
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAllosteric regulation of rotational, optical and catalytic properties within multicomponent machinery†
Suchismita
Saha
a,
Amit
Ghosh
a,
Thomas
Paululat
b and
Michael
Schmittel
 *a
*a
aCenter of Micro- and Nanochemistry and Engineering, Department Chemie – Biologie, Organische Chemie I, Adolf–Reichwein–Str. 2, D-57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de
bDepartment Chemie – Biologie, Organische Chemie II, Adolf–Reichwein–Str. 2, D-57068 Siegen, Germany
First published on 18th June 2020
Abstract
The reversible transformation of multicomponent nanorotors (ROT-1, k298 = 44 kHz or ROT-2, k298 = 61 kHz) to the “dimeric” supramolecular structures (DS-1 or DS-2, k298 = 0.60 kHz) was triggered by a stoichiometric chemical stimulus. Simple coordination changes at the central phenanthroline of the molecular device by altering metal ions (Cu+ → Zn2+) or stoichiometry (Cu+, 1 equiv. → 0.5 equiv.) affected the terminal zinc(II) porphyrin units, the active sites within the machinery, changing rotational, catalytic and optical properties. In presence of added pyrrolidine, the nanorotor ROT-1 was inactive for catalysis whereas formation of the dimeric supramolecular structures DS-1 initiated a Michael addition reaction by releasing the organocatalyst from the porphyrin sites. This catalytic machinery (ROT-1 ⇄ DS-1) proved to reproducibly work over two full cycles using allosteric OFF/ON control of catalysis.
Introduction
Coordination-driven structural rearrangements1,2 of supramolecular architectures3–7 have been recently utilized to control catalytic reactions8–11 and guest uptake/release.3a,12–15 Whereas examples of allosteric regulation16–21 are abundant for controlling functions, even in supramolecular constructs, very few examples are known where a multitude of functions is tuned allosterically.22–24Most of the literature-known allosteric regulation has been performed using covalent receptors.20,25,26 However, in biological systems, a remarkable amount of proteins showing allosteric regulation of their enzymatic activity are multicomponent in nature.27,28 Some of these cases demonstrate that small molecular effectors allow or inhibit allosteric enzymatic regulation through control of dimerization (Fig. 1). For instance, the enzyme ATP phosphoribosyltransferase is active in its dimeric state and allosterically inhibited by added histidine triggering a transformation of the active dimeric into an inactive hexameric form.27,29
 | ||
| Fig. 1 Formation of the active site by the dimerization of two protein molecules.27 | ||
Herein, we demonstrate how reversible allosteric regulation30 allows parallel control of three functions: ON/OFF catalysis, OFF/ON rotor operation and UP/DOWN fluorescence changes upon moving from a multicomponent monomeric assembly to a dimeric system by addition of small molecular effectors. The key to reach such multifunctional control is the stoichiometric and reversible coordination-driven transformation of a three-component nanorotor (2 equiv.) into a three-component dimeric and dynamic supramolecule (1 equiv.). The main components of both supramolecular self-assemblies are ligand 1, a phenanthroline carrying two distal zinc(II) porphyrin units, and the bis-pyridine bipeds 2a,b (Fig. 2). Indeed, this seems to be the first example of allosterically controlling multiple functions in parallel within a multicomponent machinery requiring dimerization of structures.
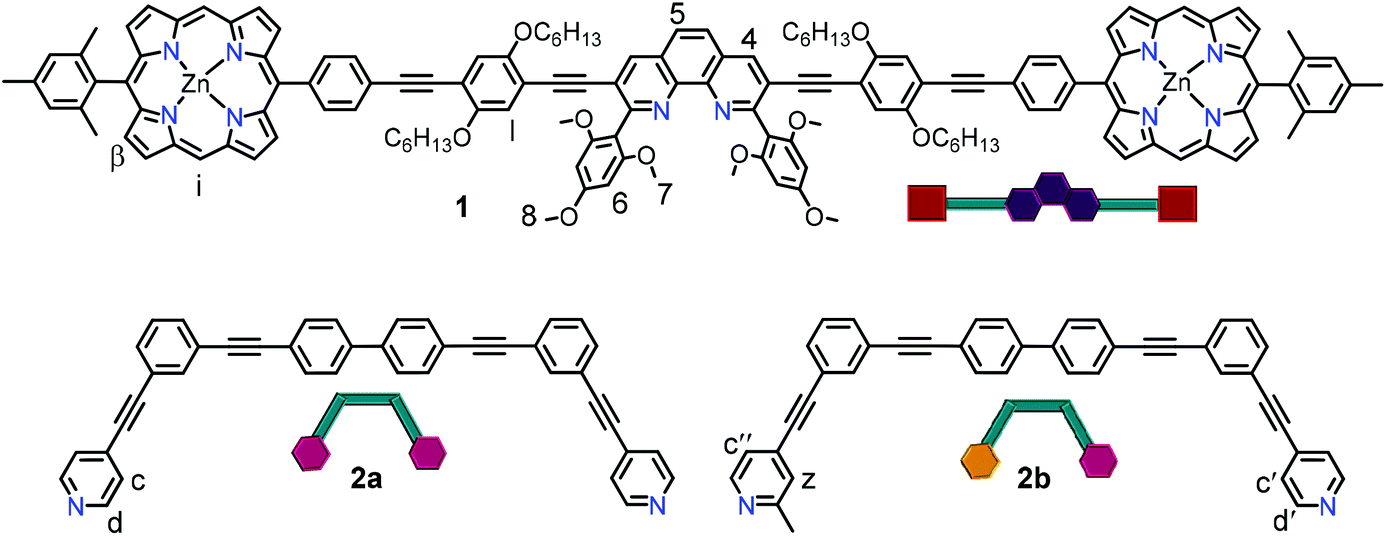 | ||
| Fig. 2 Chemical structure of ligands 1, 2a and 2b along with their cartoon representations. | ||
In detail, either by changing the stoichiometry of the metal ion (Cu+) or the metal ion itself (Cu+ → Zn2+) the coordinative situation at the central phenanthroline site in [Cu(1)(2a)]+ (ROT-1) or [Cu(1)(2b)]+ (ROT-2) was changed which regulated the binding mode at the terminal zinc(II) porphyrin (ZnPor) units providing quantitative and reversible interconversion of nanorotor (ROT-1/ROT-2) ⇄ “dimeric” supramolecule (DS-1/DS-2) (Fig. 3). This device-to-dimer transformation was used for controlling the liberation of a catalyst from the ZnPor binding site into the solution hence regulating OFF/ON catalysis. The switchable ensemble thus constitutes catalytic machinery.
 | ||
| Fig. 3 Cartoon representation of the reversible transformations between nanorotor ROT-1/ROT-2 → dimeric supramolecules DS-1/DS-2. | ||
Results and discussion
Design
For the required stoichiometry-dependent self-sorting, ligand 1 was designed. It contains two terminal ZnPor sites and one central phenanthroline unit so that its metal complexes can act as stator ([Cu(1)]+) or decks ([Cu(1)2]+ and [Zn(1)2]2+) in either the rotor or the dimeric supramolecule, respectively (Fig. 3).8c,31 The heart of ligand 1 was conceived as a 2,9-bis(2,4,6-trimethoxyphenyl)-1,10-phenanthroline unit (vide infra). We have chosen the 2,4,6-trimethoxyphenyl substitution at the 2,9-position of phenanthroline because it was expected to provide the optimal electronic stabilization and steric bulk to quantitatively afford the coordinatively frustrated complex [Cu(1)]+ (with copper(I) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio), while the 2:1 mixture should furnish complex [Cu(1)2]+. Phenanthrolines without or with too bulky substituents, e.g., mesityl rings at the 2,9-position of phenanthroline, do not exhibit the plasticity of forming 2:1 and 1:1 complexes with Cu+ depending on the stoichiometry.32
:1 ratio), while the 2:1 mixture should furnish complex [Cu(1)2]+. Phenanthrolines without or with too bulky substituents, e.g., mesityl rings at the 2,9-position of phenanthroline, do not exhibit the plasticity of forming 2:1 and 1:1 complexes with Cu+ depending on the stoichiometry.32
Model studies
In order to ascertain the final design of ligands 1 and 2, we have performed model studies to prove the orthogonality between the [Cu(1)2]+ complexation and the Npy → ZnPor binding. For this purpose, ligands 3, 4, 5, and [Cu(CH3CN)4]PF6 (2:1:1:1) were dissolved in CD2Cl2 in an NMR tube (Fig. 4a). The comparison of characteristic 1H NMR signals of the phenanthroline (3′-, 4′-, 5′-, 6′-H), pyridine (2-H) and porphyrin (i′-, β′-H) units with those in the individual complexes sustained quantitative formation of the two non-interfering complexes [Cu(3)2]+ and 4·5 (Fig. 4b).
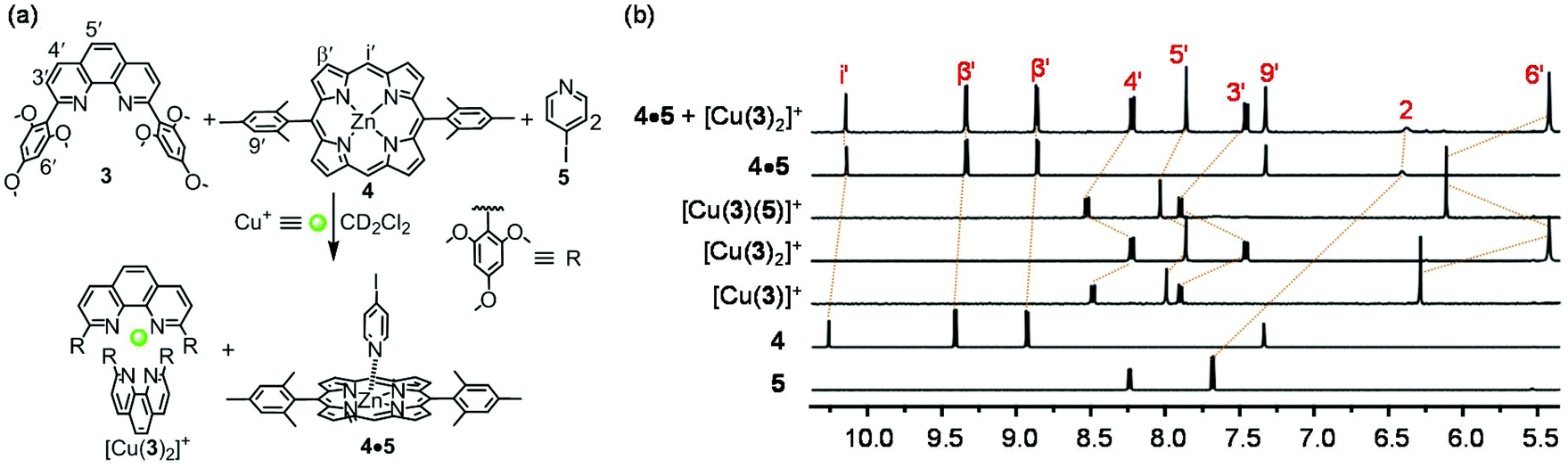 | ||
| Fig. 4 (a) Orthogonality between complexes [Cu(3)2]+ and 4·5. (b) Partial 1H NMR (400 MHz, CD2Cl2, 295 K) of 5, 4, [Cu(3)]+, [Cu(3)2]+, [Cu(3)(5)]+, 4·5 and 4·5 + [Cu(3)2]+. | ||
Synthesis and characterization of 1, 2a and 2b
Ligands 1, 2a and 2b were prepared in multistep approaches (see ESI†). The Sonogashira coupling reaction of 3,8-diethynyl-2,9-bis(2,4,6-trimethoxyphenyl)-1,10-phenanthroline and zinc(II) 5-iodophenyl-15-mesitylporphyrin afforded ligand 1 in 75% yield. The symmetric biped 2a was synthesized by Sonogashira coupling between 4,4′-bis((3-ethynylphenyl)ethynyl)biphenyl and 4-iodopyridine in the final step. The dissymmetric biped 2b was prepared by stepwise Sonogashira couplings of 4,4′-bis((3-ethynylphenyl)ethynyl)biphenyl with 4-iodopyridine and 4-bromopicoline, respectively. All the ligands were unambiguously characterized by 1H NMR, 1H–1H COSY, 13C NMR, ESI-MS and elemental analysis (see ESI†).Preparation of metal complexes, rotors and dimeric supramolecules
Alike the model compound 3, ligand 1 can form both the coordinatively frustrated Cu+ complex [Cu(1)]+ and the bishomoleptic complex [Cu(1)2]+ based on the relative stoichiometry. Significant chemical shifts in the 1H NMR signal of protons 6-H, l-H and other phenanthroline protons in ligand 1, [Cu(1)]+ and [Cu(1)2]+ supported quantitative formation of the complexes (Fig. 5a and ESI, Fig. S49†). Considerable upfield shift of protons 6-H, 7-H, 8-H and l-H in complex [Cu(1)2]+ were attributed to the π-ring current arising from the 2,4,6-trimethoxyphenyl units. | ||
| Fig. 5 (a) Partial 1H NMR (400 MHz, CD2Cl2, 295 K) of 2a, 1, [Cu(1)]+, [Cu(1)2]+, ROT-1 and DS-1. (b) UV-vis spectra of 1, ROT-1 and DS-1 in CH2Cl2 at 298 K (c = 10−5 M). | ||
Due to the precise stoichiometry-dependent self-sorting, ROT-1 = [Cu(1)(2a)]+ (Fig. 3a) was quantitatively formed by mixing 1, 2a and [Cu(CH3CN)4]PF6 (1:1:1) in CD2Cl2, irrespective of the sequence. In nanorotor ROT-1, [Cu(1)]+ acts as the stator holding the rotator 2avia Npy → [Cu(phenAr2)]+ (= HETPYP-I: ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) eroleptic
eroleptic ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif)
![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) ridine and henanthroline complexation)33 and Npy → ZnPor binding. Upfield shifts of proton signals i-H, 6-H and l-H from 10.34 to 10.28 ppm, 6.40 to 6.32 ppm and 6.70 to 6.64 ppm in the 1H NMR unambiguously attested the binding of 2a to [Cu(1)]+ (Fig. 5a). As monitored in the UV-vis spectrum, the red shift of the Q-band of the ZnPor from 537 to 540 nm when going from 1 to ROT-1 = [Cu(1)(2a)]+ manifested the axial binding of one pyridine foot of 2a to the ZnPor units of [Cu(1)]+ (Fig. 5b). Moreover, a single peak in the ESI-MS at m/z = 1457.8 and a single set of 1H-DOSY signals corroborated the formation of ROT-1 (ESI, Fig. S76 & S60†).
ridine and henanthroline complexation)33 and Npy → ZnPor binding. Upfield shifts of proton signals i-H, 6-H and l-H from 10.34 to 10.28 ppm, 6.40 to 6.32 ppm and 6.70 to 6.64 ppm in the 1H NMR unambiguously attested the binding of 2a to [Cu(1)]+ (Fig. 5a). As monitored in the UV-vis spectrum, the red shift of the Q-band of the ZnPor from 537 to 540 nm when going from 1 to ROT-1 = [Cu(1)(2a)]+ manifested the axial binding of one pyridine foot of 2a to the ZnPor units of [Cu(1)]+ (Fig. 5b). Moreover, a single peak in the ESI-MS at m/z = 1457.8 and a single set of 1H-DOSY signals corroborated the formation of ROT-1 (ESI, Fig. S76 & S60†).
Quantitative formation of the dimeric supramolecular structure DS-1 = [Cu(1)2(2a)2]+ was achieved by mixing 1, 2a and [Cu(CH3CN)4]PF6 in a 1:1:0.5 ratio in CD2Cl2 (Fig. 3a). In the 1H NMR, protons 6-H and l-H along with all other phenanthroline protons appeared at the same position as in the homoleptic complex [Cu(1)2]+ that represents the deck for both bipeds in DS-1 (Fig. 5a). Upfield shift of proton signal i-H (10.34 to 10.22 ppm), d-H (8.60 to 2.35 ppm) and c-H (7.41 to 5.52 ppm) when going from [Cu(1)2]+ to DS-1 confirmed binding of 2a (Npy → ZnPor) (Fig. 5a and ESI, Fig. S49†). In the UV-vis the red shift of the Q-band of the ZnPor unit from 537 to 543 nm (for 1 → DS-1) supported the Npy → ZnPor axial binding of 2a in [Cu(1)2]+ (Fig. 5b). Formation of DS-1 was further confirmed by ESI-MS and 1H DOSY NMR (ESI, Fig. S77 and S61†).
In ROT-1, one of the pyridine feet of the rotating biped is always attached to the central copper(I) phenanthroline thereby representing an axle for the other pyridine site that oscillates between both degenerate zinc porphyrins of the stator (cf. 50% loading of the ZnPor units). In contrast, in DS-1 all ZnPor sites are constantly occupied by the pyridine sites of the bipeds due to the pyridine:ZnPor ratio of 1:1. The strong Npy → ZnPor axial binding in DS-1 was clearly supported by the upfield shift of proton signal i-H (10.28 to 10.22 ppm) in the 1H NMR and the red shift of the Q-band (Fig. 5) of the ZnPor sites (540 → 543 nm) in the UV-vis when going from ROT-1 (50% loading of the ZnPor sites by the pyridine foot of 2a) to DS-1 (full loading of the ZnPor sites by the pyridine foot of 2a).
Similarly, the 1:1:1 mixture of 1, [Cu(CH3CN)4]PF6 and 2b led to quantitative formation of ROT-2 = [Cu(1)(2b)]+ where the picoline site of 2b was bound selectively to the copper(I) phenanthroline unit of [Cu(1)]+ through HETPYP-I33 interaction (Fig. 3b). The selective binding originated from the ortho-methyl substitution at the pyridine which increased the strength of the HETPYP-coordination to logKpic = 5.86 (ESI, Fig. S83†) and weakened the Npic → ZnPor interaction (logKpic = 2.72) due to steric crowding.8b,34 The upfield shift in the 1H NMR of proton signals 6-H, l-H and i-H from [Cu(1)]+ → [Cu(1)(2b)]+ proved the interaction between 2b and [Cu(1)]+ which was further verified by the red-shift of the Q-band (from 537 to 540 nm) (Fig. 6 and ESI, Fig. S86†). Formation of ROT-2 was additionally confirmed by 1H DOSY and ESI-MS data (ESI, Fig. S62 and S78†).
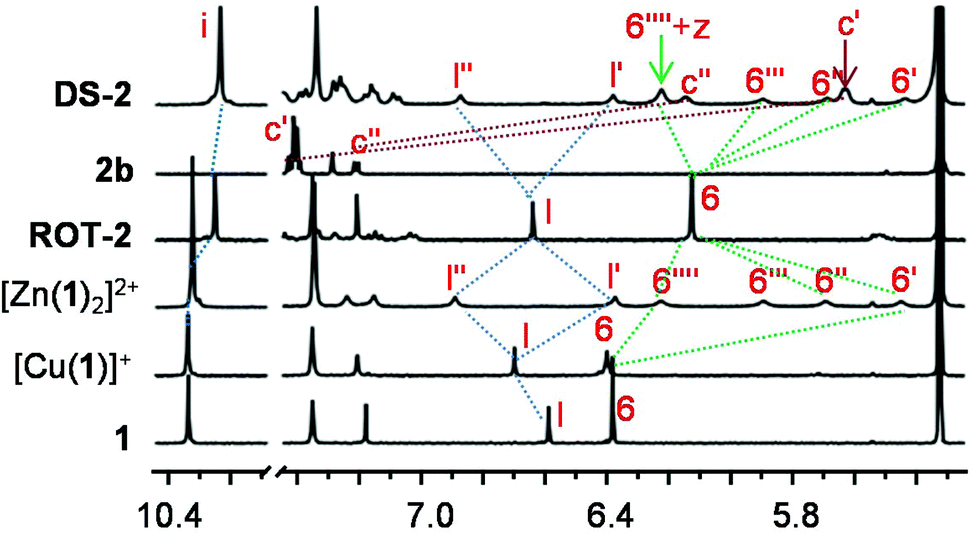 | ||
| Fig. 6 Partial 1H NMR (400 MHz, CD2Cl2, 295 K) of 1, [Cu(1)]+, [Zn(1)2]2+, ROT-2, 2b and DS-2. | ||
Unlike DS-1, mixing of 1, 2b and Cu+ in 1:1:0.5 ratio did not quantitatively furnish the dimeric supramolecular structure [Cu(1)2(2b)2]+. Alike, using the model ligands there was no 100% orthogonality between [Cu(3)2]+ and Npic → ZnPor binding. In comparison with ROT-2, the picoline group of biped 2b has to sacrifice in [Cu(1)2(2b)2]+ the strong HETPYP binding at the [Cu(phenAr2)]+ unit in ROT-2 (logKpic = 5.86) for the weak one at the ZnPor site in [Cu(1)2(2b)2]+ (logKpic = 2.72). Such loss in driving force cannot be compensated by formation of the central [Cu(1)2]+ unit. The thermodynamics, however, can be remedied by using Zn2+ instead of Cu+ owing to its stronger bishomoleptic complexes with ligands of type 3 (ESI, Fig. S41†). The self-assembly DS-2 = [Zn(1)2(2b)2]2+ was indeed quantitatively furnished by mixing 1, 2b and Zn2+ in 1:1:0.5 ratio (Fig. 3b). Its formation was unambiguously proved by 1H NMR, ESI-MS and elemental analysis (ESI, page S29–30†). For instance, in the 1H NMR (CD2Cl2), the splitting of proton signal 6-H into four singlets (6′, 6′′, 6′′′ and 6′′′′-H) and l-H into two singlets (l′ and l′′-H) like [Zn(1)2]2+, the upfield shifts of c′′-H (7.22 to 6.15 ppm) and c′-H (7.42 to 5.63 ppm) from free 2b verified the formation of DS-2 (Fig. 6).
Dynamic VT-NMR
After successfully realizing quantitative formation of new dynamic multicomponent structures, we were interested to determine their exchange processes, exchange frequencies and activation parameters. The single set of protons for both porphyrin units in rotors ROT-1 and ROT-2 in the 1H NMR attested fast rotation on the 1H NMR timescale.VT 1H NMR of ROT-1 was performed from 25 °C to −70 °C and changes in the signal of proton i-H served for analyzing the kinetic data (Fig. 7a). It appeared as a sharp singlet at 25 °C (10.28 ppm) but split into a 1:1 set at −70 °C (10.33 and 10.22 ppm). The signal at 10.22 ppm was assigned to the pyridine-coordinated ZnPor whereas the signal at 10.33 ppm was allocated to the free ZnPor unit. Rotational exchange frequencies (k) at different temperatures were calculated using WinDNMR35 which provided the exchange frequency at room temperature k298 = 44 kHz. Activation parameters were derived from the Eyring plot (ESI, Fig. S57†) and are provided in Table 1.
 | ||
| Fig. 7 VT-1H NMR (600 MHz, CD2Cl2) of (a) ROT-1 and (b) ROT-2 displaying the splitting of proton signal i-H into 1:1 ratio. (c) VT-1H NMR (600 MHz, CD2Cl2) of DS-2 showing the splitting (two sets: 1:1) of proton signal β-H. (d) Cartoon representation showing reversible transformation of the high-speed nanorotor to the dynamic low-speed DS-2. | ||
| Nanomachines | k 298/kHz | ΔH‡/kJ mol−1 | ΔS‡/J mol−1 K−1 | ΔG‡298/kJ mol−1 |
|---|---|---|---|---|
| ROT-1 | 44 | 46.5 ± 1.2 | 0.6 ± 0.2 | 46.3 ± 1.2 |
| ROT-2 | 61 | 46.1 ± 0.5 | 1.4 ± 0.9 | 45.7 ± 0.2 |
| DS-2 | 0.60 | 68.9 ± 3.6 | 43.1 ± 3.6 | 57.2 ± 0.2 |
The VT 1H NMR of ROT-2, recorded from 25 °C to −50 °C, exhibited a splitting of the sharp singlet of proton i-H into a set of two singlets (1:1) with a coalescence temperature at −30 °C (Fig. 7b). As before, the corresponding activation parameters were determined (Table 1). The exchange frequency at room temperature was calculated as k298 = 61 kHz.
As in DS-1 all binding sites (Npy → ZnPor) are identical, even after freezing, the exchange cannot be monitored. To determine the kinetic parameters of such a process, the dissymmetric arm 2b was used in DS-2 with pyridine at one terminal and picoline at the other end.
The single set of all porphyrin protons in DS-2 manifested fast exchange on the NMR time scale at room temperature. Upon lowering the temperature, the VT 1H-NMR of DS-2 (Fig. 7c) exhibited a splitting of proton signal β-H (1:1). Exchange frequencies (k) at different temperatures provided the activation parameters (Table 1) and k298 = 0.60 kHz.
The much lower speed in the dynamic dimeric supramolecule than that of the rotors can be easily understood by their distinct mechanisms. In the rotors ROT-1 and ROT-2, the rate determining step for rotation comprises the single detachment of the Npy axial coordination from the ZnPor unit,8c leading to an exchange rate kex = ½kdiss (½ is a statistical factor recognizing that one out of two re-attachments will lead to exchange of both site). In contrast, the mechanism of exchange in DS-2 is quite different. Firstly, in DS-2 all sites are loaded and, secondly, only the exchange of pyridine by a picoline at a given ZnPor site or vice versa is detected by NMR. Based on the highly positive activation entropy and the high activation enthalpy there is a possibility that the exchange proceeds by full dissociation/re-association of one biped.
Interconversion
After the preparation of the individual parts of the machinery (ROT-1/ROT-2 and DS-1/DS-2) with their highly different dynamic properties, our next target was to interconvert them using chemical input. As their 1H NMR and fluorescence spectra proved to be different, interconversion was monitored by both emission and 1H NMR spectroscopy.At first, the dimeric supramolecule DS-1 was prepared by mixing ligands 1, 2a and Cu+ (1:1:0.5). Its formation was confirmed by 1H NMR and fluorescence spectroscopy. Further addition of 0.5 equiv. of [Cu(CH3CN)4]PF6 to DS-1, hence changing the stoichiometry of Cu+:1 from 0.5:1 to 1:1, quantitatively generated nanorotor ROT-1 as validated by spectroscopic data. The transformation of the dimeric supramolecule to a “monomeric” rotor was possible by changing the tetracoordinated homoleptic copper(I) center at the central phenanthroline to a tricoordinated HETPYP-I center. Addition of 0.5 equiv. of cyclam to ROT-1 regenerated DS-1 by removing a stoichiometric amount of Cu+ as cyclam is a stronger chelating agent for Cu+ than phenanthroline.3b,8b Three complete cycles interconverting DS-1 ⇄ ROT-1 were performed and monitored by fluorescence and NMR spectroscopy (Fig. 8a, c and ESI, Fig. S50†).
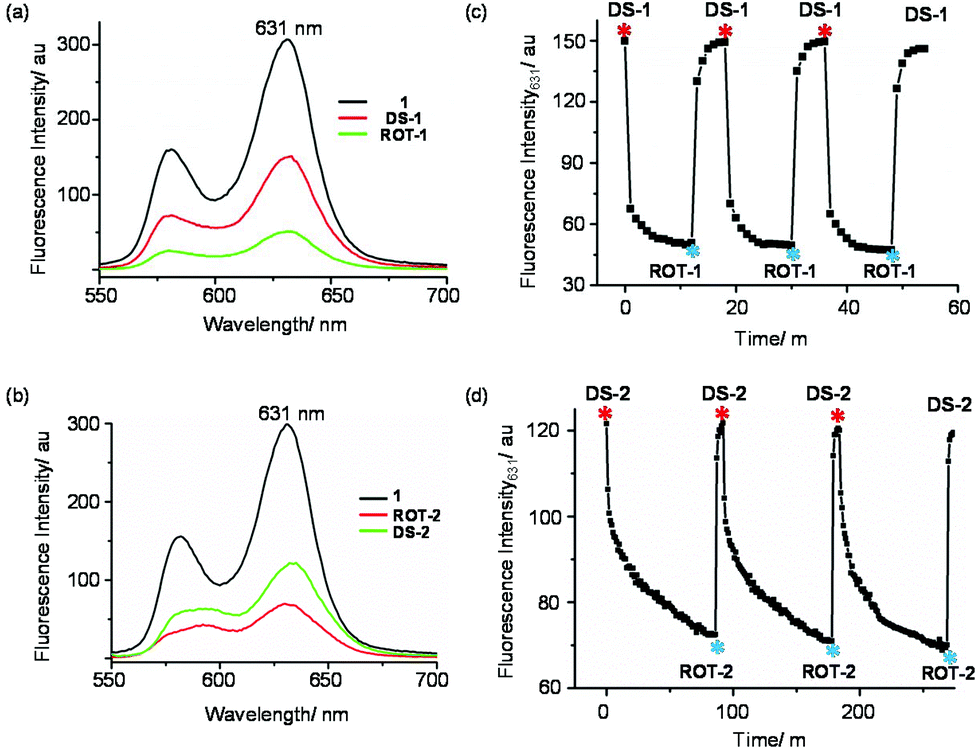 | ||
| Fig. 8 Fluorescence spectra of (a) 1, ROT-1, DS-1 and (b) 1, ROT-2, DS-2 in CH2Cl2 at 298 K (λexc = 540 nm, c = 10−5 M). Fluorescence intensity changes (λem = 631 nm) vs. time over three cycles for (c) DS-1 → ROT-1 and (d) DS-2 → ROT-2 in CH2Cl2 at 298 K (λexc = 540 nm, c = 10−5 M). Copper(I) and cyclam additions are shown by red and indigo asterisk, respectively, in Fig. 8c. Additions of hexacyclen + Cu+ (1:2) and cyclam + Zn2+ (2:1) are shown by red and indigo asterisk, respectively, in Fig. 8d. | ||
The kinetics of the interconversion was followed by fluorescence. Ligand 1, DS-1 and ROT-1 exhibit typical ZnPor emission patterns with different intensity. In the free ligand, the fluorescence intensity is highest followed by that in DS-1 and in ROT-1 (Fig. 8a). Conversion of DS-1 to ROT-1 took 12 min whereas ROT-1 to DS-1 was finished within 6 min at room temperature, c = 10−5 M (Fig. 8c).
In order to investigate the interconversion of DS-2 → ROT-2, we first prepared DS-2 by mixing 1, 2b and Zn2+ (1:1:0.5). Addition of 0.5 equiv. of hexacyclen and 1.0 equiv. of Cu+ transformed the dimeric supramolecule DS-2 into the monomeric nanorotor ROT-2 by selective interchange of the metal ion at the central phenanthroline station.8a,c,22 Now, addition of 1.0 equiv. of cyclam and 0.5 equiv. of Zn2+ regenerated DS-2 by “dimerization” of the nanorotor. Three complete interconversion cycles were successfully carried out in a highly reproducible manner as demonstrated by fluorescence and 1H NMR data (Fig. 8b, d and ESI, Fig. S56†).
Equally, the kinetic course of the interconversion was evaluated by fluorescence (Fig. 8d). The conversion of DS-2 to ROT-2, triggered by addition of 0.5 equiv. of hexacyclen and 1.0 equiv. of Cu+, took around 85 min whereas the transformation ROT-2 → DS-2 was completed within 6 min after addition of 1.0 equiv. of cyclam and 0.5 equiv. of Zn2+ at room temperature.
Catalysis
The stoichiometry-driven interconversion of the supramolecular rotor to dimeric structure goes along with distinct changes in nanomechanical motions and optical properties. Our focus though was to set up catalytic machinery requiring control of the ON/OFF catalytic activity of the interconverting system by allosteric release and capture of a catalytically active guest at the zinc(II) porphyrin center. Hereunto, the key is the structural rearrangement at the central phenanthroline site that triggers conformational/constitutional changes as the ZnPor sites. After several tests, we identified the parent pyrrolidine (6) as the best candidate because binding of 6 to ZnPor is strong enough (logKpyrr = 4.90 ± 0.20) to be seized by ROT-1 (1:1 ratio of ROT-1 and 6) whereas it is freed in DS-1 for catalysis (ESI, Fig. S84†). Model reactions of Michael acceptor 7 (100 mol%) and nucleophile 8 (2000 mol%) in CD2Cl2:CDCl3 (20:1) with 6 (10 mol%) as catalyst provided (42 ± 1)% of product 9 after 4 h (at 50 °C) (Fig. 9a and ESI, Fig. S89†). In contrast, a (1:1) mixture of model zinc porphyrin 4 and catalyst 6 (10 mol%) furnished no product under identical reaction conditions due to the strong binding of the catalyst in the ZnPor complex 4·6 (ESI, Fig. S90†). When 10 mol% of 6·ROT-1 (1:1 mixture of ROT-1 and 6) were used as catalyst in the above Michael addition reaction, similarly no product formed, as 6 was sufficiently bound at the ZnPor unit of ROT-1 (ESI, Fig. S91†). This state we denote as the catalytic OFF state. For the catalytic ON state now DS-1 (5 mol%) + 2 × 6 were used which furnished (39 ± 1)% of product 9 under identical reaction conditions (ESI, Fig. S92†). Importantly, we observe basically identical catalytic activity with DS-1 (39%) as with the free catalyst in the model system (42%). Apparently, all added catalyst 6 is available for catalysis in DS-1.
 | ||
| Fig. 9 (a) Representation of the OFF/ON regulation of the pyrrolidine-catalyzed Michael addition involving the reversible interconversion within the catalytic machinery ROT-1 ⇄ DS-1 by removal and addition of Cu+. (b) Two complete catalytic cycles were performed between ROT-1 and DS-1 in presence of pyrrolidine (6). The instant of cyclam addition is shown by green asterisk, whereas Cu+ and consumed substrate addition are indicated by blue arrow. | ||
After testing in separate steps the function of the catalytic ON and OFF states, allosteric catalytic cycles were performed starting with the OFF state composed of 10 mol% 6·ROT-1, 7 (100 mol%) and 8 (2000 mol%) affording no product 9 after 4 h at 50 °C (Fig. 9b and ESI, Fig. S93†). Addition of cyclam (5 mol%) should remove an equimolar amount of Cu+ from the system furnishing 5 mol% of DS-1 which was expected to allosterically release the guest catalyst 6 from the ZnPor stations into solution because all ZnPor sites are now intrasupramolecularly loaded with biped 2a. Indeed, addition of 5 mol% of cyclam to the OFF state provided 39% of product 9 after 4 h at 50 °C. After realizing this ON catalytic state, consumed amount of substrates and 5 mol% of [Cu(CH3CN)4]PF6 were added. Heating at 50 °C for 4 h did not provide any further product formation, hence proving that this is an OFF state. Addition of 5 mol% of [Cu(CH3CN)4]PF6 to the ON state converted the dimeric supramolecule DS-1 to nanorotor ROT-1 with one ZnPor station free to capture all the guest catalyst 6, making it unavailable for the catalytic reaction. Two complete OFF/ON catalytic cycles were performed to demonstrate the ability of the full catalytic machinery in allosterically controlling the uptake/release of the guest catalyst (Fig. 9b).
In conclusion we have demonstrated the stoichiometry and coordination-driven reversible transformation of a multicomponent nanorotor to dimeric supramolecule by changing the coordination mode at the central phenanthroline unit. As a result of this interconversion within the machinery, allosteric tuning of OFF/ON rotor formation, UP/DOWN fluorescence and ON/OFF of Michael addition catalysis was achieved over two cycles.
This work demonstrates that self-sorting36 in combination with allosteric tuning of binding events is a powerful tool for realizing multifunctional machinery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to the University of Siegen and the Deutsche Forschungsgemeinschaft for continued support under Schm 647/20-2. Dedicated to Prof. Dr Günther von Bünau (Siegen) on the occasion of his 90th birthday.References
- W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618–1629 CrossRef CAS PubMed.
- (a) I. Paul, D. Samanta, S. Gaikwad and M. Schmittel, Beilstein J. Org. Chem., 2019, 15, 1371–1378 CrossRef CAS PubMed; (b) N. Mittal, M. L. Saha and M. Schmittel, Chem. Commun., 2016, 52, 8749–8752 RSC; (c) M. L. Saha and M. Schmittel, Inorg. Chem., 2016, 55, 12366–12375 CrossRef CAS.
- R. Zhu, J. Lübben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796–2800 CrossRef CAS PubMed.
- V. E. Campbell, X. de Hatten, N. Delsuc, B. Kauffmann, I. Huc and J. R. Nitschke, Nat. Chem., 2010, 2, 684–687 CrossRef CAS PubMed.
- K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300–5301 CrossRef CAS PubMed.
- J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed.
- (a) A. Goswami, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 15656–15663 CrossRef CAS PubMed; (b) N. Mittal, M. S. Özer and M. Schmittel, Inorg. Chem., 2018, 57, 3579–3586 CrossRef CAS PubMed; (c) A. Goswami, S. Pramanik and M. Schmittel, Chem. Commun., 2018, 54, 3955–3958 RSC; (d) S. Gaikwad, A. Goswami, S. De and M. Schmittel, Angew. Chem., Int. Ed., 2016, 55, 10512–10517 CrossRef CAS PubMed.
- G.-H. Ouyang, Y.-M. He, Y. Li, J.-F. Xiang and Q.-H. Fan, Angew. Chem., Int. Ed., 2015, 54, 4334–4337 CrossRef CAS PubMed.
- J. T. Foy, D. Ray and I. Aprahamian, Chem. Sci., 2015, 6, 209–213 RSC.
- C. M. McGuirk, J. Mendez-Arroyo, A. M. Lifschitz and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 16594–16601 CrossRef CAS.
- S. Bandi, A. K. Pal, G. S. Hanan and D. K. Chand, Chem. – Eur. J., 2014, 20, 13122–13126 CrossRef CAS PubMed.
- N. Kishi, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS PubMed.
- N. C. Gianneschi, M. S. Masar and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825–837 CrossRef CAS PubMed.
- S. Hiraoka, K. Harano, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2005, 44, 2727–2731 CrossRef CAS PubMed.
- V. Marti-Centelles, R. L. Spicer and P. J. Lusby, Chem. Sci., 2020, 11, 3236–3240 RSC.
- (a) C. M. McGuirk, J. Mendez-Arroyo, A. I. d'Aquino, C. L. Stern, Y. Liu and C. A. Mirkin, Chem. Sci., 2016, 7, 6674–6683 RSC; (b) C. M. McGuirk, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 4689–4696 CrossRef CAS PubMed; (c) J. Mendez-Arroyo, J. Barroso-Flores, A. M. Lifschitz, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 10340–10348 CrossRef CAS PubMed; (d) H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed; (e) N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508–10509 CrossRef CAS PubMed.
- S. Freye, J. Hey, A. Torras-Galan, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed.
- M. Schmittel, S. Pramanik and S. De, Chem. Commun., 2012, 48, 11730–11732 RSC.
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161–3187 CrossRef CAS PubMed.
- M. Ayabe, A. Ikeda, S. Shinkai, S. Sakamoto and K. Yamaguchi, Chem. Commun., 2002, 1032–1033 RSC.
- A. Goswami, I. Paul and M. Schmittel, Chem. Commun., 2017, 53, 5186–5189 RSC.
- A. Faulkner, T. van Leeuwen, B. L. Feringa and S. J. Wezenberg, J. Am. Chem. Soc., 2016, 138, 13597–13603 CrossRef CAS PubMed.
- M. Ikeda, M. Takeuchi, S. Shinkai, F. Tani, Y. Naruta, S. Sakamoto and K. Yamaguchi, Chem. – Eur. J., 2002, 8, 5541–5550 CrossRef CAS.
- V. Marcos, A. J. Stephens, J. Jaramillo-Garcia, A. L. Nussbaumer, S. L. Woltering, A. Valero, J.-F. Lemonnier, I. J. Vitorica-Yrezabal and D. A. Leigh, Science, 2016, 352, 1555–1559 CrossRef CAS PubMed.
- F. G. A. Lister, B. A. F. Le Bailly, S. J. Webb and J. Clayden, Nat. Chem., 2017, 9, 420–425 CrossRef CAS.
- R. A. Laskowski, F. Gerick and J. M. Thornton, FEBS Lett., 2009, 583, 1692–1698 CrossRef CAS.
- D. Suplatov and V. Švedas, Acta Naturae, 2015, 7, 34–45 CrossRef CAS.
- S. Pedreño, J. P. Pisco, G. Larrouy-Maumus, G. Kelly and L. P. S. de Carvalho, Biochemistry, 2012, 51, 8027–8038 CrossRef PubMed.
- (a) C. Kremer and A. Lützen, Chem. – Eur. J., 2013, 19, 6162–6196 CrossRef CAS PubMed; (b) S. Zahn, W. Reckien, B. Kirchner, H. Staats, J. Matthey and A. Lützen, Chem. – Eur. J., 2009, 15, 2572–2580 CrossRef CAS PubMed.
- I. Paul, A. Goswami, N. Mittal and M. Schmittel, Angew. Chem., Int. Ed., 2018, 57, 354–358 CrossRef CAS PubMed.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- (a) S. Neogi, Y. Lorenz, M. Engeser, D. Samanta and M. Schmittel, Inorg. Chem., 2013, 52, 6975–6984 CrossRef CAS PubMed; (b) S. Neogi, G. Schnakenburg, Y. Lorenz, M. Engeser and M. Schmittel, Inorg. Chem., 2012, 51, 10832–10841 CrossRef CAS PubMed.
- A. Ghosh, I. Paul, S. Saha, T. Paululat and M. Schmittel, Org. Lett., 2018, 20, 7973–7976 CrossRef CAS PubMed.
- H. J. Reich, NMR Spectrum Calculations: WinDNMR, Version 7.1.13, Department of Chemistry, University of Wisconsin Search PubMed.
- M. Schmittel, H. Ammon, V. Kalsani, A. Wiegrefe and C. Michel, Chem. Commun., 2002, 2566–2567 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0dt01961e |
| This journal is © The Royal Society of Chemistry 2020 |