
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation effects on dynamics of ligand-benzylated formazanate boron and aluminium complexes†
Ranajit
Mondol
 and
Edwin
Otten
*
and
Edwin
Otten
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: edwin.otten@rug.nl
First published on 15th June 2020
Abstract
The dynamic processes present in ligand-benzylated formazanate boron and aluminium complexes are investigated using variable temperature NMR experiments and lineshape analyses. The observed difference in activation parameters for complexes containing either organic countercations (NBu4+) or alkali cations is rationalized on the basis of a different degree of ion-pairing in the ground state, and the data are in all cases consistent with a mechanism that involves pyramidal inversion at the nitrogens in the heterocyclic ring rather than homolytic N–C(benzyl) bond cleavage.
Introduction
Alkali cations have important roles in a biological context1–3 and synthetic chemistry.4–6 The noncovalent interactions of organic moieties with alkali–metal cations can have a pronounced effect on the (electronic) structure and reactivity of complexes, as exemplified by features such as geometry,7,8 redox potentials,9 N2 cleavage,10 reaction rates,11–14 and even selectivity of chemical transformations.15–17 Of particular relevance to the work described here is a series of reduced iron complexes with a redox-active formazanate ligand that are paired with alkali–metal countercations as reported by Broere, Holland and co-workers.18 In the presence of a crown ether to sequester the alkali cation, the reduced iron complex was obtained as a monomer, whereas in absence of a crown ether the reduced iron complex was isolated in dimeric form (Chart 1, A).18 It was demonstrated that in these dimeric compounds, the binding mode of counter cations to the ligand is dependent on the nature of the alkali–metal (Chart 1), and that this has a pronounced effect on the structure and dynamics of these compounds.18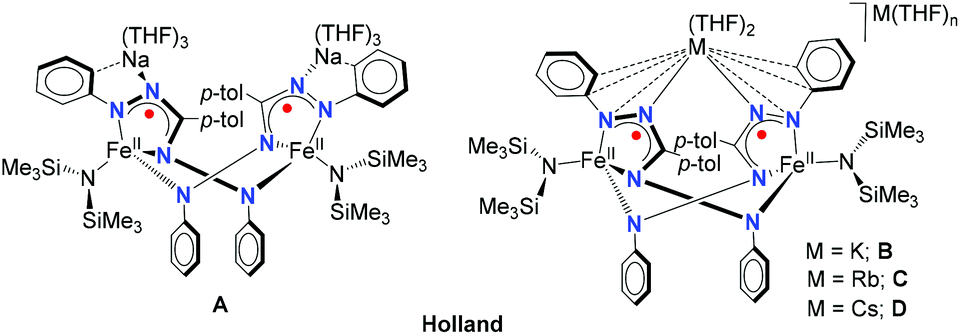 | ||
| Chart 1 | ||
The formazanate ligands used in this study have received increasing attention in the last decade following a report by Hicks and coworkers that described ligand-centered redox-reactions in a formazanate boron compound.19 In 2014, our group has started to investigate the coordination chemistry, ligand-centered reductions and reactivity of compounds with formazanate ligands.20–27 Concurrent with our work, the Gilroy group28–37 and others38–40 have synthesized a variety of compounds with formazanate ligands, and studied their optical and electrochemical properties.41 Previously, we described that main group (B and Al) complexes with these ligands can accept up to two electrons, and that these reduction products subsequently react with electrophiles (e.g., Bn+ or H+) to form new N–C and N–H bonds at the formazanate ligand (Scheme 1).42–45 Furthermore, we have demonstrated that the [2e−/E+]-equivalent ‘stored’ at the ligand could be converted to E˙ radicals via homolytic cleavage of the N–C(Bn) and N–H bonds, respectively.43 In a recent paper, the structural features and homolytic bond dissociation energies of ligand-benzylated B and Al complexes were reported.45 These studies suggested that the increased ionic character in the Al analogues leads to more facile bond homolysis, while the rate of benzyl transfer to TEMPO is independent on the nature of the countercation.
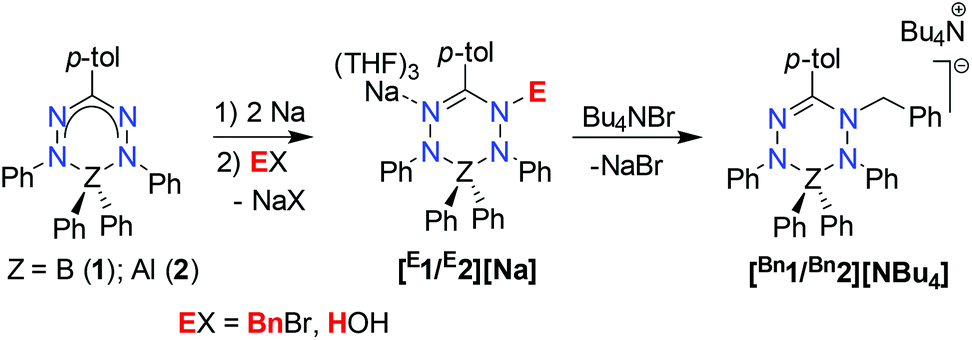 | ||
| Scheme 1 Ligand-based storage of [2e−/E+]-equivalent in B and Al complexes with formazanate ligands. | ||
Here, we report a study on the dynamics of anionic, ligand-benzylated B and Al complexes by NMR lineshape analysis for the resonances of the (diastereotopic) N-benzyl group. Analysis of the empirical exchange rates for a series of compounds with different countercations suggests that the dynamic features are due to inversion at the nitrogen atom rather than homolytic N–C bond cleavage, and that the rate-determining step involves the formation of a separated ion-pair. The results presented herein emphasize the importance of coulombic and/or cation-π interactions in modulating structure and reactivity.
Results and discussion
Treatment of the two-electron reduced formazanate boron diphenyl compound 12− with benzyl bromide afforded the ligand-benzylated anionic complex [Bn1][Na] (Scheme 1) as reported previously.43 The 1H NMR spectrum recorded in THF-d8 solution at room temperature showed broadened resonances for the protons at the benzylic position, which upon cooling to −5 °C became a sharp set of mutually coupled signals (δ 3.79 and 3.38 ppm; 2JHH = 15.3 Hz) due to the diastereotopic benzyl-CH2 group. Conversely, measurement of the NMR spectrum at 65 °C or above showed that these resonances coalesce to a singlet (δ 3.69 ppm), indicating a dynamic process that exchanges the two diastereotopic sites. Similar dynamic features are also observed for the BPh2 groups. In order to probe the nature of these dynamics, 1H NMR spectra were collected in the temperature range between −5 and +75 °C (Fig. 1) and lineshape analysis was performed to obtain exchange rate constants. The rate of chemical exchange for the benzylic protons is found to be identical to that of the BPh2 groups across the entire temperature range, indicating that a single dynamic process is involved. Eyring analysis afforded activation parameters for the exchange in [Bn1][Na] as ΔH‡ = 80.4 ± 1.7 kJ mol−1 and ΔS‡ = 59.3 ± 5.6 J mol−1 K−1 (Fig. 2 and Table 1; see ESI† for details).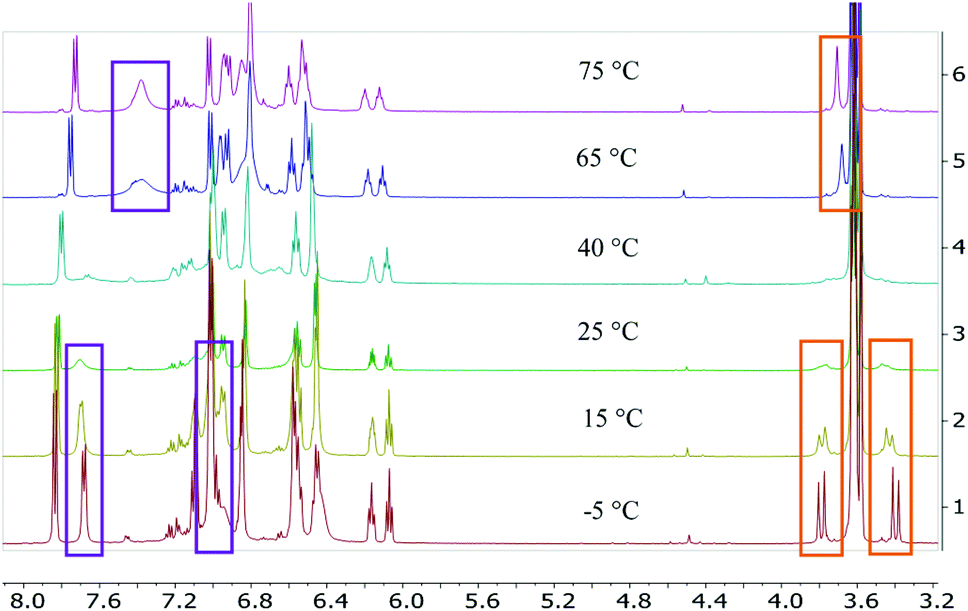 | ||
| Fig. 1 Selected variable temperature 1H NMR spectra of [Bn1][Na] in THF-d8. The resonances indicated in the orange boxes (δ 3.4–3.8 ppm) are for the benzyl-CH2 group, those in the aromatic range (δ 7.0–7.8 ppm, purple box) are for the BPh2 groups. | ||
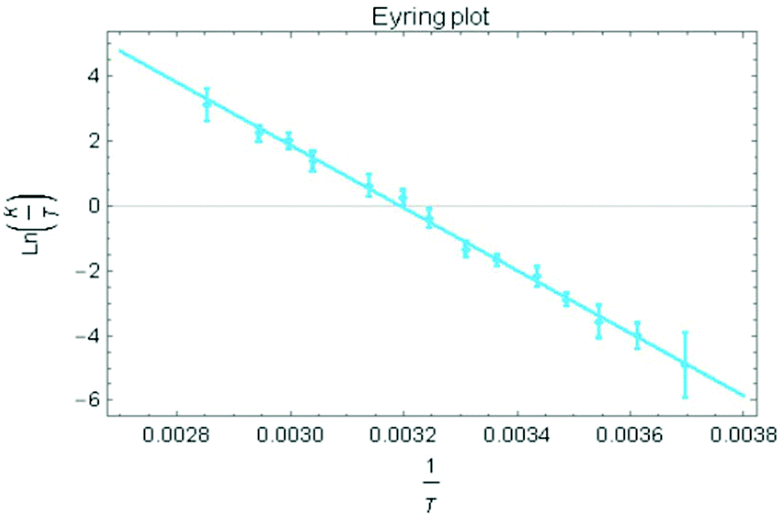 | ||
| Fig. 2 Eyring plot for the determination of activation parameters for benzyl-CH2 exchange in [Bn1][Na]. | ||
| ΔH‡ (kJ mol−1) | ΔS‡ (J mol−1 K−1) | ΔG‡298K (kJ mol−1) | |
|---|---|---|---|
| [Bn1]− | |||
| NBu4+ | 66.7 ± 1.6 | −4.2 ± 5.0 | 68.0 |
| Na+ | 80.4 ± 1.7 | 59.3 ± 5.6 | 62.7 |
| K+ | 68.4 ± 2.3 | 59.3 ± 7.3 | 50.7 |
| Rb+ | 72.7 ± 5.5 | 42.3 ± 18.0 | 60.1 |
| [Bn2]− | |||
| NBu4+ | 68.4 ± 1.3 | −19.7 ± 3.9 | 74.3 |
| Na+ | 88.9 ± 2.5 | 56.5 ± 7.3 | 72.1 |
The large, positive entropy of activation suggests a substantial degree of bond-breaking in the rate-determining step. Although homolytic N–C(benzyl) bond cleavage to generate Bn˙ and 1˙− (Scheme 2A) is a potential mechanism for exchange of the diastereotopic environments in [Bn1][Na] and is consistent with the empirical value for ΔS‡, our previous data for the N–C(benzyl) bond dissociation energy in [Bn1][Na] (BDE = 121 ± 5 kJ mol−1; measured via Bn˙ transfer to TEMPO)43 is significantly higher than the activation enthalpy for exchange (ΔH‡ = 80.4 (1.7) kJ mol−1). This rules out that bond homolysis is operative as a mechanism for site exchange in [Bn1][Na].
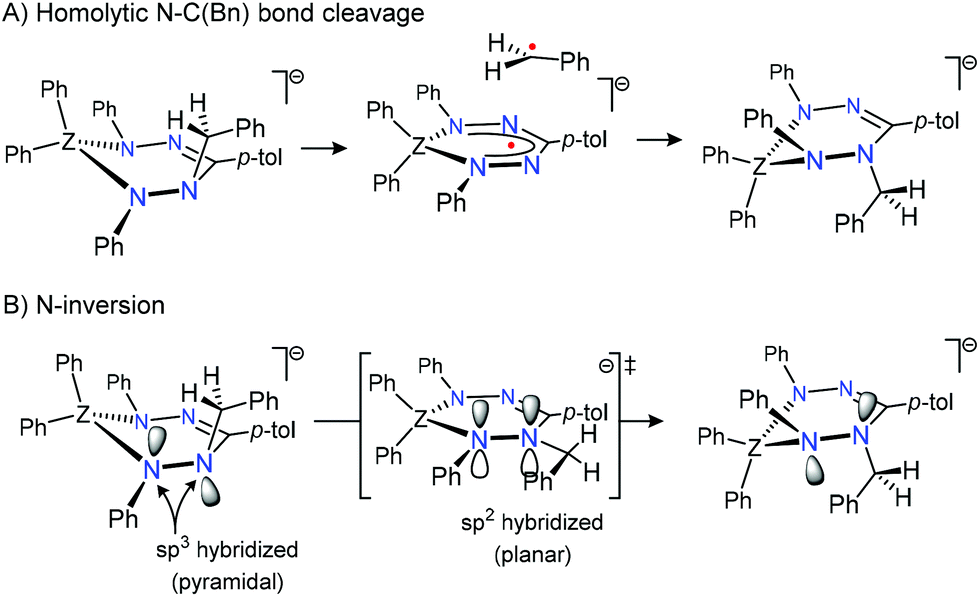 | ||
| Scheme 2 Schematic representation of possible mechanisms to account for exchange between the diastereotopic benzyl CH2 resonances in anions [Bn1/2]−. | ||
Alternatively, the observed dynamics in [Bn1][Na] could be due to pyramidal inversion via a planar transition state with sp2-hybridized, trigonal planar geometries around the hydrazine nitrogen atoms (Scheme 2B). Pyramidal nitrogen inversion is a facile, low-barrier process in the majority of N-containing compounds, but substantial activation energies have been reported when the N-atom is part of a (hetero)cyclic ring.46–50 However, nitrogen inversion is an intra-molecular process that would be expected to have an activation entropy close to zero, or (in the case of highly strained systems) a slightly negative value.46–48
Although both homolytic N–C bond cleavage and N-inversion are plausible mechanisms to explain the dynamics observed for [Bn1][Na], neither of the two can be reconciled with the activation parameters determined experimentally. It thus appears that the rate-determining step as probed by the NMR lineshape analysis may precede the actual exchange of the diastereotopic sites. To investigate whether (partial) dissociation of the Na+ cation in [Bn1][Na] could play a role, we subsequently examined the exchange rate in the corresponding tetrabutylammonium salt [Bn1][NBu4].45 In this compound, the organic cation Bu4N+ does not interact with the [Bn1]− moiety other than through electrostatic interactions (i.e., it forms a solvent-separated ion pair in solution; Scheme 1).45 While the low-temperature 1H NMR spectra for both compounds are very similar, the extent of line-broadening for the diastereotopic benzyl-CH2 group at a given temperature is quite different between the two, which indicates that these compounds have distinct exchange rates. Lineshape analysis on NMR spectral data collected between 25 and 85 °C allowed the activation parameters for the exchange process in [Bn1][NBu4] to be determined as ΔH‡ = 66.7 ± 1.6 kJ mol−1 and ΔS‡ = −4.2 ± 5.0 J mol−1 K−1 (Table 1; see ESI† for details). A comparison between these values to the ones measured for the sodium salt [Bn1][Na] reveals striking differences, despite the fact that the anionic boron complex undergoing exchange is identical: both the entropy and enthalpy of activation are significantly lower in the tetrabutylammonium salt, with values for ΔS‡ changing from positive (+59.3 ± 5.6 J mol−1 K−1 in [Bn1][Na]) to slightly negative (−4.2 ± 5.0 J mol−1 K−1 for [Bn1][NBu4]). The latter value is fully consistent with an intramolecular nitrogen inversion process.46–48 Thus, we conclude that in the absence of a coordinating countercation in [Bn1][NBu4], the activation parameters reflect the intrinsic values for the exchange process, i.e. pyramidal N-inversion, whereas in [Bn1][Na] this step is obscured by the involvement of cation–anion dissociation as part of the rate-determining step.
Although the overall barrier to nitrogen inversion (ΔG‡ (298 K) = 68.0 kJ mol−1 for [Bn1][NBu4]) is higher than that in simple amines (21–42 kJ mol−1),51–53 or in bicyclic hydrazines (e.g., 49.4 kJ mol−1 in 2,3-dimethyl-2,3-diazabicyclo[2.2.2]octane),54 it should be noted that in the six-membered boron heterocycle [Bn1]− the N-inversion occurs concurrent with exchange of the BPh2 environments, indicating a correlated (‘geared’) movement of the entire heterocycle (and its substituents). In support of this interpretation we note that Neugebauer and Mannschreck reported slow N-inversion in related all-organic, neutral 1,2,3,4-tetrahydro-s-tetrazines (leucoverdazyls), which were found to have activation free energies between ca. 52 and 83 kJ mol−1 based on determination of coalescence temperatures.55 Our previously published X-ray crystal structure for [Bn1][NBu4]45 provides additional insight in the origin of the high barrier: simultaneous inversion of both hydrazine-type nitrogens (the adjacent N–Ph and N–Bn atoms) via a planar transition state likely causes substantial steric hindrance in this densely substituted heterocycle.
To further corroborate the importance of ion-pair dissociation in these systems, we investigated complexes with heavier alkali cations. Synthesis of salts of [Bn1]− with K+ and Rb+ counterions was achieved by treatment of 1 with 2 equiv. of KC8 or RbC8, followed by addition of benzyl bromide. Similar to [Bn1][Na],43 the UV/Vis spectra of the isolated ligand-benzylated compounds [Bn1][K] and [Bn1][Rb] have absorption maxima at 396 nm (Fig. S1†), and the 1H NMR spectra show diastereotopic signals for the benzyl-CH2 group and line-broadening indicative of exchange (see ESI† for details). Overall, the 1H NMR spectra in this series are comparable,45 although there are noticeable differences in chemical shifts (in particular for the Rb+-salt) that suggest that the heavier alkali metals interact differently with the anion [Bn1]−. A similar trend was observed in anionic formazanate iron complexes,18 and a possible explanation may be the different stability for π- vs. σ-bonding between the alkali metal cation and these anionic heterocycles.
The aluminum analogue [Bn2]− was investigated to probe the effect of the central group 13 element on the observed dynamics. Both the sodium salt [Bn2][Na] and the tetrabutylammonium analogue [Bn2][NBu4] were synthesized according to the published procedures,45 and a variable NMR spectroscopic study reveals that chemical exchange also occurs in these compounds (Fig. S14 and S13†). Lineshape analysis afforded activation parameters for [Bn2][Na] as ΔH‡ = 88.9 (2.5) kJ mol−1 and ΔS‡ = 56.5 ± 7.3 J mol−1 K−1, whereas the corresponding values for [Bn2][NBu4] are ΔH‡ = 68.4 ± 1.3 kJ mol−1 and ΔS‡ = −19.7 ± 3.9 J mol−1 K−1 (Table 1; see ESI† for details). Thus, the change in countercation from Na+ to Bu4N+ results again in markedly different activation entropies for these two compounds: ΔS‡ is large/positive when the cation (Na+) is bound to the ligand backbone, whereas it is negative when this is not the case (Table 1). As in the boron analogues [Bn1]−, we interpret this as a changeover in the rate-determining step from a genuine N-inversion step to dissociation of the coordinating cation. Comparison of the Bu4N+ salts of the boron and aluminum complexes shows similar values for ΔH‡, indicating that the intrinsic effect of the central element on nitrogen inversion is small. The increase in ΔH‡ for the sodium salt of the Al heterocycle [Bn2][Na] compared to the boron analogue ([Bn2][Na]) is consistent with our previous results that bonding in the Al complex is significantly more ionic in nature: the benzylated formazanate ligand in [Bn2]− bears more negative charge,45 and thus has stronger electrostatic interactions with the Na+ cation.
Conclusions
The NMR study presented here shows that the dynamics in ligand-benzylated formazanate boron and aluminium compounds (i.e., Bn1− and Bn2−) is due to nitrogen inversion rather than homolytic N–C bond cleavage. The observation that the activation parameters for exchange in these anions are highly dependent on the nature of the countercation highlights the importance of alkali cation in modulating rates of reactions as simple as nitrogen inversion. Specifically, we conclude that the divergent activation parameters of salts of [Bn1/2]− with either alkali or organic cations is due to a difference in ion-pairing in the ground state. When there is no specific interaction between the anionic group 13 complex and the cation (i.e., in the solvent-separated ion pairs with NBu4+), the activation parameters reflect the intrinsic barrier for nitrogen inversion, whereas for the compounds with alkali cations there is a significant contribution from dissociation of the ion pair.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Netherlands Organisation for Scientific Research (NWO) (VIDI grant to EO) is gratefully acknowledged.Notes and references
- The Alkali Metal Ions: Their Role for Life, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Springer International Publishing, Cham, 2016, vol. 16 Search PubMed.
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- R. Oukaci, J. C. S. Wu and J. G. Goodwin, J. Catal., 1987, 107, 471–481 CrossRef CAS.
- F. J. Williams, A. Palermo, M. S. Tikhov and R. M. Lambert, Surf. Sci., 2001, 482–485, 177–182 CrossRef CAS.
- G. P. Connor and P. L. Holland, Catal. Today, 2017, 286, 21–40 CrossRef CAS PubMed.
- S. F. McWilliams, K. R. Rodgers, G. Lukat-Rodgers, B. Q. Mercado, K. Grubel and P. L. Holland, Inorg. Chem., 2016, 55, 2960–2968 CrossRef CAS PubMed.
- G. González-Riopedre, M. R. Bermejo, M. I. Fernández-García, A. M. González-Noya, R. Pedrido, M. J. Rodríguez-Doutón and M. Maneiro, Inorg. Chem., 2015, 54, 2512–2521 CrossRef PubMed.
- A. H. Reath, J. W. Ziller, C. Tsay, A. J. Ryan and J. Y. Yang, Inorg. Chem., 2017, 56, 3713–3718 CrossRef CAS PubMed.
- K. Grubel, W. W. Brennessel, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 16807–16816 CrossRef CAS PubMed.
- M. R. Kita and A. J. M. Miller, Angew. Chem., Int. Ed, 2017, 56, 5498–5502 Search PubMed.
- M. R. Kita and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14519–14529 CrossRef CAS PubMed.
- C. M. Moore, B. Bark and N. K. Szymczak, ACS Catal., 2016, 6, 1981–1990 CrossRef CAS.
- H. Bosch, J. G. Van Ommen and P. J. Gellings, Appl. Catal., 1985, 18, 405–408 CrossRef CAS.
- R. Y. Rohling, E. J. M. Hensen and E. A. Pidko, ChemPhysChem, 2018, 19, 446–458 CrossRef CAS PubMed.
- J. Sivaguru, A. Natarajan, L. S. Kaanumalle, J. Shailaja, S. Uppili, A. Joy and V. Ramamurthy, Acc. Chem. Res., 2003, 36, 509–521 CrossRef CAS PubMed.
- L. M. Jackman and B. C. Lange, Tetrahedron, 1977, 33, 2737–2769 CrossRef CAS.
- D. L. J. Broere, B. Q. Mercado, E. Bill, K. M. Lancaster, S. Sproules and P. L. Holland, Inorg. Chem., 2018, 57, 9580–9591 CrossRef CAS PubMed.
- J. B. Gilroy, M. J. Ferguson, R. McDonald, B. O. Patrick and R. G. Hicks, Chem. Commun., 2007, 126–128 RSC.
- M. C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., Int. Ed., 2014, 53, 4118–4122 CrossRef CAS PubMed.
- M.-C. Chang and E. Otten, Chem. Commun., 2014, 50, 7431–7433 RSC.
- R. Travieso-Puente, M. C. Chang and E. Otten, Dalton Trans., 2014, 43, 18035–18041 RSC.
- R. Travieso-Puente, J. O. P. Broekman, M. C. Chang, S. Demeshko, F. Meyer and E. Otten, J. Am. Chem. Soc., 2016, 138, 5503–5506 CrossRef CAS PubMed.
- M. C. Chang and E. Otten, Organometallics, 2016, 35, 534–542 CrossRef CAS.
- M. C. Chang, A. Chantzis, D. Jacquemin and E. Otten, Dalton Trans., 2016, 45, 9477–9484 RSC.
- M. C. Chang, P. Roewen, R. Travieso-Puente, M. Lutz and E. Otten, Inorg. Chem., 2015, 54, 379–388 CrossRef CAS PubMed.
- M.-C. Chang and E. Otten, Inorg. Chem., 2015, 54, 8656–8664 CrossRef CAS PubMed.
- S. M. Barbon, J. T. Price, P. A. Reinkeluers and J. B. Gilroy, Inorg. Chem., 2014, 53, 10585–10593 CrossRef CAS PubMed.
- M. Hesari, S. M. Barbon, V. N. Staroverov, Z. Ding and J. B. Gilroy, Chem. Commun., 2015, 51, 3766–3769 RSC.
- S. M. Barbon, J. T. Price, U. Yogarajah and J. B. Gilroy, RSC Adv., 2015, 5, 56316–56324 RSC.
- S. M. Barbon, J. V. Buddingh, R. R. Maar and J. B. Gilroy, Inorg. Chem., 2017, 56, 12003–12011 CrossRef CAS PubMed.
- S. Novoa and J. B. Gilroy, Polym. Chem., 2017, 8, 5388–5395 RSC.
- S. M. Barbon, S. Novoa, D. Bender, H. Groom, L. G. Luyt and J. B. Gilroy, Org. Chem. Front., 2017, 4, 178–190 RSC.
- S. M. Barbon, V. N. Staroverov and J. B. Gilroy, Angew. Chem., Int. Ed., 2017, 129, 8285–8289 CrossRef.
- R. R. Maar, A. Rabiee Kenaree, R. Zhang, Y. Tao, B. D. Katzman, V. N. Staroverov, Z. Ding and J. B. Gilroy, Inorg. Chem., 2017, 56, 12436–12447 CrossRef CAS PubMed.
- J. S. Dhindsa, R. R. Maar, S. M. Barbon, M. Olivia Avilés, Z. K. Powell, F. Lagugné-Labarthet and J. B. Gilroy, Chem. Commun., 2018, 54, 6899–6902 RSC.
- A. Van Belois, R. R. Maar, M. S. Workentin and J. B. Gilroy, Inorg. Chem., 2019, 58, 834–843 CrossRef CAS PubMed.
- A. Mandal, B. Schwederski, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 8126–8135 CrossRef CAS PubMed.
- G. Mu, L. Cong, Z. Wen, J. I. C. Wu, K. M. Kadish and T. S. Teets, Inorg. Chem., 2018, 57, 9468–9477 CrossRef CAS PubMed.
- E. Kabir, D. Patel, K. Clark and T. S. Teets, Inorg. Chem., 2018, 57, 10906–10917 CrossRef CAS PubMed.
- J. B. Gilroy and E. Otten, Chem. Soc. Rev., 2020, 49, 85–113 RSC.
- R. Mondol, D. A. Snoeken, M.-C. Chang and E. Otten, Chem. Commun., 2017, 53, 513–516 RSC.
- R. Mondol and E. Otten, Inorg. Chem., 2018, 57, 9720–9727 CrossRef CAS PubMed.
- R. Mondol and E. Otten, Inorg. Chem., 2019, 58, 6344–6355 CrossRef CAS PubMed.
- R. Mondol and E. Otten, Dalton Trans., 2019, 48, 13981–13988 RSC.
- F. A. L. Anet, R. D. Trepka and D. J. Cram, J. Am. Chem. Soc., 1967, 89, 357–362 CrossRef CAS.
- J. H. Hall and W. S. Bigard, J. Org. Chem., 1978, 43, 2785–2788 CrossRef CAS.
- J. E. Anderson and J. M. Lehn, J. Am. Chem. Soc., 1967, 89, 81–87 CrossRef CAS.
- A. Rauk, L. C. Allen and K. Mislow, Angew. Chem., Int. Ed. Engl., 1970, 9, 400–414 CrossRef CAS.
- (a) A. R. Katritzky, R. C. Patel and F. G. Riddell, J. Chem. Soc., Chem. Commun., 1979, 674 RSC; (b) A. R. Katritzky, R. C. Patel and F. G. Riddell, Angew. Chem., Int. Ed., 1981, 521–529 CrossRef.
- A. Rauk, L. C. Allen and K. Mislow, Angew. Chem., Int. Ed. Engl., 1970, 9, 400–414 CrossRef CAS.
- J. B. Lambert and W. L. Oliver, J. Am. Chem. Soc., 1969, 91, 7774–7775 CrossRef CAS.
- C. D. Montgomery, J. Chem. Educ., 2013, 90, 661–664 CrossRef CAS.
- J. E. Anderson and J. M. Lehn, J. Am. Chem. Soc., 1967, 89, 81–87 CrossRef CAS.
- F. A. Neugebauer and A. Mannschreck, Tetrahedron, 1972, 28, 2533–2538 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0dt01918f |
| This journal is © The Royal Society of Chemistry 2020 |