
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative N–H bond activation by amido-Ge(II) cations†
Xueer
Zhou
a,
Petra
Vasko
 ab,
Jamie
Hicks
a,
M. Ángeles
Fuentes
a,
Andreas
Heilmann
a,
Eugene L.
Kolychev
a and
Simon
Aldridge
*a
ab,
Jamie
Hicks
a,
M. Ángeles
Fuentes
a,
Andreas
Heilmann
a,
Eugene L.
Kolychev
a and
Simon
Aldridge
*a
aDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: simon.aldridge@chem.ox.ac.uk
bDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P. O. Box 35, FI-40014 University of Jyväskylä, Finland
First published on 25th June 2020
Abstract
N-heterocyclic carbene (NHC) and tertiary phosphine-stabilized germylium-ylidene cations, [R(L)Ge:]+, featuring tethered amido substituents at R have been synthesized via halide abstraction. Characterization in the solid state by X-ray crystallography shows these systems to be monomeric, featuring a two-coordinate C,N- or P,N-ligated germanium atom. The presence of the strongly Lewis acidic cationic germanium centre and proximal amide function allows for facile cleavage of N–H bonds in 1,2-fashion: the products resulting from reactions with carbazole feature a tethered secondary amine donor bound to a three-coordinate carbazolyl-GeII centre. In each case, addition of the components of the N–H bond occurs to the same face of the germanium amide function, consistent with a coordination/proton migration mechanism. Such as sequence is compatible with the idea that substrate coordination via the pπ orbital at germanium reduces the extent of N-to-Ge π donation from the amide, thereby enhancing the basicity of the proximal N-group.
Introduction
The metal-mediated activation of N–H bonds (particularly those in ammonia) is a challenging fundamental chemical step with potential significance to a number of important transformations of industrial relevance.1 The scarcity of transition metal systems capable of effecting N–H cleavage via oxidative addition, in a manner familiar for a plethora of other E–H bonds, reflects the competing tendency of ammonia to form classical Werner complexes at unsaturated metal centres.2Within p-block chemistry, a number of systems have been reported in the last 15 years which will cleave ammonia to give a derivative containing the E(H)(NH2) function,3,4 including several carbene and related heavier group 14 species in the +2 oxidation state.3 The presence of a low-lying formally vacant pπ orbital in such systems allows for simple coordination of amines (akin to d-block metal complexes); facile N-to-E proton transfer, however, has been proposed to offer a route to generate an amido hydride species without the need for amine dissociation.3e,h,5,6 Moreover, in addition to single site N–H oxidative addition, heavier group 14 analogues of carbenes have also been shown to offer a number of alternative (cooperative) pathways for N–H cleavage involving H-atom transfer to a ligand site (Scheme 1). The relative propensity for different modes of activation has been shown to reflect the identity of the group 14 element/supporting ligand set (and the associated EII/EIV redox potential). In the case of germylene systems, for example, both single site (1,1 addition) and ligand-assisted 1,4 activation modes have been reported, depending not only the basicity of ligand backbone sites, but also the ability of the donor set to promote formation of the GeIV oxidation state.3b–f
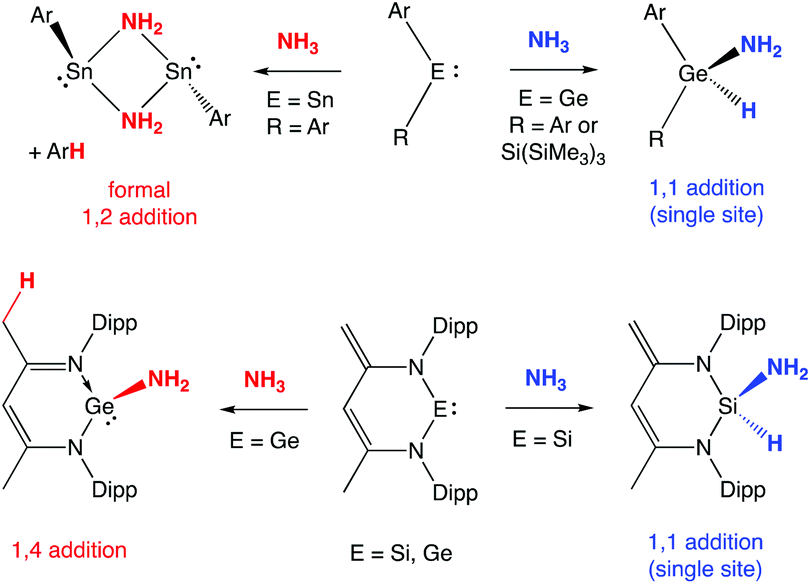 | ||
| Scheme 1 Different modes of N–H activation previously reported for germylene and related systems (Ar = 2,6,-C6H3Mes2; Mes = 2,4,6-Me3C6H2; Dipp = 2,6-iPr2C6H3).3b–e,i | ||
While charge-neutral tetrelenes of the type EX2 have been investigated in some depth,3 isoelectronic cations of the type [R(L)E]+ have been less extensively studied,7–12 despite the fact that the net positive charge should promote initial coordination of ammonia/amines, enhance the acidity of the NH bond and thereby promote N-to-E proton migration. We have recently examined the chemistry of N-nacnac supported tetrylium-ylidene cations towards N–H containing substrates, with the isolation of products derived from oxidative addition or simple amine adduct formation being found to be dependent on the group 14 element (Scheme 2).13
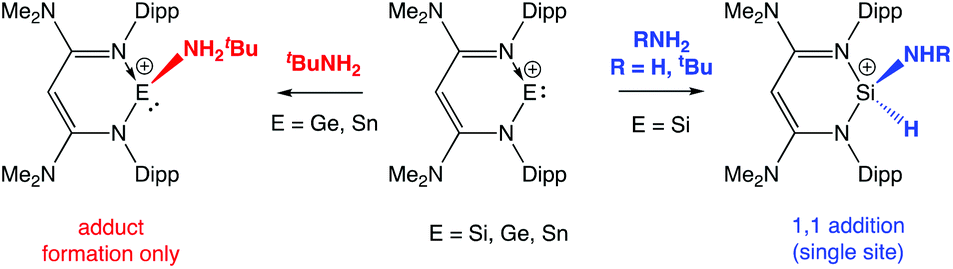 | ||
| Scheme 2 N–H activation vs. amine coordination: reactions of N-nacnac stabilized silylium-, germylium- and stannylium-ylidenes with NH3 and tBuNH2.13 | ||
Given the lack of productivity in N–H activation exhibited by germylium-ylidene systems stabilized by these β-diketiminate (amido/imine) systems we were interested in (i) exploring the possibilities for the synthesis of two-coordinate amidogermylium-ylidene species stabilized by alternative (strong) donor sets (e.g. carbenes14 and phosphines) which might promote the formation of GeIV products; and (ii) exploring the mode(s) of reactivity of such systems towards N–H bonds. These studies are reported in this manuscript.
Results and discussion
Germylium-ylidene synthesis
We initially targeted halo-germylene precursors featuring amido/NHC ligand [L1]− or amido/phosphine ligand [L2]− (Scheme 3). NHC-ligated bromo-germylene precursor 1 can be synthesized via one of two routes: (i) the reaction between protio-ligand [(L1)H2]Br and one equivalent of the germanium(II) bis amide Ge{N(SiMe3)2}2,14,15 or (ii) in situ double deprotonation of [(L1)H2]Br (e.g. with nBuLi) followed by metathesis with GeCl2·dioxane. In our hands, route (i) is preferable, leading to yields of ca. 90%. By contrast, chloro-germylene complex 2 is most readily synthesized by deprotonation of (L2)H, followed by reaction of the lithiated ligand with GeCl2·dioxane. The overall yield for the two steps combined is typically in the region of 50%. Both 1 and 2 have been characterized by standard spectroscopic and analytical methods, and by X-ray crystallography (Fig. 1). The structures of both compounds in the solid state are in line with related complexes, featuring angles at the germanium centre which are close to 90° (e.g. 90.0(1)–98.9(1)° for 1) consistent with the expected (low) degree of ns/np mixing for n = 4.16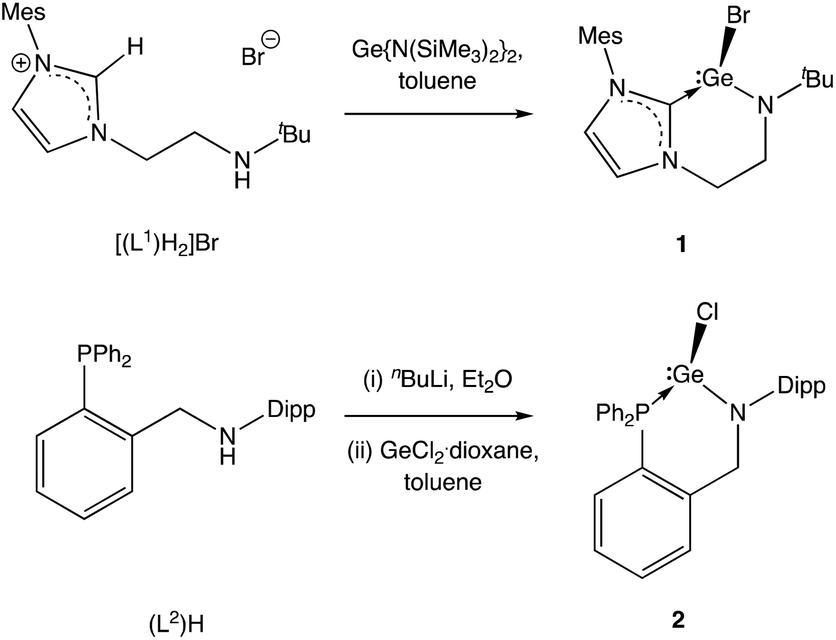 | ||
| Scheme 3 Syntheses of halo-germylene precursors (L1)GeBr (1) and (L2)GeCl 2. | ||
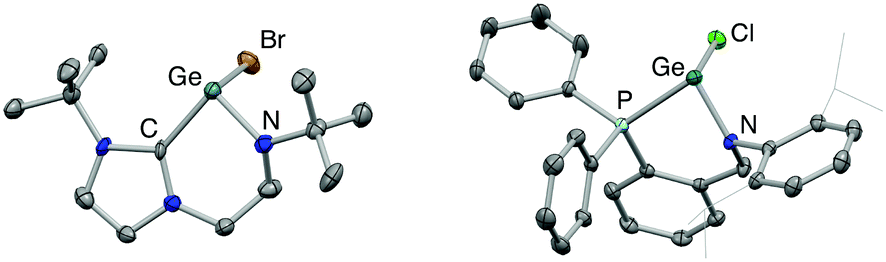 | ||
| Fig. 1 Molecular structures of (L1)GeBr (1, left) and (L2)GeCl (2, right) as determined by X-ray crystallography. Thermal ellipsoids set at the 40% probability level and hydrogen atoms omitted for clarity. Key bond lengths (Å) and angles (°): (for 1) Ge–C 2.07(1), Ge–N 1.866(9), Ge–Br 2.609(2), Br–Ge–C 94.3(3), Br–Ge–N 98.5(3), C–Ge–N 90.5(4); (for 2) Ge–P 2.446(1), Ge–N 1.889(1), Ge–Cl 2.333(1), Cl–Ge–P 90.5(1), Cl–Ge–N 99.0(1), P–Ge–N 84.4(1). | ||
From these precursors, two-coordinate NHC- or phosphine-stabilized GeII cations (germylium-ylidenes) can be synthesised by halide abstraction, most conveniently using Li[Al(OC(CF3)3)4] as a weakly coordinating anion (WCA) source (Scheme 4).17 Treatment of 1 or 2 with Li[Al(OC(CF3)3)4] in bromobenzene at room temperature leads to the formation of the respective cationic species 3 and 4 in reasonable yields (30–40%).18 In both cases, the 1H and 13C NMR spectra reveal distinct changes from the respective bromogermylene precursor: for 3 the NtBu signal is shifted from δH = 1.42 to 0.98 ppm, and the carbenic 13C resonance is shifted upfield from δC = 169.5 to 165.6 ppm. In the case of phosphine-ligated system 4, the 31P resonance is shifted from δP = −24.4 (for 2) to −2.2 ppm.
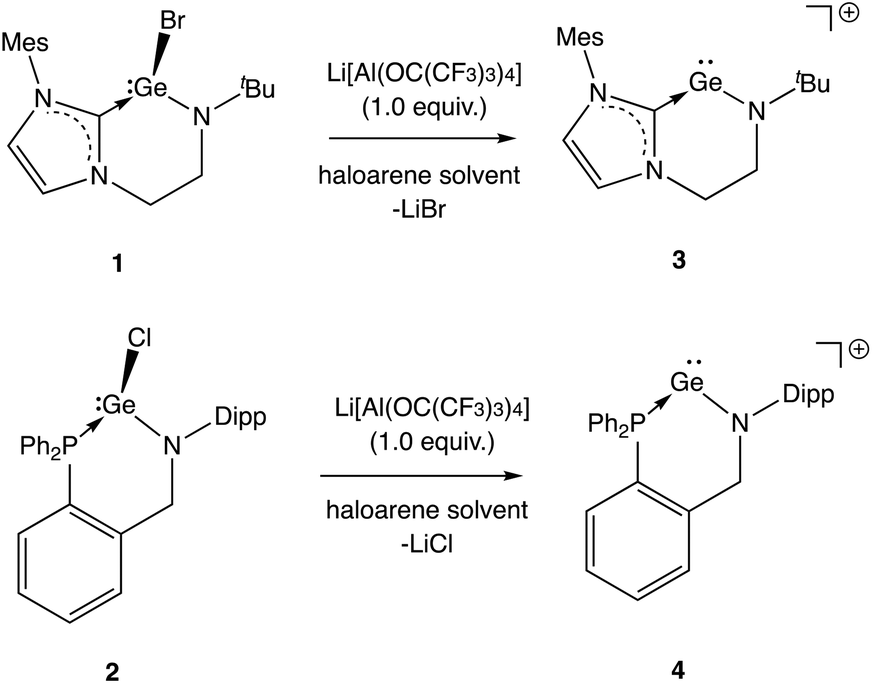 | ||
| Scheme 4 Generation of cationic species via halide abstraction from 1 and 2 (anions omitted for clarity). | ||
Both 3 and 4 could be obtained as single crystals suitable for X-ray diffraction, to allow for unambiguous confirmation of the monomeric two-coordinate structures in the solid state (Fig. 2). Cation formation is reflected in marked shortening of the Ge–N bonds compared to precursors 1 and 2, presumably due to enhanced possibilities for N-to-Ge π bonding in the two-coordinate systems (e.g. d(Ge–N) = 1.889(1), 1.811(3) Å for 2 and 4, respectively). Consistently, in both cases, the geometry around the amido nitrogen is significantly more planar than in the halo-germylene precursor (e.g. 359.7° for 3vs. 350.0° for 1). The distances from germanium to the neutral NHC or phosphine donor, on the other hand, are much less affected by halide abstraction (e.g. d(Ge–P) = 2.446(1), 2.449(1) Å for 2 and 4, respectively). In each cation, the angle subtended at germanium is relatively narrow (91.9(2) and 88.5(1)° for 3 and 4, respectively) reflecting the constraints of the six-membered chelate ring.12 The effect of the differing strengths of the neutral donor (i.e. NHC vs. phosphine) on the Ge–N moiety appear not to be statistically significant: the Ge–N bond lengths for 3 and 4 are 1.829(4) and 1.811(3) Å, respectively.
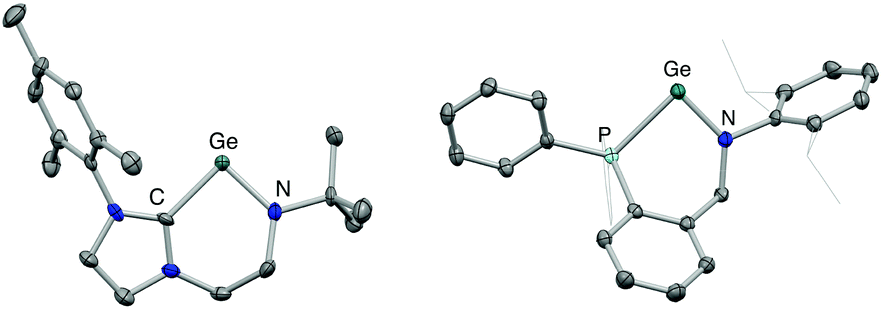 | ||
| Fig. 2 Molecular structures of the cationic component of [(L1)Ge][Al(OC(CF3)3)4] (3, left) and one of the cationic components in the asymmetric unit of [(L2)Ge][Al(OC(CF3)3)4] (4, right) as determined by X-ray crystallography. Thermal ellipsoids set at the 40% probability level. Anions, second component of the asymmetric unit (for 4) and hydrogen atoms omitted, and selected substituents shown in wireframe format for clarity. Key bond lengths (Å) and angles (°): (for 3) Ge–N 1.829(4), Ge–C 2.040(4), C–Ge–N 91.9(2); (for 4) Ge–N 1.811(3), Ge–P 2.449(1), P–Ge–N 88.5(1). | ||
These studies also reveal that the product obtained is strongly dependent on the conditions employed. In the case of 3, clean product formation requires the use of a haloarene solvent (fluoro- or bromobenzene), while the use of benzene leads to the formation of different products arising from incomplete halide abstraction.14 If the reaction is carried out in benzene using 0.5 equiv. of Li[Al(OC(CF3)3)4] the bromide-bridged digermanium system [{(L1)Ge}2(μ-Br)][Al(OC(CF3)3)4] (5) is obtained, via trapping of the [(L1)Ge]+ cation by unreacted 1 (Scheme 5 and Fig. 3).
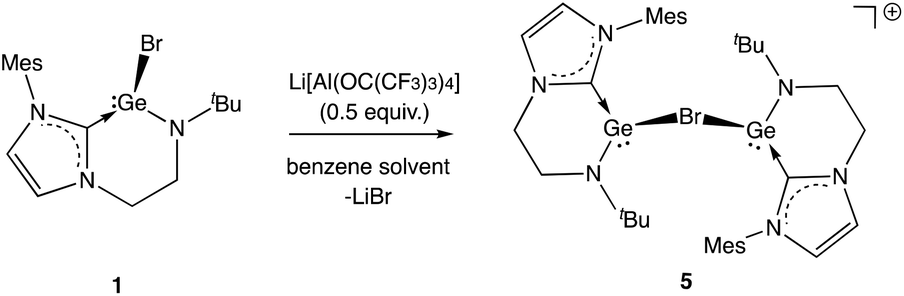 | ||
| Scheme 5 Generation of monocationic digermanium species [{(L1)Ge}2(μ-Br)][Al(OC(CF3)3)4] (5) via incomplete halide abstraction from 1 in benzene solution (anion omitted for clarity). | ||
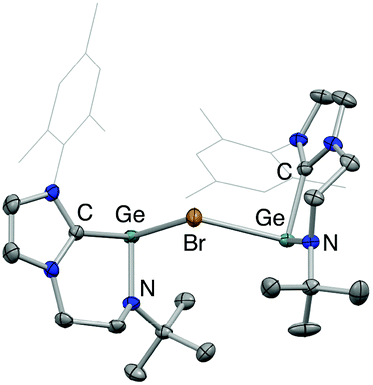 | ||
| Fig. 3 Molecular structure of the cationic component of [{(L1)Ge}2(μ-Br)][Al(OC(CF3)3)4] (5) as determined by X-ray crystallography. Thermal ellipsoids set at the 40% probability level; anion and hydrogen atoms omitted and selected substituents shown in wireframe format for clarity. Key bond lengths (Å) and angles (°): Ge–N 1.857(1), 1.854(1), Ge–C 2.057(2), 2.062(2), Ge–Br 2.729(1), 2.768(1), Br–Ge–C 91.7(1), 88.6(1), Br–Ge–N 95.6(1), 97.9(1), C–Ge–N 90.2(1), 91.0(1). | ||
Reactivity studies – activation of N–H bonds
Mechanistically, the pathways for activation of E–H bonds by tetrelene and related systems are known to be dependent on the nature of E. For H2, mechanisms have been advanced for carbene and silylene systems which involve simultaneous interaction of the substrate with the C/Si centred lone pair and the orthogonal, formally vacant, pπ orbital.3a,19 The orientation of the H2 molecule in the transition state then reflects the relative importance of the donor and acceptor capabilities of the tetrelene. Such mechanistic proposals emphasize the importance of the n-to-pπ energy gap (which is often equivalent to the HOMO–LUMO gap) in facilitating the activation of H2.20On the other hand, protic substrates, such as ammonia, have been shown to be activated by an alternative coordination/proton migration pathway.3e,h,5,6,21 This sequence involves initial coordination of the NH3 molecule, with the tetrelene acting as an electrophile. Subsequent N-to-E proton migration (facilitated, for example, by a second molecule of NH3) then completes the formal N–H oxidative addition process.3e,h,5,6 In this case it is the energy of the vacant pπ orbital of the tetrelene, and its consequent ability to coordinate and activate the NH3 substrate that is thought to be important in bond cleavage.
In the cases of cationic systems 3 and 4, the presence of strongly π-donor amido α-substituents would be expected to lead to significant elevation of the Ge-centred pπ orbital, and this, taken together with the relative narrow angle at germanium in each case (and the associated high degree of 4s character in the lone pair) would be expected to lead to a wide n-to-pπ energy separation.3i,22 Consistently, DFT calculations (PBE1PBE, Def2-TZVP level of theory), exemplified for 3 (Fig. 4) reveal that this separation is >400 kJ mol−1. The LUMO features significant Ge pπ character, with some delocalization onto the carbene carbon, and the expected anti-bonding phase relationship with the N pπ orbital (Fig. 4). The germanium-centred lone pair is relatively low in energy, being associated with the HOMO−3.
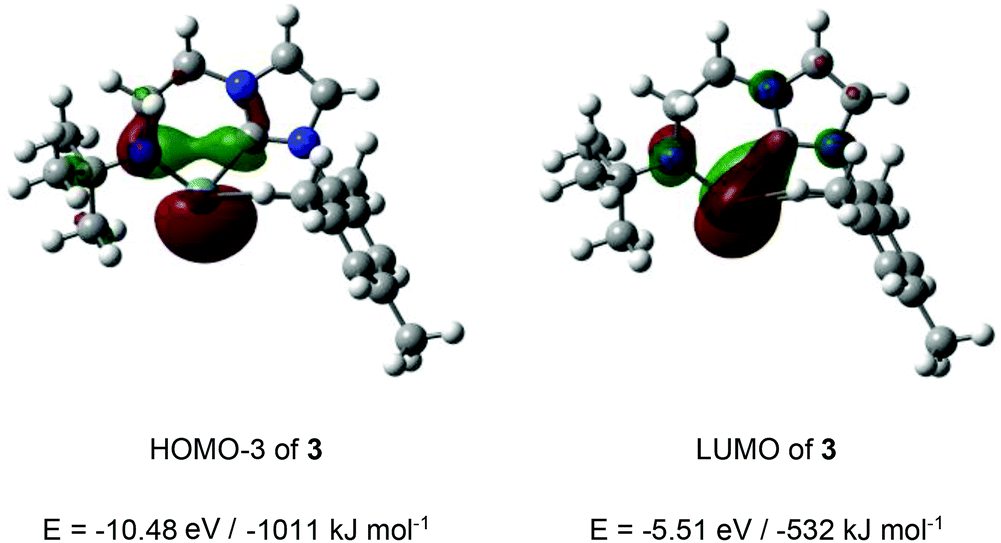 | ||
| Fig. 4 Key germanium-centred frontier orbitals for the cationic component of 3. | ||
Unsurprisingly then, we find that neither 3 and 4 shows any hint of reactivity towards H2, or the hydridic E–H bonds present in PhSiH3, Et3SiH or Me3N·BH3, for which more-or-less concerted oxidative activation would be expected. On the other hand, the low-lying nature of the orbital manifold (and the implied high Lewis acidity) for both systems would appear to be better suited to the activation of polar bonds, such as N–H linkages. Accordingly, the cleavage of N–H bonds can be demonstrated explicitly through the reactions of 3 and 4 with carbazole (Scheme 6). The corresponding reactions with ammonia are much more difficult to control in terms of stoichiometry,23 and invariably result in the presence of protonated ligand among the products formed. Carbazole, by contrast, can easily be added stoichiometrically and its planar structure proves to be critical in isolating the reaction product by crystallization.
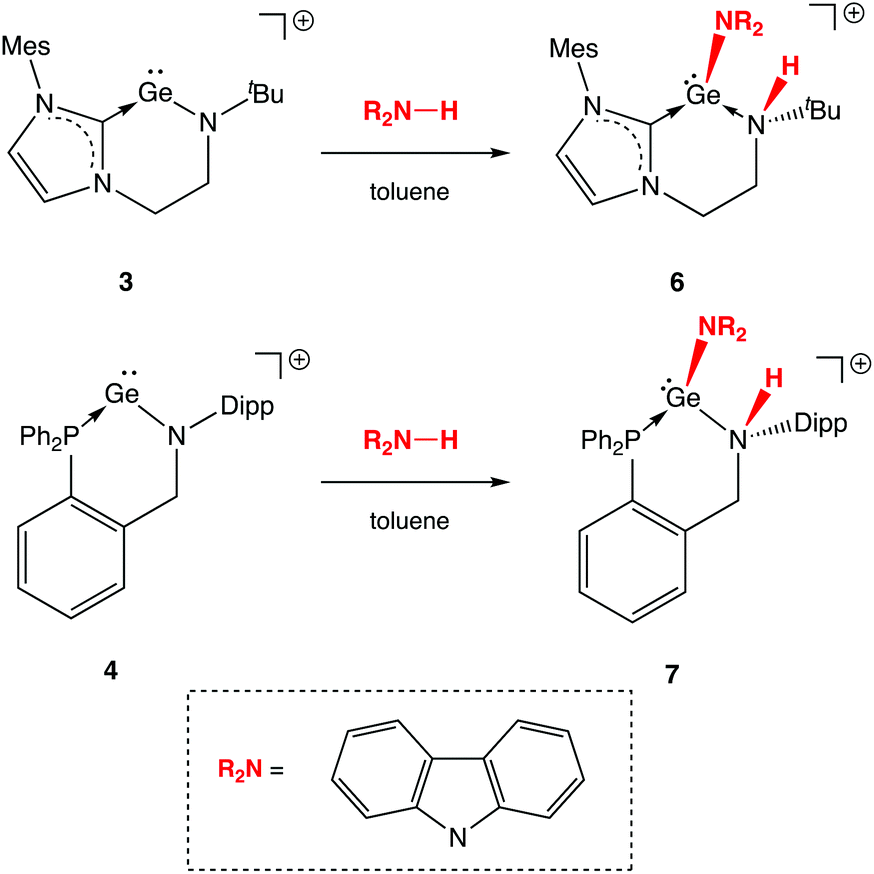 | ||
| Scheme 6 Activation of N–H bonds in 1,2-fashion by NHC and phosphine stabilized germylium-ylidenes (anions omitted for clarity). | ||
In contrast to two-coordinate diaryl germylene and cationic β-diketiminate silylium-ylidene complexes (Schemes 1 and 2), for which single-site N–H activation processes result in net oxidative addition at the group 14 element, the mode of activation in the cases of 3 and 4 involves 1,2-addition across the amido Ge–N bond (Scheme 6). As such, products 6 and 7 are generated, in which the GeII oxidation state is retained, the amido donor is protonated (to generate a secondary amine) and coordination of the anionic carbazolyl conjugate base increases the germanium coordination number from two to three.
N–H bond formation is signalled in each case by the appearance of an additional signal in the respective 1H NMR spectrum, at δH = 3.72 and 6.02 ppm (for 6 and 7, respectively). In the case of 7, the signal in question is a doublet with a 3JHP coupling of ca. 11 Hz to the germanium-bound phosphine donor. In addition (notwithstanding the problems associated with the definitive location of hydrogen atoms by X-ray crystallography), both the pyramidalization of the heavy atom skeleton at N and the lengthening of the Ge–N bond [1.829(4) to 2.134(7) Å for 3/6 and 1.811(3) to 2.137(4) Å for 4/7] are also consistent with the conversion of an anionic amido donor to a charge neutral secondary amine ligand (Fig. 5). In addition, the location of the carbazolyl substituent and the H atom on the same face of the resulting cations 6 and 7 is consistent with a mechanistic hypothesis involving initial N-coordination at the highly Lewis acidic germanium centre followed by proton migration to the proximal amido ligand.3e,h,5,6 Precedent for the formation of an initial donor/acceptor adduct of this sort comes from a recently reported β-diketiminate supported germylium-ylidene cation, which can be isolated due to the presence of a less acidic N–H bond and a less basic amido ligand.13 Subsequent proton transfer to a basic ligand site has previously been reported for Nacnac-derived germanium and aluminium/gallium systems,3d,g,24 and finds more general precedent in the pyridine-derived ligand systems pioneered by Milstein and co-workers.25
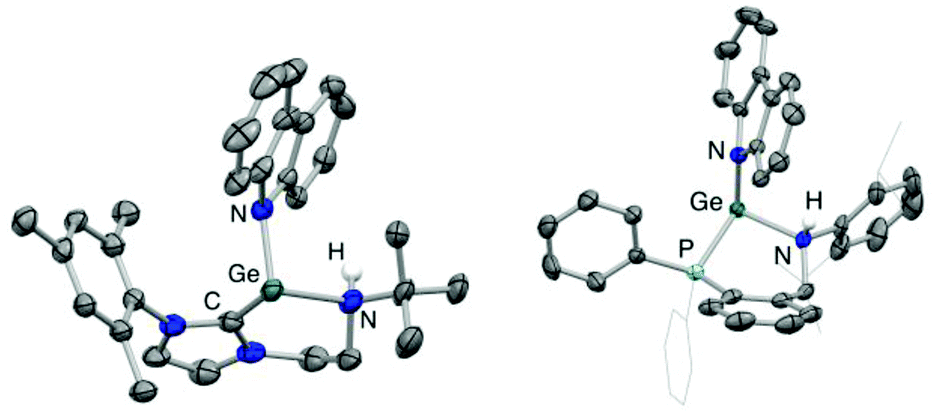 | ||
| Fig. 5 Molecular structures of the cationic components of the N–H bond activation products [(L1H)Ge(NC12H8)][Al(OC(CF3)3)4] (6, left) and [(L2H)Ge(NC12H8)][Al(OC(CF3)3)4] (7, right), as determined by X-ray crystallo-graphy. Thermal ellipsoids set at the 40% probability level; cations and most hydrogen atoms omitted and selected substituents shown in wireframe format. Key bond lengths (Å) and angles (°): (for 6) Ge–N 2.134(7), Ge–C 2.030(9), Ge–Ncarb 1.935(7), C–Ge–N 87.5(3); (for 7) Ge–N 2.137(4), Ge–P 2.481(1), Ge–Ncarb 1.923(3), P–Ge–N 86.8(1). | ||
Conclusions
NHC- and phosphine-stabilized germylium-ylidene cations, featuring tethered amido substituents have been isolated for the first time and shown definitively to be two-coordinate in the solid state by X-ray crystallography. The presence of the strongly Lewis acidic cationic germanium centre and proximal amide function allows for facile cleavage of protic E–H bonds in cooperative (1,2-) fashion (exemplified by the N–H bond in carbazole), leading to the formation of a tethered secondary amine donor bound to a three-coordinate GeII centre. By analogy with chemistry reported for neutral stannylene and germylene systems,3e,h,5,6 and consistent with structural results which imply that addition of the components of the E–H bond happen at one face of the Ge–N linkage, we propose that this chemistry proceeds via coordination of the substrate at the highly electrophilic germanium centre, followed by proton migration (i.e. intramolecular deprotonation) involving the nearby amide group. Such a sequence is consistent with the idea that substrate coordination via the pπ orbital at germanium markedly reduces the extent of N-to-Ge π donation from the amide, thereby enhancing the basicity of the proximal N-group. As such, the presence of the highly Lewis acidic site in cations of this sort is key to cooperative activation of the substrate across the germanium-nitrogen bond. Differences in the regiochemistry of N–H addition compared to other GeII systems (1,2- vs. 1,1- (single site) or 1,4-addition, for example),3d,e can then be rationalized on the basis of the location of the most accessible basic site within an initially formed amine adduct. Consistent with these hypotheses, we find that the HOMO of the model adduct 3·NH3 (at −9.18 eV/−886 kJ mol−1) is characterized as the amide N lone pair: this orbital is elevated significantly from its counterpart in the free cation 3 (the HOMO-2 at −10.22 eV/−986 kJ mol−1). The germanium-centred lone pair in the adduct 3·NH3 is found in the HOMO−1 (at −9.44 eV/−911 kJ mol−1) (see ESI†).Experimental
General considerations
All manipulations were carried out using standard Schlenk line or dry-box techniques under an atmosphere of argon. Toluene and hexane were degassed by sparging with argon and dried by passing through a column of the appropriate drying agent using a commercially available Braun SPS and stored over potassium; fluorobenzene and bromobenzene were dried by refluxing over CaH2 and stored over molecular sieves. Benzene-d6 was dried using a potassium mirror and bromobenzene-d5 dried using CaH2 and stored over molecular sieves. NMR samples were prepared under argon in 5 mm Wilmad 507-PP tubes fitted with J. Young Teflon valves. NMR spectra were measured on Bruker Avance III HD Nanobay or Bruker AVII spectrometers operating at 400 or 500 MHz, respectively (for 1H measurements); 1H and 13C NMR spectra were referenced internally to residual protio-solvent (1H) or solvent (13C) resonances and are reported relative to tetramethylsilane. 19F and 27Al NMR spectra were referenced with respect to CFCl3 and [Al(H2O)6]3+, respectively. Chemical shifts are quoted in δ (ppm) and coupling constants in Hz. Elemental analyses were carried out at London Metropolitan University. Protio-ligands [(L1)H2]Br and (L2)H,15 and metal precursors Ge{N(SiMe3)2}226 and Li[Al(OC(CF3)3)4]17 were prepared via literature methods. Li(L2) was prepared from (L2)H and nBuLi as described in the ESI.†DFT calculations
All computational work reported here was carried out using density functional theory (DFT) within the Gaussian16 (Revision C.01) program package.27 Geometry optimizations were performed with the PBE1PBE exchange correlation functional,28–30 using the Def2-TZVP basis set with an ultrafine integration grid and Grimme's empirical dispersion correction (GD3BJ).31 The nature of stationary points found (minimum) was confirmed by full frequency calculations (no imaginary frequencies).Crystallography
Single-crystal X-ray diffraction data for all compounds were collected using an Oxford Diffraction Supernova dual-source diffractometer equipped with a 135 mm Atlas CCD area detector. Crystals were selected under Paratone-N oil, mounted on MiTeGen Micromount loops and quench-cooled using an Oxford Cryosystems open flow N2 cooling device.32 Data were collected at 150 K using mirror monochromated Cu Kα radiation (λ = 1.5418 Å; Oxford Diffraction Supernova). Data collected were processed using the CrysAlisPro package, including unit cell parameter refinement and inter-frame scaling (which was carried out using SCALE3 ABSPACK within CrysAlisPro).33 Equivalent reflections were merged and diffraction patterns processed with the CrysAlisPro suite.33 Structure were solved ab initio from the integrated intensities using SHELXT34 and refined on F2 using SHELXL34 with the graphical interface Olex235 or X-Seed.36 Full details are given in the supplementary deposited CIF files (CCDC 1952091–1952095, 1952097 and 2005217–2005219†).Syntheses of novel compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 928.28(18) Å3, Z = 8, ρc = 1.478 g cm−3, T = 150 K, λ = 1.54184 Å, R1 = 0.0406 for 3229 observed unique reflections [I > 2σ(I)], wR2 = 0.0983 for all 4070 unique reflections. Max. and min. residual electron densities 0.94, −0.43 e Å−3. CCDC 1952092.†
928.28(18) Å3, Z = 8, ρc = 1.478 g cm−3, T = 150 K, λ = 1.54184 Å, R1 = 0.0406 for 3229 observed unique reflections [I > 2σ(I)], wR2 = 0.0983 for all 4070 unique reflections. Max. and min. residual electron densities 0.94, −0.43 e Å−3. CCDC 1952092.†
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.1837(3), b = 11.0396(4), c = 14.5439(5) Å, α = 111.357(3)°, β = 90.475(3)°, γ = 90.475(3)°, V = 1371.34(9) Å3, Z = 2, ρc = 1.353 g cm−3, T = 150 K, μ(CuKα) = 3.105 mm−1, λ = 1.54184 Å, R1 = 0.0250 for 5273 observed unique reflections [I > 2σ(I)], wR2 = 0.0672 for all 5691 unique reflections. Max. and min. residual electron densities 0.50, −0.25 e Å−3. CCDC 2005217.†
, a = 12.1911(3), b = 15.3762(6), c = 29.9659(10) Å, α = 83.214(3)°, β = 88.694(2)°, γ = 87.381(2)°, V = 5571.2(3) Å3, Z = 4, ρc = 1.777 g cm−3, T = 150 K, μ(CuKα) = 2.718 mm−1, λ = 1.54184 Å, R1 = 0.0647 for 19220 observed unique reflections [I > 2σ(I)], wR2 = 0.1824 for all 24700 unique reflections. Max. and min. residual electron densities 1.84, −0.86 e Å−3. CCDC 2005219.†
, a = 11.0633(1), b = 18.7791(2), c = 18.8201(2) Å, α = 61.968(1), β = 80.591(1), γ = 80.424(1)°, V = 3386.38(7) Å3, Z = 2, ρc = 1.727 g cm−3, T = 150 K, λ = 1.54184 Å, R1 = 0.0265, for 13008 observed unique reflections [I > 2σ(I)], wR2 = 0.0681 for all 14100 unique reflections. Max. and min. residual electron densities 0.66, −0.51 e Å−3. CCDC 1952095.†
309.3(7) Å3, Z = 16, ρc = 1.616 g cm−3, T = 150 K, λ = 1.54184 Å, R1 = 0.1155 for 12751 observed unique reflections [I > 2σ(I)], wR2 = 0.3316 for all 13230 unique reflections. Max. and min. residual electron densities 0.63, −0.39 e Å−3. CCDC 1952097.†
281.4(7) Å3, Z = 8, ρc = 1.658 g cm−3, T = 150 K, μ(CuKα) = 2.356 mm−1, λ = 1.54184 Å, R1 = 0.0669 for 10222 observed unique reflections [I > 2σ(I)], wR2 = 0.1958 for all 13689 unique reflections. Max. and min. residual electron densities 0.737, −0.595 e Å−3. CCDC 2005218.†
, a = 9.1837(3), b = 11.0396(4), c = 14.5439(5) Å, α = 111.357(3)°, β = 90.475(3)°, γ = 90.475(3)°, V = 1371.34(9) Å3, Z = 2, ρc = 1.353 g cm−3, T = 150 K, μ(CuKα) = 3.105 mm−1, λ = 1.54184 Å, R1 = 0.0250 for 5273 observed unique reflections [I > 2σ(I)], wR2 = 0.0672 for all 5691 unique reflections. Max. and min. residual electron densities 0.50, −0.25 e Å−3. CCDC 2005217.†
, a = 12.1911(3), b = 15.3762(6), c = 29.9659(10) Å, α = 83.214(3)°, β = 88.694(2)°, γ = 87.381(2)°, V = 5571.2(3) Å3, Z = 4, ρc = 1.777 g cm−3, T = 150 K, μ(CuKα) = 2.718 mm−1, λ = 1.54184 Å, R1 = 0.0647 for 19220 observed unique reflections [I > 2σ(I)], wR2 = 0.1824 for all 24700 unique reflections. Max. and min. residual electron densities 1.84, −0.86 e Å−3. CCDC 2005219.†
, a = 11.0633(1), b = 18.7791(2), c = 18.8201(2) Å, α = 61.968(1), β = 80.591(1), γ = 80.424(1)°, V = 3386.38(7) Å3, Z = 2, ρc = 1.727 g cm−3, T = 150 K, λ = 1.54184 Å, R1 = 0.0265, for 13008 observed unique reflections [I > 2σ(I)], wR2 = 0.0681 for all 14100 unique reflections. Max. and min. residual electron densities 0.66, −0.51 e Å−3. CCDC 1952095.†
309.3(7) Å3, Z = 16, ρc = 1.616 g cm−3, T = 150 K, λ = 1.54184 Å, R1 = 0.1155 for 12751 observed unique reflections [I > 2σ(I)], wR2 = 0.3316 for all 13230 unique reflections. Max. and min. residual electron densities 0.63, −0.39 e Å−3. CCDC 1952097.†
281.4(7) Å3, Z = 8, ρc = 1.658 g cm−3, T = 150 K, μ(CuKα) = 2.356 mm−1, λ = 1.54184 Å, R1 = 0.0669 for 10222 observed unique reflections [I > 2σ(I)], wR2 = 0.1958 for all 13689 unique reflections. Max. and min. residual electron densities 0.737, −0.595 e Å−3. CCDC 2005218.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the Academy of Finland (project number 314794, PV), the Oxford-SCG Centre of Excellence and the Leverhulme Trust (grant number RP-2018-246, JH), the EPSRC (grant number EP/K014714/1, MAF) and the EU 7th Framework Program, Marie Skłodowska-Curie actions (grant number PIEF-GA-2013-626441 for an IEF Fellowship, EK). We also thank Oxford University Advanced Research Computing (ARC) facilities for computational resources.Notes and references
- See, for example; (a) J. Haggin, Chem. Eng. News, 1993, 71, 23 CrossRef; (b) J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86–95 CrossRef CAS PubMed.
- For examples of transition metal activation of NH3 by a range of mechanisms: (a) E. G. Bryan, B. F. G. Johnson and J. Lewis, J. Chem. Soc., Dalton Trans., 1977, 1328–1330 RSC; (b) G. L. Hillhouse and J. E. Bercaw, J. Am. Chem. Soc., 1984, 106, 5472–5478 CrossRef CAS; (c) A. L. Casalnuovo, J. C. Calabrese and D. Milstein, Inorg. Chem., 1987, 26, 973–976 CrossRef; (d) M. M. Banaszak Holl, P. T. Wolczanski and G. D. Van Duyne, J. Am. Chem. Soc., 1990, 112, 7989–7994 CrossRef CAS; (e) J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed; (f) Y. Nakajima, H. Kameo and H. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 950–952 CrossRef CAS PubMed; (g) C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318–10319 CrossRef CAS PubMed; (h) E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef CAS PubMed; (i) I. Mena, M. A. Casado, P. García-Orduña, V. Polo, F. J. Lahoz, A. Fazal and L. A. Oro, Angew. Chem., Int. Ed., 2011, 50, 11735–11738 CrossRef CAS PubMed; (j) T. Kimura, N. Koiso, K. Ishiwata, S. Kuwata and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 8880–8883 CrossRef CAS PubMed; (k) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed; (l) Y.-H. Chang, Y. Nakajima, H. Tanaka, K. Yoshizawa and F. Ozawa, J. Am. Chem. Soc., 2013, 135, 11791–11794 CrossRef CAS PubMed; (m) R. M. Brown, J. Borau Garcia, J. Valjus, C. J. Roberts, H. M. Tuononen, M. Parvez and R. Roesler, Angew. Chem., Int. Ed., 2015, 54, 6274–6277 CrossRef CAS PubMed; (n) E. Despagnet-Ayoub, M. K. Takase, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2015, 137, 10500–10503 CrossRef CAS PubMed; (o) M. J. Bezdek, S. Guo and P. Chirik, Science, 2016, 354, 730–733 CrossRef CAS PubMed; (p) G. Margulieux, M. J. Bezdek, Z. R. Turner and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 6110–6113 CrossRef CAS PubMed; (q) E. A. LaPierre, W. E. Piers and C. Gendy, Dalton Trans., 2018, 47, 16789–16797 RSC.
- For examples of NH3 activation by carbenes and heavier group 14 analogues, see for example: (a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed; (b) Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed; (c) A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef CAS PubMed; (d) A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Inorg. Chem., 2009, 48, 798–800 CrossRef CAS PubMed; (e) Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS PubMed; (f) A. Meltzer, S. Inoue, C. Präsang and M. Driess, J. Am. Chem. Soc., 2010, 132, 3038–3046 CrossRef CAS PubMed; (g) W. Wang, S. Inoue, S. Yao and M. Driess, Organometallics, 2011, 30, 6490–6494 CrossRef CAS; (h) A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed; (i) M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem. – Eur. J., 2016, 22, 11685–11698 CrossRef CAS PubMed; (j) D. Wendel, T. Szilvási, D. Henschel, P. J. Altmann, C. Jandl, S. Inoue and B. Rieger, Angew. Chem., Int. Ed., 2018, 57, 14575–14579 CrossRef CAS PubMed. See also: (k) C. Präsang, M. Stoelzel, S. Inoue, A. Meltzer and M. Driess, Angew. Chem., Int. Ed., 2010, 49, 10002–10005 CrossRef PubMed.
- For other examples of main group compounds capable of NH3 activation see, for example: (a) Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2031–2034 CrossRef CAS PubMed; (b) S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef CAS PubMed; (c) T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed; (d) J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef CAS PubMed; (e) W. Zhao, S. M. McCarthy, T. Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS PubMed; (f) J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed; (g) T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- M. E. Alberto, N. Russo and E. Sicilia, Chem. – Eur. J., 2013, 19, 7835–7846 CrossRef CAS PubMed.
- For a related mode of coordination/activation of hydrazine see: Z. Brown, J.-D. Guo, S. Nagase and P. P. Power, Organometallics, 2012, 31, 3768–3772 CrossRef CAS.
- For relevant reviews see: (a) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (b) V. S. V. S. N. Swamy, S. Pal, S. Khan and S. S. Sen, Dalton Trans., 2015, 44, 12903–12923 RSC; (c) T. A. Engesser, M. R. Lichenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC; (d) H. Fang, Z. Wang and X. Fu, Coord. Chem. Rev., 2017, 344, 214–237 CrossRef CAS.
- A quasi one-coordinate GeII cation: J. Li, C. Schenk, F. Winter, H. Scherer, N. Trapp, A. Higelin, S. Keller, R. Pöttgen, I. Krossing and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 9557–9561 CrossRef CAS PubMed.
- Cyclopentadienyl supported GeII cations: (a) J. G. Winter, P. Portius, G. Kociok-Kohn, R. Steck and A. C. Filippou, Organometallics, 1998, 17, 4176–4182 CrossRef CAS For a related example featuring neutral arene donor stabilization see, for example: (b) T. Probst, O. Steigelmann, J. Riede and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1990, 29, 1397–1398 CrossRef.
- N-Donor supported GeII cations: (a) H. V. R. Dias and Z. Wang, J. Am. Chem. Soc., 1997, 119, 4650–4655 CrossRef CAS; (b) M. Stender, A. D. Phillips and P. P. Power, Inorg. Chem., 2001, 40, 5314–5315 CrossRef CAS PubMed; (c) F. Cheng, J. M. Dyke, F. Ferrante, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Dalton Trans., 2010, 39, 847–856 RSC; (d) Y. Xiong, S. Yao, S. Inoue, A. Berkefeld and M. Driess, Chem. Commun., 2012, 48, 12198–12200 RSC.
- Carbodiphosphorane-supported GeII cations: S. Khan, G. Gopakumar, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2013, 52, 5644–5647 CrossRef CAS PubMed.
- Carbene-supported GeII cations: (a) P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138–15139 CrossRef CAS PubMed; (b) Y. Xiong, S. Yao, G. Tan, S. Inoue and M. Driess, J. Am. Chem. Soc., 2013, 135, 5004–5007 CrossRef CAS PubMed; (c) K. Inomata, T. Watanabe and H. Tobita, J. Am. Chem. Soc., 2014, 136, 14341–14344 CrossRef CAS PubMed; (d) K. Inomata, T. Watanabe, Y. Miyazaki and H. Tobita, J. Am. Chem. Soc., 2015, 137, 11935–11937 CrossRef CAS PubMed; (e) A. Rit, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2016, 55, 378–382 CrossRef CAS PubMed; (f) M. Roy, S. Fujimori, M. Ferguson, R. McDonald, N. Tokitoh and E. Rivard, Chem. – Eur. J., 2018, 24, 14392–14399 CrossRef CAS PubMed; (g) R. J. Mangan, A. Rit, C. P. Sindlinger, R. Tirfoin, J. Campos, J. Hicks, K. E. Christensen, H. Niu and S. Aldridge, Chem. – Eur. J., 2020, 26, 306–315 CrossRef CAS PubMed See also: (h) A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022–1026 CrossRef CAS PubMed.
- D. C. H. Do, A. V. Protchenko, M. Á. Fuentes, J. Hicks, P. Vasko and S. Aldridge, Chem. Commun., 2020, 56, 4684–4687 RSC.
- Hahn and Glorius have reported attempts to synthesize germylium-ylidene systems featuring amido substituents and stabiliized by NHC donors, although no structural or reactivity data was reported: D. Paul, F. Heins, S. Krupski, A. Hepp, C. G. Daniliuc, K. Klahr, J. Neugebauer, F. Glorius and F. E. Hahn, Organometallics, 2017, 36, 1001–1008 CrossRef CAS.
- (a) P. L. Arnold, S. A. Mungur, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2003, 42, 5981–5984 CrossRef CAS PubMed; (b) B. Liu, D. Cui, J. Ma, X. Chen and X. Jing, Chem. – Eur. J., 2007, 13, 834–845 CrossRef CAS PubMed.
- M. Kaupp, in The Chemical Bond: Fundamental Aspects of Chemical Bonding, eds. G. Frenking and S. Shaik, Wiley-VCH, Weinheim, 2014, pp. 1–24 Search PubMed.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS.
- Similar chemistry can be used to access compounds analogous to 1 and 3 featuring an imidazole bound tBu group (see ESI†).
- (a) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed; (b) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568–571 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- For key papers on the activation of related E–H bonds by germylenes, see, for example ref. 3f, and (a) J. W. Dube, Z. D. Brown, C. A. Caputo, P. P. Power and P. J. Ragogna, Chem. Commun., 2014, 50, 1944–1946 RSC; (b) Z. D. Brown, J. D. Erickson, J. C. Fettinger and P. P. Power, Organometallics, 2013, 32, 617–622 CrossRef CAS; (c) M. M. Juckel, J. Hicks, D. Jiang, L. Zhao, G. Frenking and C. Jones, Chem. Commun., 2017, 53, 12692–12695 RSC; (d) F. Diab, F. S. W. Aicher, C. P. Sindlinger, K. Eichele, H. Schubert and L. Wesemann, Chem. – Eur. J., 2019, 25, 4426–4434 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed.
- Attempts to use thf solutions of ammonia invariably led to problems stemming from the strongly Lewis acidic nature of 3/4.
- J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed.
- (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (b) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- D. H. Harris and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1974, 895–896 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105–107 CrossRef CAS.
- CrysAlisPro, Agilent Technologies, Version 1.171.35.8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic and characterizing data, representative NMR spectra of new compounds, xyz file for DFT optimized structure; CIFs available from the CCDC, references 1952091–1952095, 1952097 and 2005217–2005219. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt01960g |
| This journal is © The Royal Society of Chemistry 2020 |