obara, M. Itabishi, F. Okuda, S. Tamaki, Y. Tanabe, Y. Ishii, K. Nozaki and M.-A. Haga, Inorg. Chem., 2006, 45, 8907 CrossRef CAS PubMed; (b) M. Ashikawa, L. Yang, K. Kobayashi, H. Sato, A. Yamagishi, F. Okuda, T. Harada, R. Kuroda and M.-A. Haga, Dalton Trans., 2009, 1700 RSC; (c) J. Kuwabara, T. Namekawa, M.-A. Haga and T. Kanbara, Dalton Trans., 2012, 21 Search PubMed.
- (a) B. Tong, H. Y. Ku, I. J. Chen, Y. Chi, H. C. Kao, C. C. Yeh, C. H. Chang, S. H. Liu, G. H. Lee and P. T. Chou, J. Mater. Chem. C, 2015, 3, 3460 RSC; (b) Y. Chi, T. K. Chang, P. Ganesan and P. Rajakannu, Coord. Chem. Rev., 2017, 346, 91 CrossRef CAS.
- (a) C. Y. Kuei, W. L. Tsai, B. Tong, M. Jiao, W. K. Lee, Y. Chi, C. C. Wu, S. H. Liu, G. H. Lee and P. T. Chou, Adv. Mater., 2016, 28, 2795 CrossRef CAS PubMed; (b) J. L. Liao, P. Rajakannu, P. Gnanasekaran, S. R. Tsai, C. H. Lin, S. H. Liu, C. H. Chang, G. H. Lee, P. T. Chou, Z. N. Chen and Y. Chi, Adv. Opt. Mater., 2018, 6, 1800083 CrossRef; (c) L. Y. Hsu, D. G. Chen, S. H. Liu, T. Y. Chiu, C. H. Chang, A. K. Y. Jen, P. T. Chou and Y. Chi, ACS Appl. Mater. Interfaces, 2020, 12, 1084 CrossRef CAS.
- E. V. Puttock, J. D. Fradgley, D. S. Yufit and J. A. G. Williams, Dalton Trans., 2019, 48, 15012 RSC.
- Similar ligands in combination with Fe(III) have been shown to form tetranuclear grids in which the pyrimidine rings bridge two metal ions: Y.-T. Wang, A.-L. Cui, D.-Z. Shen and H.-Z. Kou, Polyhedron, 2013, 52, 970 CrossRef CAS.
- (a) A. Sarkar and S. Pal, Polyhedron, 2006, 25, 1689 CrossRef CAS; (b) D. Ramakrishna and B. R. Bhat, Inorg. Chem. Commun., 2010, 13, 195 CrossRef CAS; (c) W. Cao, Y. Liu, T. Zhang and J. Jia, Polyhedron, 2018, 147, 62 CrossRef CAS.
- J. L. Schmitt, A. M. Stadler, N. Kyritsakas and J. M. Lehn, Helv. Chim. Acta, 2003, 86, 1598 CrossRef CAS.
- K. L. Garner, L. F. Parkes, J. D. Piper and J. A. G. Williams, Inorg. Chem., 2010, 49, 476 CrossRef CAS.
- J. P. Collin, I. M. Dixon, J. P. Sauvage, J. A. G. Williams, F. Barigelletti and L. Flamigni, J. Am. Chem. Soc., 1999, 121, 5009 CrossRef CAS.
- D. L. Rochester, S. Develay, S. Záliš and J. A. G. Williams, Dalton Trans., 2009, 1728 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS.
- (a) E. I. Solomon, Comments Inorg. Chem., 1984, 3, 225 CAS; (b) H. Yersin and D. Donges, Top. Curr. Chem., 2001, 214, 81 CrossRef; (c) A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef.
- M. Z. Shafikov, R. Daniels and V. N. Kozhevnikov, J. Phys. Chem. Lett., 2019, 10, 7015 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and characterisation data for H2L7 and [Ir(dpyx)HL7]PF6; additional figures showing molecular structures and crystal packing in the crystals; table of key bond lengths and angles determined crystallographically; additional emission spectra. CCDC 2006844–2006849. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt01964j |
| ‡ Absorption and emission spectra and data for mononuclear complex [Ir(dpyx)HL7]PF6 (the dinuclear analogue of which could not be isolated in high purity) are provided in the ESI.† |
| This journal is © The Royal Society of Chemistry 2020 |