
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,3,4-Azadiphospholides as building blocks for scorpionate and bidentate ligands in multinuclear complexes†
Riccardo
Suter
 *a,
Mona
Wagner
a,
Lorenzo
Querci
a,
Riccardo
Conti
a,
Zoltán
Benkő
b and
Hansjörg
Grützmacher
*a
*a,
Mona
Wagner
a,
Lorenzo
Querci
a,
Riccardo
Conti
a,
Zoltán
Benkő
b and
Hansjörg
Grützmacher
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, CH-8093 Zurich, Switzerland. E-mail: hgruetzmacher@ethz.ch; riccardo.suter@bluewin.ch
bBudapest University of Technology and Economics, Szent Gellért tér 4., 1111 Budapest, Hungary
First published on 27th May 2020
Abstract
Annulated oxy-substituted 1,3,4-azadiphospholides such as the anion in Na[1] are readily accessible phosphorus heterocycles made from the phosphaethynolate anion (OCP)− and 2-chloropyridines. The sodium salt Na[1] reacts with oxophilic element halides such as OPCl3, PhSiCl3, PhBCl2 and CpTiCl3 at room temperature to form exclusively the oxygen bound tris-substituted compounds E(1)3 (with E = OP, PhSi, PhB− or CpTi). Six equivalents of Na[1] with group four metal chlorides MCl4 (M = Ti, Zr, Hf) form cleanly the hexa-substituted dianions (Na2[M(1)6]) which are isolated in excellent yields. The titanium complexes are deeply coloured species due to ligand to metal charge transfer (LMCT) excitations. In all complexes, the phosphorus atoms of the azadiphosphole moieties are able to coordinate to a soft metal center as shown in their reactions with [Mo(CO)3Mes], yielding complexes in which the Mo(CO)3 binds in a fac manner. Functionalization of the oxy group with amino phosphanes allows isolation of tridentate ligands, which have been used as synthons for macrocyclic molybdenum carbonyl complexes.
Introduction
One of the world's most famous dyes is Prussian blue, an iron cyanide complex with the formula [Fe(CN)6]3Fe4.1 The different oxidation states of the iron centers in the polynuclear complex allow charge transfer processes which cause the dark blue color of the complex. Polynuclear metal complexes often show luminescence2 or even phosphorescence making them potential triplet emitters in light emitting diodes.3 Assemblies of metal complexes with unpaired electrons can form single molecule magnets.4–10 Not only small linear linkers11 such as cyanates12–14 or azides15 have been applied but also anionic rings such as triazoles,16 tetrazoles17 or phosphorus containing heterocycles18 have been used to generate scorpionate type ligands which serve as ligands in a very broad range of metal complexes19–21 and as components in opto-electronic materials.22 The phosphaethynolate anion (OCP)− is an excellent building block for a wide variety of organophosphorus compounds.23–26 Due to its ambident nucleophilic character, it binds to soft centers27–32via the phosphorus atom (M–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) and hard centers via the oxygen atom (M–O–C
CO) and hard centers via the oxygen atom (M–O–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P).33–39 Furthermore, phosphaketenes ([M]–PCO) with [M] = main group element or transition metal fragment show a tendency to lose carbon monoxide which allows to prepare mono- and polynuclear metal phosphides.23,33,40–47 There are very few examples which show a bridging OCP-unit between two metal centers.42,48–51 A high yield multigram synthesis of annulated oxy functionalized 1,3,4-azadiphospholides such as Na[1] (Scheme 1) from Na(OCP) and 2-chloropyridines was developed.52,53 These anionic heterocycles contain a OCP-functionality stabilized within the aromatic scaffold of the 1,3,4-azadiphosphole and therefore no elimination of carbon monoxide is expected.
P).33–39 Furthermore, phosphaketenes ([M]–PCO) with [M] = main group element or transition metal fragment show a tendency to lose carbon monoxide which allows to prepare mono- and polynuclear metal phosphides.23,33,40–47 There are very few examples which show a bridging OCP-unit between two metal centers.42,48–51 A high yield multigram synthesis of annulated oxy functionalized 1,3,4-azadiphospholides such as Na[1] (Scheme 1) from Na(OCP) and 2-chloropyridines was developed.52,53 These anionic heterocycles contain a OCP-functionality stabilized within the aromatic scaffold of the 1,3,4-azadiphosphole and therefore no elimination of carbon monoxide is expected.
 | ||
| Scheme 1 Synthesis of main group substituted scorpionates 2, Na[3] and 4 from Na[1] and the corresponding molybdenum complexes 5 and Na[6]. | ||
Results and discussion
In this paper, we report the use of Na[1] as anionic building block to form tridentate scorpionate-type ligands containing low-coordinated λ3,σ2-phosphorus atoms as donor sites. The electronic properties of these polydentate ligands can be modified easily by varying the central oxophilic linker.To form a neutral tridentate ligand, three equivalents of the azadiphospholide Na[1] were added to the phenyl trichlorosilane PhSiCl3 in THF. The reaction mixture turned yellow immediately and a white precipitate was formed. Isolation of the pure compound 2 was possible by recrystallization. In CD2Cl2 as solvent, the product exhibits two doublet resonances with very similar chemical shifts (31P δ = 118.8 and 123.0 ppm, 1JPP = 442.3 Hz). The similarly tris(substituted) but anionic boron analogue Na[3] has been isolated in the reaction of three equivalents of Na[1] with PhBCl2. However, similar reactions with PCl3 only formed a mixture of products and copious amounts of a brown precipitate, which could not be identified. The reaction of three equivalents of Na[1] with phosphoryl chloride POCl3 on the other hand formed one major product, which was unambiguously identified by X-ray crystallography as the expected tris(substituted) phosphine oxide compound 4 (Fig. 1). The 31P{1H}-NMR spectrum in CD2Cl2 shows three different phosphorus resonances. The resonance for the PO atom is observed at low frequency (31P δ = −20.8 ppm), comparable to (Ph-O)3PO (31P δ = −17.3 ppm in CDCl3),54 and does not show any coupling to the phosphorus atoms of the azadiphospholide substituents. The 1JPP coupling in the azadiphosphole ring (1JP1,P2 = 446.5 Hz) is comparable to that in the silicon species 2 (1JPP = 442.3 Hz) and significantly larger than that in the sodium salt Na[1] (1JP1,P2 = 424.7 Hz).
 | ||
| Fig. 1 Crystal structure of compound 4. Anisotropic thermal displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms and the solvent molecules are omitted for clarity reasons. Average interatomic distances (Å) summarized in Table 1. | ||
| 4 | σ | 5 | σ | Na[6] | σ | 7 | σ | Na 2 [ 8 Ti ] | σ | 9 | σ | Na2[10Hf] | Σ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Significant disorder around the phosphine oxide unit resulting in a low bond accuracy and potentially a large error. | ||||||||||||||
| P1 P2 | 2.1092 | 0.0014 | 2.100 | 0.003 | 2.1004 | 0.0017 | 2.118 | 0.002 | 2.1223 | 0.0014 | 2.1029 | 0.0014 | 2.1024 | 0.0006 |
| P1 C1 | 1.710 | 0.004 | 1.705 | 0.009 | 1.710 | 0.005 | 1.732 | 0.005 | 1.741 | 0.004 | 1.730 | 0.003 | 1.7242 | 0.0018 |
| P2 C2 | 1.742 | 0.003 | 1.746 | 0.009 | 1.744 | 0.005 | 1.741 | 0.005 | 1.730 | 0.004 | 1.752 | 0.003 | 1.7413 | 0.0018 |
| N1 C1 | 1.369 | 0.004 | 1.386 | 0.010 | 1.391 | 0.006 | 1.379 | 0.006 | 1.397 | 0.005 | 1.388 | 0.004 | 1.384 | 0.002 |
| N1 C2 | 1.395 | 0.004 | 1.401 | 0.010 | 1.400 | 0.006 | 1.397 | 0.006 | 1.400 | 0.005 | 1.398 | 0.004 | 1.397 | 0.002 |
| O1 C1 | 1.44 | 0.09 | 1.358 | 0.009 | 1.340 | 0.006 | 1.332 | 0.005 | 1.304 | 0.005 | 1.322 | 0.005 | 1.306 | 0.002 |
| COCarbonyl |
— | — | 1.144 | 0.011 | 1.269 | 0.006 | — | — | — | — | 1.154 | 0.004 | 1.148 | 0.002 |
| E⋯Mo | — | — | 4.498 | — | 4.485 | — | — | — | — | — | 4.817 | — | 5.087 | — |
| P⋯Mo | — | — | 2.467 | 0.002 | 2.475 | 0.0012 | — | — | — | — | 2.4813 | 0.0009 | 2.472 | 0.0005 |
To study the suitability of these compounds as ligands in transition metal complexes, one equivalent of the molybdenum complex [Mo(Mes)(CO)3] dissolved in THF was added to a solution of 2, Na[3], or 4, respectively. Upon standing at room temperature for 16 hours, dark yellow single crystals suitable for X-ray crystal diffraction studies of the silicon complex 5 and boron species Na[6], grew from the reaction mixtures (Fig. 2). The complexes show the expected κ3P-coordination mode of the tridentate ligands 2 and Na[3]. No clean product was isolated from the reaction of [Mo(Mes)(CO)3] with 4.
 | ||
| Fig. 2 Solid state structure of 5 (a) and the anion in Na[6] (b), anisotropic thermal displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms and the sodium ion in [6]− are omitted for clarity. Average interatomic distances are summarized in Table 1. | ||
In the next step, the use of oxophilic transition metal halides as central connecting node was tested. Therefore CpTiCl3 and three equivalents of Na[1] were suspended in toluene to instantly form a deep purple solution. The mixture was left at room temperature for 72 hours without stirring to give a dark purple product 7 as crystalline solid. This procedure only produces a low yield due to the formation of several side products. When a large excess of Na[1] was added to CpTiCl3, a dark green solution was obtained from which dark green crystals were isolated and identified as the dianionic hexa-substituted compound Na2[8Ti] (Scheme 2). In this reaction not only the chloride but also the cyclopentadienide (Cp−) became substituted by a azadiphospholide group. Both compounds, 7 and Na2[8Ti], have been unambiguously identified by single crystal X-ray diffraction methods and plots are shown in Fig. 5.
 | ||
| Scheme 2 Synthesis of compounds 7 and Na2[8Ti]. | ||
The titanium species Na2[8Ti] can be synthesized directly in the reaction of six equivalents of the sodium salt of Na[1] with TiCl4(THF)2. This route gives very good yields (>95%) and pure and single crystalline material can be isolated on a multi-gram scale. The analogous dianions (Na2[8Zr] and Na2[8Hf]) with the heavier group four metals zirconium and hafnium were synthesized using a similar procedure (ESI† for details).
The salts containing the heavier group four metals in the center show a significant blue shift of the longest wave absorption compared to the titanium species (Fig. 3).
 | ||
| Fig. 3 UV-VIS absorption spectra of the complexes Na2[8Ti], Na2[8Zr], Na2[8Hf] in THF. The wavelength in nm is plotted against the absorption in arbitrary units. | ||
Solutions of 7 are dark purple and Na2[8Ti] are coloured dark green, whereas the zirconium and hafnium complexes yield yellow to orange solutions, respectively. To elucidate the excitation processes, TD-DFT calculations (on the B3LYP/6-31G* level of theory) were performed on optimized structures of the titanium compound 7 and [8Ti]2− including a PCM solvent model with THF. Complex 7 exhibits an absorption maximum at λmax = 498 nm in THF, which agrees well with the calculated values. The most intense absorption of the ones at lower energies is calculated to be at 464 nm with weaker absorptions up to 504 nm. Analogously [8Ti]2− possesses a λmax of 520 nm in THF with a broad shoulder up to 700 nm in the UV-VIS spectrum (Fig. 3) which matches well with the absorptions found by TD-DFT at λmax = 544–547 nm with weaker ones up to 608 nm. Contours of the frontier molecular orbitals for compound [8Ti]2− are depicted in Fig. 4 and show that the occupied orbitals are mainly ligand centered. On the other hand, the unoccupied acceptor orbitals are metal centered d-orbitals. Consequently, these absorptions are best described as ligand to metal charge transfer (LCMT) bands and the fact that these occur at rather low energies support the fact that azadiphospholides Na[1] are electron rich molecules. A related charge transfer process was observed for the anions of the cyano-substituted azadiphospholides of Na[1], which have absorption maxima ranging from λmax = 525 to 596 nm.52 In these anions, the azadiphospholide acts as a donor and the cyano substituent as the electron accepting group causing strongly coloured species. The absorption maxima of the heavier group four metals dianions, Na2[8Zr] (λmax = 422.5 nm) and Na2[8Hf] (λmax = 409.0 nm) are blue shifted compared to Na2[8Ti] which is explained by the lack of LCMT bands (Fig. 3). The reason for this are the energetically higher lying acceptor orbitals at the heavier group four metals resulting in a poor overlap with the occupied ligand orbitals.
 | ||
| Fig. 4 Selected molecular orbitals of complex [8Ti]2− which are involved in the absorption processes at low energies. Energies are given in eV and orbitals are depicted at an isovalue of 0.04. | ||
In analogy to the reaction with compound 5, compound 7 was added to a THF solution of the molybdenum precursor complex [Mo(Mes)(CO)3]. After 24 hours dark red crystals were obtained from the reaction mixture and the product was identified as the bimetallic complex 9 (Fig. 5). Due to the low solubility in common organic solvents, no NMR spectra could be recorded but the purity of the complex was proven by elemental analysis. Reactions of Na2[8Ti] with two equivalents of [Mo(Mes)(CO)3] did form an insoluble microcrystalline solid. In the IR-spectrum, two distinct CO stretching vibrations at ν = 1925 cm−1 and ν = 1829 cm−1 are observed indicating a similar coordination environment around the molybdenum center as observed in compounds 5 or 9, respectively. Addition of two equivalents of [Mo(Mes)(CO)3] to a solution containing 18-crown-6 (18C6) and Na2[8Hf] in DCM yielded single crystals suitable for X-ray diffraction analysis. This species was identified as the trinuclear heterobimetallic complex Na2[10Hf] which contains one Hf4+ in the center and two Mo(CO)3 units bound to the outer-sphere via the six phosphorus donor sites of the azadiphospholide units (Fig. 5). Once crystallized, compound Na2[10Hf] is also only sparingly soluble in common organic solvents.
 | ||
| Fig. 5 Plots of the structures of (a): 7; (b): 9, (c): Na2[8Ti], and (d): Na2[10Hf]. Anisotropic thermal displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms, solvent molecules are omitted for clarity. The two sodium cations in Na2[8Ti] and Na2[10Hf] are not shown. Average interatomic distances are summarized in Table 1. | ||
The three compounds 4 (Fig. 1), 7 and Na2[8Ti] (Fig. 5) which do not contain a Mo(CO)3 fragment show similar characteristics in the solid state. The “hard” P(V) or Ti(IV) centers are, as expected, bound to the oxygen atoms of the azadiphospholide unit which for steric reasons adopt a paddle wheel arrangement around the oxophilic centers. Upon complexation to Mo(CO)3, the azadiphospholide units are rotated inwards such that in every case the P1 centers form a tridentate pocket to which the Mo(CO)3 can bind in a facial fashion; that is every CO group is positioned trans to a P1 center. This feature can be seen in complexes 5 and Na[6] (Fig. 2). The coordination sphere around the molybdenum centers is slightly distorted octahedral with Mo–P bond distances (Mo–Pav 2.475 Å) in the expected range.55 A comparison between the structures of 7 and 9 indicates that the structural parameters of the azadiphospholide rings does not change significantly upon coordination to Mo(CO)3 and specifically the P1–P2 distance varies very little (7: 2.118 Å; 9: 2.1029 Å).
The only remarkable structural differences between these complexes are associated with the P1–C1, P2–C2, and C1–O1 distances. In compounds 4, 5, and Na[6], which do not contain a transition metal in the center, the P1–C1 bond adjacent to the C1–O1 group (av. 1.345 Å) is in the range of 1.705–1.710 Å. This is significantly shorter than the P2–C2 bond (1.742–1.746 Å). This feature is opposite to what is observed in the salt Na[1] – with the “free” azadiphospholide anion – where the P1–C1 bond [1.760(3) Å] is longer than the P2–C2 distance [1.725(4) Å] and the short C1–O1 bond [1.268(3) Å] indicates significant C–O multiple bond character.52 In the compounds 7, Na2[8Ti], 9, and Na2[10Hf] with a M(IV) center (M = Ti, Hf) the P1–C1 and P2–C2 bonds are almost equally long and the C1–O1 bond slightly shortened (1.302–1.332 Å) with respect to 4, 5, and Na[6]. We attribute these effects to the more pronounced polarization of the C1δ+–O1δ− bond which makes the azadiphospholide units in the Ti(IV) and Hf(IV) compounds more “anion like” such that adopt a structure which is closer to [1]−. The partial charge transfer from the azadiphospholide units to the Ti(IV), which is the reason for the red shifted long wave absorptions, is not reflected in the structural parameters. In all compounds containing Mo(CO)3 fragments, the distance between the Mo centers and the central atom is far above 4 Å and excludes any electronic through space interactions.
In order to evaluate the electronic properties of the new tris(azadiphospholide) ligands Na[3], 4 as well as the titanium compound 7 and hafnium derivative Na2[8Hf], Table 2 lists the carbonyl stretching frequencies, νCO [cm−1], of the Mo(CO)3 complexes synthesized in this work. For comparison, the νCO of a number of [Mo(L)x(CO)3] complexes including [Mo(CO)6] are given. As expected, all complexes with a facial arrangement of the three CO groups show two νCO stretching vibrations. Strongly σ-electron donating and poor π-accepting ligands like PMe3 or especially the anionic tris(pyrazolyl)borate shift the νCO to lower wave numbers below 1900 cm−1 and 1800 cm−1, respectively, for the latter. Strong σ-donation increases the electron density at the Mo center which consequently increases the electron back donation in the π* orbital of the CO groups. On the contrary, a strong π-acceptor ligand like 1,3,5-tri(methyl)benzene (i.e. mesitylene = Mes) diminishes the M → CO back donation and hence the νCO increases. As Table 2 shows, the neutral tris(azadiphospholes) 2 and 7 have a smaller σ-donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :π-acceptor ratio when compared to phosphanes like PMe3 or triphos (Me-C{CH2PPh2}3).56 That is they behave as comparatively weaker σ-donors but stronger π-acceptors, a property which is well established for phosphinines, PC5R5, the phosphorus analogues of pyridines.57–59 As expected the monoanionic ligand [3]− or dianionic ligand [8Hf]2− give the lowest νCO in the series of complexes described in this paper and are coming very close to [Mo(PMe3)3(CO)3].60 It is, however, remarkable that replacement of the PhSi group in 5 by the CpTi unit in 9 has a significant influence on νCO and shifts the frequency 16 cm−1 to lower wavenumbers. This shows that the electron donation in the tridentate azadiphospholide ligands can be sensitively tuned by the remotely placed connecting node in the center of the ligand.
:π-acceptor ratio when compared to phosphanes like PMe3 or triphos (Me-C{CH2PPh2}3).56 That is they behave as comparatively weaker σ-donors but stronger π-acceptors, a property which is well established for phosphinines, PC5R5, the phosphorus analogues of pyridines.57–59 As expected the monoanionic ligand [3]− or dianionic ligand [8Hf]2− give the lowest νCO in the series of complexes described in this paper and are coming very close to [Mo(PMe3)3(CO)3].60 It is, however, remarkable that replacement of the PhSi group in 5 by the CpTi unit in 9 has a significant influence on νCO and shifts the frequency 16 cm−1 to lower wavenumbers. This shows that the electron donation in the tridentate azadiphospholide ligands can be sensitively tuned by the remotely placed connecting node in the center of the ligand.
Finally, we studied the suitability of the azadiphospholide salt Na[1] as building block for bidentate ligands. Earlier work showed that related alkoxy functionalized phosphinines63 react with chlorophosphanes, R2PCl, to form such bidentate ligands which could be successfully used in the synthesis of photo-luminescent complexes.64,65 However, reactions of Na[1] with chloro-phosphanes such as PCl3, PhPCl2 and Ph2PCl only formed brown insoluble precipitates. On the other hand, the reaction of Na[1] with bis(diisopropylamino)chlorophosphane, (iPr2N)2PCl, proceeded cleanly to form compound 11 (Scheme 3). This substitution is accompanied by a colour change of the reaction mixture from orange to dark yellow and the precipitation of an off-white solid. 31P{1H} NMR spectroscopic analysis revealed the formation of one major product with three inequivalent phosphorus nuclei, forming a AMX spin system: 31P δ (ppm) = 134.4 (dd, 1JP3,P1 = 121.7 Hz, 2JP2,P3 = 19.1 Hz), 121.7 (dd, 1JP2,P1 = 425.6 Hz, 2JP2,P3 = 19.1 Hz), 109.0 (dd, 1JP1,P2 = 425.6 Hz, 1JP1,P3 = 121.7 Hz). The coupling constant between the two phosphorus atoms in the azadiphosphole ring (JPP = 426 Hz) is comparable to the one in Na[1] (JP,P = 424.7 Hz) indicating that the substitution took place at the oxygen atom. The coupling constant JP1,P3 = 121.7 Hz is relatively large for a 3J-through bond coupling, and may be caused by the alignment of the two lone pairs at P1 and P3 causing a strong through space coupling.66 This assignment was supported by a structure determination through X-ray diffraction methods using a single crystal of 11 (Fig. 6).
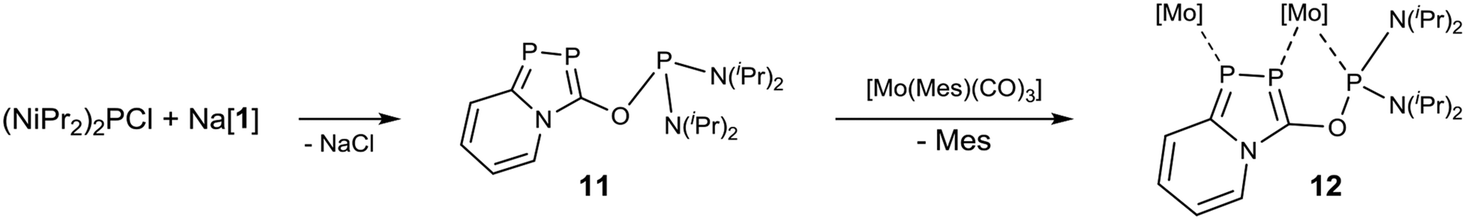 | ||
| Scheme 3 Synthesis of 11 and the reaction with [Mo(Mes)(CO)3] to form complex 12. | ||
 | ||
| Fig. 6 Crystal structure of compound 11. Anisotropic thermal displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms and the solvent molecules are omitted for clarity. Average interatomic distances (Å): P1 P2 2.1284(5), P2 C2 1.7373(15), P1 C1 1.7308(14), O1 C1 1.3496(16), N1 C1 1.3773(17), N1 C2 1.4074(17), P3 P1 3.299. | ||
In 11, the two phosphorus atoms P1 and P3 form a bidentate binding site (P1⋯P3 3.299 Å) which is ideally suited to bind a metal center in a κ2-fashion. Furthermore, the phosphorus center P2 may bind to an additional metal center which opens the possibility of preparing multinuclear complexes. This idea was tested in the reaction of equimolar amounts of 11 and [Mo(Mes)(CO)3] which upon loss of mesitylene opens up three coordination sites. Dark orange crystals were isolated directly from the reaction mixture after about 12 hours at room temperature and the product was analysed by X-ray diffraction analysis. Indeed, a tetranuclear molybdenum complex of the composition [Mo4(11)4(CO)12] (12) was formed (Fig. 7). Elemental analysis proved the purity of the compound, which is insoluble in all common deuterated solvents. The IR spectrum exhibits two major carbonyl bands at νCO [cm−1] = 1950, 1849 in the same range as observed in the complexes mentioned above.
 | ||
| Fig. 7 Crystal structure of the organometallic macrocycle 12. Anisotropic thermal displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms and three DCM molecules are omitted for clarity reasons. Average interatomic distances (Å): Mo1 P1 2.4816(17), Mo1 P3 2.5357(17), P2 Mo2 2.5042(15), P2 P1 2.100(2), P2 C2 1.740(7), P1 C1 1.722(7), C1 O1 1.347(8), C1 N1 1.372(9), C2 N1 1.397(8). | ||
Complex [Mo4(11)4(CO)12] crystallizes in the orthorhombic space group Fdd2, with two azadiphosphole and molybdenum centers per asymmetric unit. The individual bond lengths and angles within the azadiphosphole units are comparable to the ones observed in the complexes discussed above. Upon coordination to Mo, the distance between the two phosphorus atoms P1 and P3 is slightly diminished to 3.132 Å. In the solid state, the [Mo4(11)4(CO)12] macrocycles stack above each other such that channels with a diameter of about 5.6 Å are formed (diagonal Mo–Mo distance 7.276 Å).
Conclusions
The salt Na[1] containing the azadiphospholide anion is a versatile reagent for the synthesis of a range of new multidentate ligands. These are accessed via nucleophilic substitutions reactions with various main group and transition metal halides and in all cases studied so far, the “hard” oxygen center in [1]− binds to the electrophilic center E in RnEClm (E = B, Si, P, Ti, Zr, Hf). The azadiphospholides form strongly coloured complexes with d0-titanium centers which is explained by LMCT excitations which indicate that the 1,3,4-azadiphospholes are rather electron rich heterocycles. The two adjacent phosphorus centers in the azadiphospholide ring possess each a lone pair of electrons which can be further engaged in the coordination to a transition metal center. This property was exploited in reactions [Mo(Mes)(CO)3] and allowed to synthesize bi- and trinulecar heterobimetallic complexes. The reaction between (iPr2N)2PCl and Na[1] leads cleanly to a molecule with a P–O–C–P scaffold which is suitable as ligand for the synthesis of polynuclear macromolecular metalloheterocycles. In the crystalline state, these compounds from channels which eventually may be used for the encapsulation or adsorption of guest molecules. The preliminary results reported in this paper let hope that an especially rich coordination chemistry can be developed with these kind of polydentate phosphorus compounds which are moreover easily synthesized on a multi-gram scale.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ETH Zürich (project 0-20406-18), and the Swiss National Science Foundation (SNF) (project 2-77199-18). Further support came from the NKFIH Grant (PD 116329), a Janos Bolyai Research Fellowship, and a UNKP Grant (UNKP-19-4-BME-422).Notes and references
- K. Itaya, I. Uchida and V. D. Neff, Acc. Chem. Res., 1986, 19, 162–168 CrossRef CAS.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–833 CrossRef CAS PubMed.
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS.
- S.-i. Ohkoshi and H. Tokoro, Acc. Chem. Res., 2012, 45, 1749–1758 CrossRef CAS PubMed.
- L. M. C. Beltran and J. R. Long, Acc. Chem. Res., 2005, 38, 325–334 CrossRef CAS PubMed.
- J. T. Culp, J.-H. Park, F. Frye, Y.-D. Huh, M. W. Meisel and D. R. Talham, Coord. Chem. Rev., 2005, 249, 2642–2648 CrossRef CAS.
- R. Lescouëzec, L. M. Toma, J. Vaissermann, M. Verdaguer, F. S. Delgado, C. Ruiz-Pérez, F. Lloret and M. Julve, Coord. Chem. Rev., 2005, 249, 2691–2729 CrossRef.
- J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563–570 CrossRef CAS PubMed.
- M. Ohba and H. Ōkawa, Coord. Chem. Rev., 2000, 198, 313–328 CrossRef CAS.
- M. Verdaguer, A. Bleuzen, V. Marvaud, J. Vaissermann, M. Seuleiman, C. Desplanches, A. Scuiller, C. Train, R. Garde, G. Gelly, C. Lomenech, I. Rosenman, P. Veillet, C. Cartier and F. Villain, Coord. Chem. Rev., 1999, 190–192, 1023–1047 CrossRef CAS.
- X. Y. Wang, Z. M. Wang and S. Gao, Chem. Commun., 2008, 281–294, 10.1039/b708122g.
- S. Derossi, D. T. Farrell, C. J. Harding, V. McKee and J. Nelson, Dalton Trans., 2007, 1762–1772, 10.1039/B617907J.
- S. Pagano, G. Montana, C. Wickleder and W. Schnick, Chem. – Eur. J., 2009, 15, 6186–6193 CrossRef CAS PubMed.
- A. S. P. Frey, F. G. N. Cloke, M. P. Coles and P. B. Hitchcock, Chem. – Eur. J., 2010, 16, 9446–9448 CrossRef CAS PubMed.
- A. Escuer and G. Aromi, Eur. J. Inorg. Chem., 2006, 4721–4736, DOI:10.1002/ejic.200600552.
- J. G. Haasnoot, Coord. Chem. Rev., 2000, 200, 131–185 CrossRef.
- G. Aromi, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485–546 CrossRef CAS.
- M. Mlatecek, L. Dostal, Z. Ruzickova, J. Honzicek, J. Holubova and M. Erben, Dalton Trans., 2015, 44, 20242–20253 RSC.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- S. Trofimenko, Polyhedron, 2004, 23, 197–203 CrossRef CAS.
- A. Otero, J. Fernandez-Baeza, A. Antinolo, J. Tejeda and A. Lara-Sánchez, Dalton Trans., 2004, 1499–1510, 10.1039/b401425a.
- V. Amendola, L. Fabbrizzi, F. Foti, M. Licchelli, C. Mangano, P. Pallavicini, A. Poggi, D. Sacchi and A. Taglietti, Coord. Chem. Rev., 2006, 250, 273–299 CrossRef CAS.
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed.
- F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed.
- G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS.
- M. Mehta, J. E. McGrady and J. M. Goicoechea, Chem. – Eur. J., 2019, 25, 5445–5450 CrossRef CAS PubMed.
- D. W. N. Wilson and J. M. Goicoechea, Chem. Commun., 2019, 55, 6842–6845 RSC.
- L. N. Grant, J. Krzystek, B. Pinter, J. Telser, H. Grützmacher and D. J. Mindiola, Chem. Commun., 2019, 55, 5966–5969 RSC.
- L. Liu, D. A. Ruiz, F. Dahcheh, G. Bertrand, R. Suter, A. M. Tondreau and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC.
- D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC.
- S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. – Eur. J., 2012, 18, 14805–14811 CrossRef CAS PubMed.
- D. W. N. Wilson, M. P. Franco, W. K. Myers, J. E. McGrady and J. M. Goicoechea, Chem. Sci., 2020, 11, 862–869 RSC.
- Y. Mei, J. E. Borger, D.-J. Wu and H. Grützmacher, Dalton Trans., 2019, 48, 4370–4374 RSC.
- L. N. Grant, B. Pinter, B. C. Manor, H. Grützmacher and D. J. Mindiola, Angew. Chem., Int. Ed., 2018, 57, 1049–1052 CrossRef CAS PubMed.
- R. J. Gilliard, D. Heift, Z. Benkő, J. M. Keiser, A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Dalton Trans., 2018, 47, 666–669 RSC.
- D. W. N. Wilson, A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 2188–2193 CrossRef CAS PubMed.
- K. M. Szkop, A. R. Jupp, R. Suter, H. Grützmacher and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 14174–14177 CrossRef CAS PubMed.
- R. Suter, Y. B. Mei, M. Baker, Z. Benkő, Z. S. Li and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 1356–1360 CrossRef CAS PubMed.
- P. Coburger, H. Grützmacher and E. Hey-Hawkins, Chem. Commun., 2019, 55, 3187–3190 RSC.
- J. Abbenseth, M. Diefenbach, A. Hinz, L. Alig, C. Würtele, J. M. Goicoechea, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2019, 58, 10966–10970 CrossRef CAS PubMed.
- G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 431–436 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, T. Szilvási, E. Ballestero-Martínez, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 4333–4336 CrossRef CAS PubMed.
- M. Joost, W. J. Transue and C. C. Cummins, Chem. Commun., 2017, 53, 10731–10733 RSC.
- L. N. Grant, B. Pinter, B. C. Manor, R. Suter, H. Grützmacher and D. J. Mindiola, Chem. – Eur. J., 2017, 23, 6272–6276 CrossRef CAS PubMed.
- D. Heift, Z. Benkő, R. Suter, R. Verel and H. Grützmacher, Chem. Sci., 2016, 7, 6125–6131 RSC.
- A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- D. W. N. Wilson, N. H. Rees and J. M. Goicoechea, Organometallics, 2019, 38, 4601–4606 CrossRef CAS.
- R. Magnall, G. Balázs, E. Lu, F. Tuna, A. J. Wooles, M. Scheer and S. T. Liddle, Angew. Chem., Int. Ed., 2019, 58, 10215–10219 CrossRef CAS PubMed.
- S. Bestgen, Q. Chen, N. H. Rees and J. M. Goicoechea, Dalton Trans., 2018, 47, 13016–13024 RSC.
- C. Camp, N. Settineri, J. Lefèvre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC.
- R. Suter, Z. Benkő, M. Bispinghoff and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 11226–11231 CrossRef CAS PubMed.
- R. Suter, Z. Benkő and H. Grützmacher, Chem. – Eur. J., 2016, 22, 14979–14987 CrossRef CAS PubMed.
- K. A. Chernyshev and L. B. Krivdin, Russ. J. Org. Chem., 2011, 47, 355–362 CrossRef CAS.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1–S83, 10.1039/DT98900000S1.
- J. Chart and H. R. Watson, J. Chem. Soc., 1961, 4980–4988 CAS.
- Y. Hou, Z. Li, Y. Li, P. Liu, C.-Y. Su, F. Puschmann and H. Grützmacher, Chem. Sci., 2019, 10, 3168–3180 RSC.
- C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 2013, 187–202 CrossRef.
- L. Nyulászi, Chem. Rev., 2001, 101, 1229–1246 CrossRef PubMed.
- C. J. Breheny, J. M. Kelly, C. Long, S. O'Keeffe, M. T. Pryce, G. Russell and M. M. Walsh, Organometallics, 1998, 17, 3690–3695 CrossRef CAS.
- J. M. Jenkins, J. R. Moss and B. L. Shaw, J. Chem. Soc. A, 1969, 2796–2800, 10.1039/j19690002796.
- S. Trofimenko, J. Am. Chem. Soc., 1969, 91, 588–595 CrossRef CAS.
- X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef CAS PubMed.
- X. Chen, Z. Li and H. Grützmacher, Chem. – Eur. J., 2018, 24, 8432–8437 CrossRef CAS PubMed.
- X. Chen, Z. Li, F. Yanan and H. Grützmacher, Eur. J. Inorg. Chem., 2016, 2016, 633–638 CrossRef CAS.
- J.-C. Hierso, A. Fihri, V. V. Ivanov, B. Hanquet, N. Pirio, B. Donnadieu, B. Rebière, R. Amardeil and P. Meunier, J. Am. Chem. Soc., 2004, 126, 11077–11087 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Containing synthetic procedures and experimental details. CCDC 1494716, 1494717, 1494718, 1494719, 1494720, 1494721, 1494722, 1494723, 1494802, 1495143, 1495146 and 1495123. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt01864c |
| This journal is © The Royal Society of Chemistry 2020 |