
Manipulating metal spin states for biomimetic, catalytic and molecular materials chemistry

Abstract
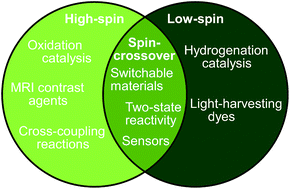
This article surveys the relationship between ligand type, coordination geometry and metal spin state in complexes of iron and other metal ions. Compounds and materials containing high-, intermediate- and low-spin metal ions differ in their molecular structures, their physical properties and their chemical reactivity. Implications and applications of these variations are summarised, including the use of base metals in light-harvesting dyes and in different forms of catalysis. Recent studies of the electronic influence of ligand substituents, or ligand conformational constraints, on metal ion spin states are described, which have revealed unexpected complexities.
- This article is part of the themed collection: 2020 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

