
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic strategies to incorporate Ru-terpyridyl water oxidation catalysts into MOFs: direct synthesis vs. post-synthetic approach†
Timofey
Liseev
a,
Andrew
Howe
ab,
Md Asmaul
Hoque
b,
Carolina
Gimbert-Suriñach
 b,
Antoni
Llobet
b and
Sascha
Ott
*a
b,
Antoni
Llobet
b and
Sascha
Ott
*a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se
bInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology (BIST), Av. Països Catalans 16, 43007, Tarragona, Spain
First published on 25th September 2020
Abstract
Incorporating molecular catalysts into metal–organic frameworks (MOFs) is a promising strategy for improving their catalytic longevity and recyclability. In this article, we investigate and compare synthetic routes for the incorporation of the potent water oxidation catalyst Ru(tda)(pyCO2H)2 (tda = 2,2′:6′,2′′-terpyridine-6,6′′-dicarboxylic acid, pyCO2H = iso-nicotinic acid) as a structural linker into a Zr-based UiO-type MOF. The task is challenging with this particular metallo-linker because of the equatorial dangling carboxylates that can potentially compete for Zr-coordination, as well as free rotation of the pyCO2H groups around the HO2Cpy⋯Ru⋯pyCO2H axis. As a consequence, all attempts to synthesize a MOF with the metallo-linker directly under solvothermal conditions led to amorphous materials with the Ru(tda)(pyCO2H)2 linker coordinating to the Zr nodes in ill-defined ways, resulting in multiple waves in the cyclic voltammograms of the solvothermally obtained materials. On the other hand, an indirect post-synthetic approach in which the Ru(tda)(pyCO2H)2 linker is introduced into a preformed edba-MOF (edba = ethyne dibenzoic acid) of UiO topology results in the formation of the desired material. Interestingly, two distinctly different morphologies of the parent edba-MOF have been discovered, and the impact that the morphological difference has on linker incorporation is investigated.
1. Introduction
Metal–organic frameworks (MOFs) have been an important part of chemical discourse for over two decades.1 Having been but an academic curiosity upon discovery, MOFs have since grown into a class of industrially promising materials with potential applications in gas separation and storage,2 sorption,3 chemical sensing4 and catalysis,5 to count a few.Alongside interest in catalytic properties of coordination polymers per se,6 MOFs have established themselves as promising scaffolds for the incorporation of molecular catalytic units.7 For many molecular catalysts, especially those based on precious metals, industrial implementation is often limited by poor long-term stability and non-straightforward recyclability. Incorporation of molecular catalysts into MOFs addresses both these issues.9–11 Additionally, MOFs are microporous materials with high internal surface area that can potentially hold a large number of active sites per geometric surface area, making them a desirable solid scaffold for catalyst incorporation.
In view of modern ecological challenges, developing MOF-borne catalysts related to artificial photosynthesis is an important topic.12–14 In recent years, many studies that apply this concept specifically to molecular water oxidation catalysts (WOCs) have been reported.10,11,15–28 Notably, insofar no attempt to incorporate one of the most active Ru-based WOC to-date, Ru(tda)(py)2 (tda = 2,2′:6′,2′′-terpyridine-6,6′′-dicarboxylic acid, py = pyridine, Fig. 1A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8,29), into a MOF has been reported.
8,29), into a MOF has been reported.
 | ||
| Fig. 1 (A) Water oxidation catalyst Ru(tda)(Py)2;8 (B) modified version, Ru(tda)(PyCO2H)2, designed to be incorporated into a MOF via the axial carboxylate groups. | ||
Related Ru-polypyridyl complexes that have been incorporated into MOFs, two of which (A and D) are also WOCs, are summarized in Fig. 2. With the exception of one case where the Ru complex was post-synthetically grafted onto MOF linkers with appended pyridine groups,10 most of these studies make use of carboxylate anchoring groups to attach the complex directly to the secondary building units (SBUs) of the target MOF. Such a strategy makes the complex an integral part of the framework without occupying the void of the MOF that provides for ion, substrate and product transport.5
 | ||
| Fig. 2 Ru-Polypyridyl complexes previously incorporated into MOFs. (A) Water oxidation catalyst Ru(bda)(Py)2;10 (B) photosensitizer Ru(cptpy)22+, cptpy is 4′-(4-carboxyphenyl)-terpyridine;30 (C) photosensitizer Ru(bpy)2(dcbpy)2+, bpy is 2,2'-bipyridine, dcbpy is 5,5′-dicarboxy-2,2′-bipyridine;31–34 (D) water oxidation catalyst Ru(tpy)(dcbpy)(OH2)2+;20,16,17 (E) Ru(dcbpy)32+ as a chemiluminophore.35 | ||
In the present work, we seek to develop a reliable synthetic approach to Ru(tda)-MOF composite materials using Ru(tda)(PyCO2H)2 (PyCO2H = iso-nicotinic acid, Fig. 1B), modified with axial carboxylates, as metallo-linker. The goal is to explore different synthetic routes and to understand factors leading to observed synthetic outcomes. We identify the following potential complications for incorporating this linker into the MOF. First, unlike traditional dicarboxylate linkers, Ru(tda)(PyCO2H)2 allows free rotation of iso-nicotinic acid ligands around the linker main axis, obstructing a pre-organization of the anchoring groups that would facilitate MOF growth.36 Second, the ruthenium centre at low oxidation states is six-coordinate, and one of the equatorial carboxylates is thus potentially available for SBU coordination. Finally, the tda ligand that is orthogonal to the linker vector is sterically demanding, posing additional challenges for undisturbed MOF growth. Together, these factors may complicate MOF formation, and motivate a systematic investigation of different synthesis approaches.
2. Methods
2.1 Electrochemical analysis
MOF-modified working electrodes (WEs) were prepared by dropcasting a suspension of the obtained materials and MWCNT in THF (MWCNT = multi-walled carbon nanotubes) onto the surface of a polished glassy carbon (GC) electrode (see ESI† for more details), followed by drying. An example SEM micrograph of the resulting MOF/MWCNT film is shown in Fig. S1.† In earlier reports, WEs were prepared by dropcasting a suspension (ink) of a MOF and mesoporous carbon in ethanolic solution of Nafion.37–40 The latter procedure was found to be incompatible with the materials investigated herein as it led to leaching of Ru complex that also adsorbed on the mesoporous carbon and contributed to the voltammetric response (see ESI Fig. S10† for further information).The homogeneous Ru(tda)(PyCO2H)2 complex was measured on a bare GC-WE for comparison. All cyclic voltammetry (CV) measurements were conducted in pH 7 phosphate buffer solution (I = 0.1 M) in a one-compartment, three electrode cell using a graphite counter electrode and a Ag/AgCl reference electrode, at a scan rate of 0.1 V s−1.
3. Results & discussion
As a target MOF, we chose the UiO-type for its hydrolytic stability, the possibilities for post-synthetic exchange processes,41,42 and its compatibility with a large variety of ditopic linkers. MOFs of the UiO family are built from 12-coordinated Zr6O4(OH)4 SBUs connected through ditopic carboxylate-based linkers, e.g., terephthalic acid in UiO-66 or biphenyl-4,4′-dicarboxylic acid in UiO-67. Likewise, by modifying the Ru(tda)py2 catalyst with two anchoring carboxylates on the axial pyridine ligands, the complex can be turned into a ditopic MOF linker (Fig. 1). Applying this idea, Ru(tda)(PyCO2H)2 was synthesized by using iso-nicotinic acid instead of pyridine in the last step of the synthesis (see Method section and ESI† for synthetic details).8With Ru(tda)(pyCO2H)2 in hand, different strategies for its incorporation into MOFs were considered. As schematically shown in Fig. 3, three general approaches were identified: (1) direct solvothermal synthesis using Ru(tda)(PyCO2H)2 as the sole linker; (2) mixed-linker solvothermal synthesis where a sterically less demanding co-linker is used together with Ru(tda)(PyCO2H)2; (3) an indirect approach where the metallo-linker is post-synthetically introduced into a pristine parent MOF with linkers of matching length. We set out to explore these three strategies to find viable ways of preparing crystalline composite Ru(tda)-MOF materials. The quality of the resulting materials was assessed by powder X-ray diffraction (PXRD), SEM and cyclic voltammetry (CV).
 | ||
| Fig. 3 Strategies of making MOFs with catalytic Ru(tda)(PyCO2H)2 linker. | ||
3.1. Direct solvothermal synthesis
The direct synthesis approach (method 1) appeals as the simplest and most straightforward way to produce a crystalline MOF material with the highest possible loading of potential catalysts. A high catalyst loading is desirable as it gives rise to the least possible distance between redox active metallo-linkers which would facilitate charge hopping through the framework.5,16Traditionally, DMF is used as the solvent in solvothermal synthesis of UiO-type MOFs. However, DMF is known to coordinate to the ruthenium centre and deactivate the catalyst. To avoid this, dimethylacetamide (DMA) was used as the solvent for the MOF synthesis, being a bulkier analogue to DMF.
Ru(tda)(PyCO2H)2 was mixed with ZrOCl2(H2O)8 in a 1:1 molar ratio in DMA (38 μmol/3 mL), and a tenfold excess of mediator (either formic acid, acetic acid or benzoic acid) was added. The mixture was sonicated for 1 h, sealed in a vial and incubated at temperatures between 80 °C and 120 °C for two days. Despite of a large number of attempts, the material that was obtained from the direct synthesis after washing and drying (material 1) was found to be mostly amorphous by PXRD analysis (Fig. 4, ESI Fig. S4†).
 | ||
| Fig. 4 PXRD patterns of amorphous materials 1 (from solvothermal synthesis) and 2 (from mixed-linker solvothermal synthesis), edba-MOF 3 and 4 and 5 and 6 (with post-synthetically introduced Ru linker), in comparison with a literature reference pattern.43 The patterns are grouped by colours: amorphous materials (black), pristine edba-MOFs (blue), Ru-edba MOFs (red), reference pattern (green, CSD identifier RUKDIM). | ||
Interestingly, upon examination of its electrochemical properties, the CV of material 1 displays distinct features of molecular species, but many more than expected for a coordinatively well-defined metallo-linker. In addition to features that could be assigned to the RuIII/II and RuIV/III couples of Ru(tda)(PyCO2H)2, numerous other waves are visible, indicating the presence of multiple Ru-complexes that differ in electronic environment around the Ru centres (Fig. 5A). We hypothesize that the given synthetic conditions allow Ru linkers to connect to SBUs through the equatorial carboxylate as well as the axial ligands. Irregular combinations of these binding modes could account for multiple waves in the CV, and explain the formation of the non-crystalline material.
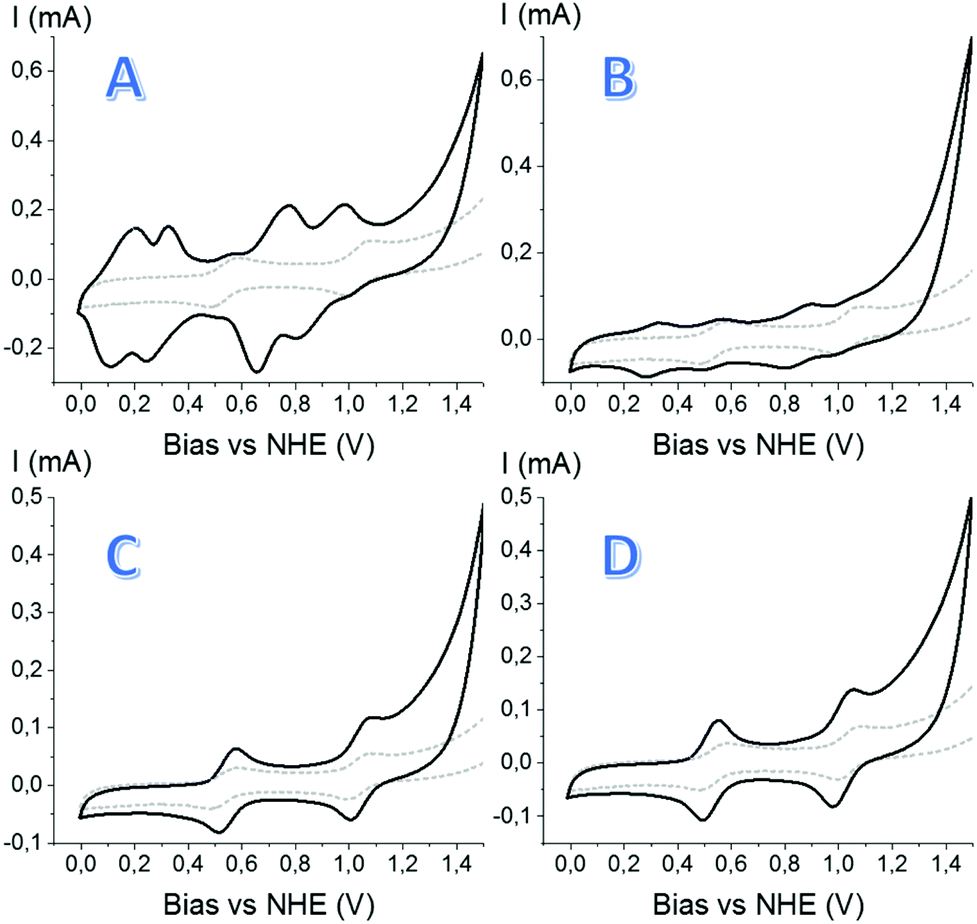 | ||
| Fig. 5 Cyclic voltammograms of materials 1, 2, 5 and 6. The grey dotted line represents the homogeneous Ru(tda)(PyCO2H)2 complex in solution as reference. (A) CV of material 1, showing two sets of sub-waves; (B) CV of co-synthesized material 2, showing multi-waves analogous to material 1; (C) CV of Ru-edba-MOF 5 (octahedral), showing two reversible single waves, qualitatively identical to the homogeneous complex; (D) CV of Ru-edba-MOF 6 (interlaced), showing two reversible single waves, analogous to Ru-edba-MOF 5. (For stability of the electrodes over multiple scans, see ESI Fig. S11†) all measurements were performed in pH 7.0 phosphate buffer, ionic strength 0,1 M vs. Ag/AgCl reference electrode with GC counter-electrode, ν = 0.1 V s−1. | ||
The non-uniform crystal growth could be further disturbed by the bulkiness of the metallo-linker. Considering the unit cell of a hypothetical UiO-type MOF composed exclusively of Ru(tda)(PyCO2H)2 linkers, the SBUs will be occupied by 12 relatively bulky tda ligands. This crowding could be responsible for the formation of the irregular coordination bonds between the linker and the SBUs. Thus, filling the MOF with less bulky co-linkers could, in principle, lead to a better crystalline material.
3.2. Direct solvothermal synthesis with an inert co-linker
The direct solvothermal synthesis was thus revisited with the idea in mind to space-out the bulky Ru(tda) linkers in the desired MOF lattice. Ethynedibenzoic acid (edba) was chosen as the co-linker due to its matching length with the Ru(tda)(PyCO2H)2 linker (13.3 Å and 13.5 Å, respectively), as well as its structural rigidity. The solvothermal synthesis conditions that were tested were analogous to those employed in the first approach, with the addition of the extra linker (edba). Three different molar ratios of Ru(tda)(PyCO2H)2:edba were tested, namely 1:1, 1:4 and 1:8. Unfortunately, all the materials obtained in this way formed amorphous materials, as evidenced by their PXRD patterns (Fig. 4 and S4†). The CVs of the obtained materials showed two sets of sub-waves, similar to those of material 1, but the waves were considerably less pronounced (Fig. 5B). The smaller currents may not be surprising, considering that the amount of metallo-linker is much smaller than in material 1, and the average distance between the redox centers much larger, thus preventing efficient charge transport.
From a synthetic viewpoint, the formation of amorphous material indicates that the steric bulk of the metallo-linker is not the deciding factor that gives rise to the inability of the material to crystallize into a regular lattice. Rather, it appears that the conformational flexibility of the Ru(tda)(PyCO2H)2 complex itself may not be compatible with ordered MOF growth under direct solvothermal synthesis conditions. In addition, the Ru(tda)(PyCO2H)2 linker may also fail to form a MOF due to potential ambiguity in SBU-coordinating sites within the Ru linker. Apart from the desired SBU-coordination through the axial carboxylates, the complex might also coordinate through one of the equatorial carboxyl groups, which in low Ru oxidation states are not coordinating the ruthenium centre (Fig. 1B). A solid material composed of zirconia SBUs irregularly connected through such 2–3-topic linkers could, indeed, account both for the amorphousness of the material and for multiple bulk electronic environments around the Ru observed by CV. In fact, we see this as the major challenge for direct incorporation of this type of complex into a MOF.
3.3. Indirect preparation
Considering the above reasoning, an indirect approach for the synthesis of crystalline materials with the Ru(tda)(PyCO2H)2 linker was pursued. In this approach, a pristine parent MOF was prepared first, followed by post-synthetic introduction of the metallo-linker. The edba-MOF43–45 was targeted as a suitable platform due to the similar length between edba and the Ru-linker. Thus, edba and ZrOCl2(H2O)8 were mixed in DMA (38 μmol/3 mL), in a 1:1 molar ratio, and 10× molar excess of formic acid (modulator) was added. The mixture was sonicated for 1 h, sealed in a vial and incubated in an oven at 135 °C for 2 days. After the washing procedure, the PXRD pattern of the produced material (Fig. 4, blue) closely matched that of the previously reported edba-MOF43 of UiO-type topology. Surprisingly, upon examination of different batches of the solvothermal syntheses by SEM, different morphologies were identified (Fig. S8†), with the two dominating being the octahedral edba-MOF 3 (Fig. 6A) and the interlaced edba-MOF 4 (Fig. 6B).
 | ||
| Fig. 6 SEM micrographs of parent MOFs obtained in method 3. (A) edba-MOF 3 of octahedral morphology; (B) edba-MOF 4 of interlaced morphology. | ||
An analogous, interlaced MOF morphology has been observed before by Cohen and co-workers for UiO-6646,47 and was explained by polymer-induced inhibition of octahedral crystal growth. In our case, one morphology tends to dominate the entire batch, i.e. a batch almost entirely consists either of octahedral crystals (edba-MOF 3) or of interlaced crystals (edba-MOF 4). Both materials exhibit identical PXRD patterns (Fig. 4), although octahedral crystals generally produced sharper reflections.
Both morphologies result from the same preparation protocol, suggesting very delicate differences in synthetic conditions that dictate the produced morphology. Upon closer inspection, it was found that the morphologies can indeed be prepared selectively by careful control of the linker:SBU stoichiometry. The octahedral geometry in 3 was preferentially formed with a 10–50% molar excess of linker, while a 10–50% excess of SBU led to the interlaced morphology (Fig. S5†). The known hygroscopic nature of ZrOCl2(H2O)8 however complicates precise control in the weighing process, and can explain the somewhat random reaction outcome in terms of morphology during the solvothermal synthesis. In analogy with Cohen's interpretation, we propose that a shortage of linkers inhibits crystal growth, forming interlaced crystals, while, apparently, a shortage of SBU is not as critical to crystal growth.
Both edba-MOF 3 and edba-MOF 4 were subjected to post-synthetic linker exchange (PSE) protocols to test the impact that the different morphologies have on the incorporation yield of the metallo-linker. Under these conditions, it is expected that existing missing linker defects are filled,48 as well as existing edba linkers are being exchanged by the Ru linker. Thus, 10 mg of 3 and 4 were separately added to 3 mL of 5 mM methanolic solution of Ru(tda)(PyCO2H)2, the resulting suspensions incubated on a shaker overnight at room temperature, and then thoroughly washed with EtOH, to give the Ru-edba-MOF 5 and 6, respectively. Framework integrity under the PSE conditions were confirmed by PXRD reflection patterns that were collected before and after the exchange with Ru(tda)(PyCO2H)2 linker (Fig. 4 and S9†). The comparison finds all major peaks intact and their positions conserved, which demonstrates overall retention of MOF structure under the PSE conditions.
:Zr and provides the metallo-linker:SBU ratio, while 1H-NMR spectroscopy quantifies introduced linker with respect to the total linker content.
For Ru-edba-MOF 5 (octahedral morphology), NMR yields 3.2% PSE extent, while ICP gives a number of 4.2%. For Ru-edba-MOF 6 (interlaced morphology), the numbers are somewhat lower with 2.1% by NMR and 2.2% by ICP. The larger Ru content in MOF 5 was somewhat unexpected due to lower external surface area compared to that of interlaced MOF 6 crystals, which should, in principle, facilitate access of the metallo-linker into the materials. However, the same rationale can be applied for the washing steps during which Ru-edba-MOF 6 yielded noticeably more coloured supernatants than Ru-edba-MOF 5.
44), 3941 m2 g−1, surpasses that of the material obtained by our synthetic method by a factor of 2 in case of edba-MOF 3 and factor of 10 for edba-MOF 4, which is consistent with less missing-linker defects in BUT-30. There is a number of reports correlating internal surface area of MOFs to the amount of defects, some of which indicate that more defects lead to higher BET surface areas,50 while others show the opposite, i.e. more defects give rise to lower surface areas.51 A notable report from Wang et al. demonstrates how the BET surface area in UiO-66 grows with increasing defects up to a threshold amount, but then begins to drop.52 It appears that the amount of missing-linker defects in our edba-MOF samples is above this threshold. This conclusion is corroborated by the fact that the BET surface areas for both morphologies increase significantly upon incorporation of the Ru(tda) linkers, and Ru-edba-MOF 5 and 6 exhibit a surface area of 1390 m2 g−1 and 800 m2 g−1, respectively. This finding also indicates that the major mechanism of incorporation is probably replacement of the modulator and solvent molecules at the nodes rather than edba struts.
 | ||
| Fig. 7 N2 sorption isotherms of edba-MOF 3 and 4, and Ru-edba-MOF 5 and 6. The octahedral edba-MOF 3 and Ru-edba-MOF 5 show overall higher adsorption profile. The interlaced edba-MOF 4 shows a hysteresis indicative of mesopores. For both morphologies, incorporation of the Ru(tda)(PyCO2H) linker increases internal surface area. | ||
4. Conclusions
Incorporation of molecular catalysts into MOFs is a beneficial, but often synthetically difficult endeavour. As an example, one of the most potent WOCs to-date, Ru(tda)(Py)2, presents a challenge due to the undesirable possibility that its equatorial ligand, tda, may coordinate to the SBUs, as well as the unrestricted rotation of axial ligands around the main axis of the complex. As a result, the direct solvothermal synthesis of the MOF using Ru(tda)(PyCO2)2 as either the sole linker or a co-linker were found not feasible. On the other hand, an indirect approach through post-synthetic incorporation into an existing MOF has successfully yielded the desired MOF-catalyst composite. The parent edba-MOF was found to form two distinct, but structurally identical morphologies (octahedral and interlaced), depending on the exact ratio of linker:SBU that was used in the solvothermal synthesis. MOFs of both morphologies were post-synthetically modified with the Ru metallo-linker and characterized. While MOFs of the interlaced morphology exhibit overall much higher missing-linker defects compared to the octahedral morphology as supported by BET measurements, the two Ru-edba-MOF materials 5 and 6 show only small differences in incorporation yield. The introduced Ru(tda)(PyCO2)2 linkers seem to occupy missing-linker sites rather than replace structural linkers. Electrochemical behaviour of Ru-edba-MOFs 5 and 6 was analogous to that of the linker in homogeneous phase, demonstrating that the indirect preparation strategy can be used to circumvent the challenges associates with linkers such as Ru(tda)(PyCO2)2. Currently under investigation are ways to activate the catalyst inside the MOF pores, as well as the development of alternative electrode platforms for more robust MOF attachment and interfacing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council through a Consolidator Grant (ERC-CoG2015-681895_MOFcat) and the EU-funded ITN eSCALED (Grant agreement ID: 765376) are gratefully acknowledged.References
- L. R. MacGillivray, Metal-Organic Frameworks, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444–1230444 CrossRef.
- A. Samokhvalov, Coord. Chem. Rev., 2018, 374, 236–253 CrossRef CAS.
- Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H. C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS.
- B. D. McCarthy, A. M. Beiler, B. A. Johnson, T. Liseev, A. T. Castner and S. Ott, Coord. Chem. Rev., 2020, 406, 213137 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- C. De Wu and M. Zhao, Adv. Mater., 2017, 29, 1609446 Search PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS.
- A. Bhunia, B. A. Johnson, J. Czapla-Masztafiak, J. Sá and S. Ott, Chem. Commun., 2018, 54, 7770–7773 RSC.
- H. Jia, Y. Yao, J. Zhao, Y. Gao, Z. Luo and P. Du, J. Mater. Chem. A, 2018, 6, 1188–1195 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- C. A. Downes and S. C. Marinescu, ChemSusChem, 2017, 10, 4374–4392 CrossRef CAS.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- P. M. Usov, B. Huffman, C. C. Epley, M. C. Kessinger, J. Zhu, W. A. Maza and A. J. Morris, ACS Appl. Mater. Interfaces, 2017, 9, 33539–33543 CrossRef CAS.
- S. Lin, Y. Pineda-Galvan, W. A. Maza, C. C. Epley, J. Zhu, M. C. Kessinger, Y. Pushkar and A. J. Morris, ChemSusChem, 2017, 10, 514–522 CrossRef CAS.
- S. Lin, A. K. Ravari, J. Zhu, P. M. Usov, M. Cai, S. R. Ahrenholtz, Y. Pushkar and A. J. Morris, ChemSusChem, 2018, 11, 464–471 CrossRef CAS.
- B. A. Johnson, A. Bhunia and S. Ott, Dalton Trans., 2017, 46, 1382–1388 RSC.
- K. Natarajan, A. K. Gupta, S. N. Ansari, M. Saraf and S. M. Mobin, ACS Appl. Mater. Interfaces, 2019, 11, 13295–13303 CrossRef CAS.
- B. A. Johnson, A. Bhunia and S. Ott, Dalton Trans., 2017, 46, 1382–1388 RSC.
- D. Micheroni, G. Lan and W. Lin, J. Am. Chem. Soc., 2018, 140, 15591–15595 CrossRef CAS.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS.
- C. Wang, Z. Xie, K. E. DeKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS.
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef.
- C. W. Kung, J. E. Mondloch, T. C. Wang, W. Bury, W. Hoffeditz, B. M. Klahr, R. C. Klet, M. J. Pellin, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2015, 7, 28223–28230 CrossRef CAS.
- X.-F. Lu, P.-Q. Liao, J.-W. Wang, J.-X. Wu, X.-W. Chen, C.-T. He, J.-P. Zhang, G.-R. Li and X.-M. Chen, J. Am. Chem. Soc., 2016, 138, 8336–8339 CrossRef CAS.
- J.-Q. Shen, P.-Q. Liao, D.-D. Zhou, C.-T. He, J.-X. Wu, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2017, 139, 1778–1781 CrossRef CAS.
- P. M. Usov, S. R. Ahrenholtz, W. A. Maza, B. Stratakes, C. C. Epley, M. C. Kessinger, J. Zhu and A. J. Morris, J. Mater. Chem. A, 2016, 4, 16818–16823 RSC.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, X. Sala and A. Llobet, Chem. Rev., 2019, 119, 3453–3471 CrossRef CAS.
- M. E. Mahmoud, H. Audi, A. Assoud, T. H. Ghaddar and M. Hmadeh, J. Am. Chem. Soc., 2019, 141, 7115–7121 CrossRef.
- W. A. Maza, R. Padilla and A. J. Morris, J. Am. Chem. Soc., 2015, 137, 8161–8168 CrossRef CAS.
- W. A. Maza, A. J. Haring, S. R. Ahrenholtz, C. C. Epley, S. Y. Lin and A. J. Morris, Chem. Sci., 2016, 7, 719–727 RSC.
- L. Y. Yao, F. Yang, W. B. Liang, G. B. Hu, Y. Yang, W. Huang, R. Yuan and D. R. Xiao, Sens. Actuators, B, 2019, 292, 105–110 CrossRef CAS.
- S. Zhang, L. Li, S. Zhao, Z. Sun and J. Luo, Inorg. Chem., 2015, 54, 8375–8379 CrossRef CAS.
- H. Shao, J. Lu, Q. Zhang, Y. Hu, S. Wang and Z. Guo, Sens. Actuators, B, 2018, 268, 39–46 CrossRef CAS.
- J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H. C. Zhou and M. Hong, J. Am. Chem. Soc., 2017, 139, 16939–16945 CrossRef CAS.
- S. Pullen, S. Roy and S. Ott, Chem. Commun., 2017, 53, 5227–5230 RSC.
- E. Mijangos, S. Roy, S. Pullen, R. Lomoth and S. Ott, Dalton Trans., 2017, 46, 4907–4911 RSC.
- D. Micheroni, G. Lan and W. Lin, J. Am. Chem. Soc., 2018, 140, 15591–15595 CrossRef CAS.
- B. Li, J. Liu, Z. Nie, W. Wang, D. Reed, J. Liu, P. McGrail and V. Sprenkle, Nano Lett., 2016, 16, 4335–4340 CrossRef CAS.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS.
- R. J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, J. Am. Chem. Soc., 2015, 137, 9527–9530 CrossRef CAS.
- X. Lv, M. Tong, H. Huang, B. Wang, L. Gan, Q. Yang, C. Zhong and J. Li, J. Solid State Chem., 2015, 223, 104–108 CrossRef CAS.
- T. Gadzikwa, B. S. Zeng, J. T. Hupp and S. B. T. Nguyen, Chem. Commun., 2008, 3672–3674 RSC.
- S. Ayala, K. C. Bentz and S. M. Cohen, Chem. Sci., 2019, 10, 1746–1753 RSC.
- S. Ayala, Z. Zhang and S. M. Cohen, Chem. Commun., 2017, 53, 3058–3061 RSC.
- M. Taddei, R. J. Wakeham, A. Koutsianos, E. Andreoli and A. R. Barron, Angew. Chem., Int. Ed., 2018, 57, 11706–11710 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- J. Ren, M. Ledwaba, N. M. Musyoka, H. W. Langmi, M. Mathe, S. Liao and W. Pang, Coord. Chem. Rev., 2017, 349, 169–197 CrossRef CAS.
- L. Yuan, M. Tian, J. Lan, X. Cao, X. Wang, Z. Chai, J. K. Gibson and W. Shi, Chem. Commun., 2018, 54, 370–373 RSC.
- K. Wang, C. Li, Y. Liang, T. Han, H. Huang, Q. Yang, D. Liu and C. Zhong, Chem. Eng. J., 2016, 289, 486–493 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0dt01890b |
| This journal is © The Royal Society of Chemistry 2020 |