
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate and product binding inside a stimuli-responsive coordination cage acting as a singlet oxygen photosensitizer†
Sonja
Pullen‡
 ,
Susanne
Löffler‡
,
André
Platzek
,
Julian J.
Holstein
and
Guido H.
Clever
*
,
Susanne
Löffler‡
,
André
Platzek
,
Julian J.
Holstein
and
Guido H.
Clever
*
Faculty of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn Str. 6, 44227 Dortmund, Germany. E-mail: guido.clever@tu-dortmund.de
First published on 15th June 2020
Abstract
An acridone-based, interpenetrated double cage [3BF4Pd4L8] acts as a photosensitizer for generating singlet oxygen which adds to 1,3-cyclohexadiene in a [2+4] hetero-Diels-Alder reaction to form 2,3-dioxabicyclo[2.2.2]oct-5-ene. Photocatalytic activity was exclusively observed for the assembled cage, whereas the free organic ligand L decomposes upon irradiation. While cage [3BF4Pd4L8] does not accept any organic guests, NMR, MS and single crystal X-ray results reveal that both substrate and product are readily encapsulated in the central pocket of its chloride-activated form [2Cl@Pd4L8]. The system combines multiple functions (photosensitization, allosteric activation and guest uptake) within a structurally complex, mechanically-bound self-assembly built up from a simple and readily accessible ligand.
Introduction
Supramolecular host compounds have garnered increasing attention with respect to applications in selective guest encapsulation1–9 and catalysis.10–19 Coordination cages, assembled from metal cations and organic ligands with two or more donor groups, represent a versatile family of hosts with tunable size and properties.20,21 In equipping such cages with distinct functionality, the introduction of addressable features into the ligand backbones has been shown to be a fruitful strategy. The resulting structures can be responsive to external stimuli such as light, pH or addition of chemical inputs.22–27 For example, the implementation of photoswitches into the ligands allows to control light-induced processes such as complete structural rearrangement28 or guest uptake and release.29,30 Chromophore-based supramolecules can also be used to catalyse reactions. For example, Fujita and coworkers presented photoactive cages for alkane oxidation31 and the demethylation of cyclopropanes through guest-to-host electron transfer.32We have reported a series of interpenetrated double cages that form quantitatively from a mixture of square-planar Pd(II) cations and banana-shaped bis-monodentate pyridyl ligands with dibenzo-suberone, phenothiazine or similar backbones.33–37 The mechanically-bound structures have three pockets, initially all filled with BF4− counter anions, of which the outer two can be exchanged by halide anions following an allosteric mechanism with strong positive cooperativity.33,38
A particularly interesting guest inclusion behaviour was observed for the derivative [3BF4@Pd4L8], assembled from acridone-based ligands (Fig. 1). Addition of chloride anions to this species leads to the quantitative conversion into [2Cl@Pd4L8] with an enlarged inner pocket that allows the encapsulation of neutral guest molecules such as benzene, cyclohexane or norbornadiene.39 This phenomenon could be explained by the compression of the catenated cage structure along the Pd4-axis due to the small chloride anions replacing larger BF4− species, resulting in the expansion of the central pocket, hence activating the host to accept a variety of guests.
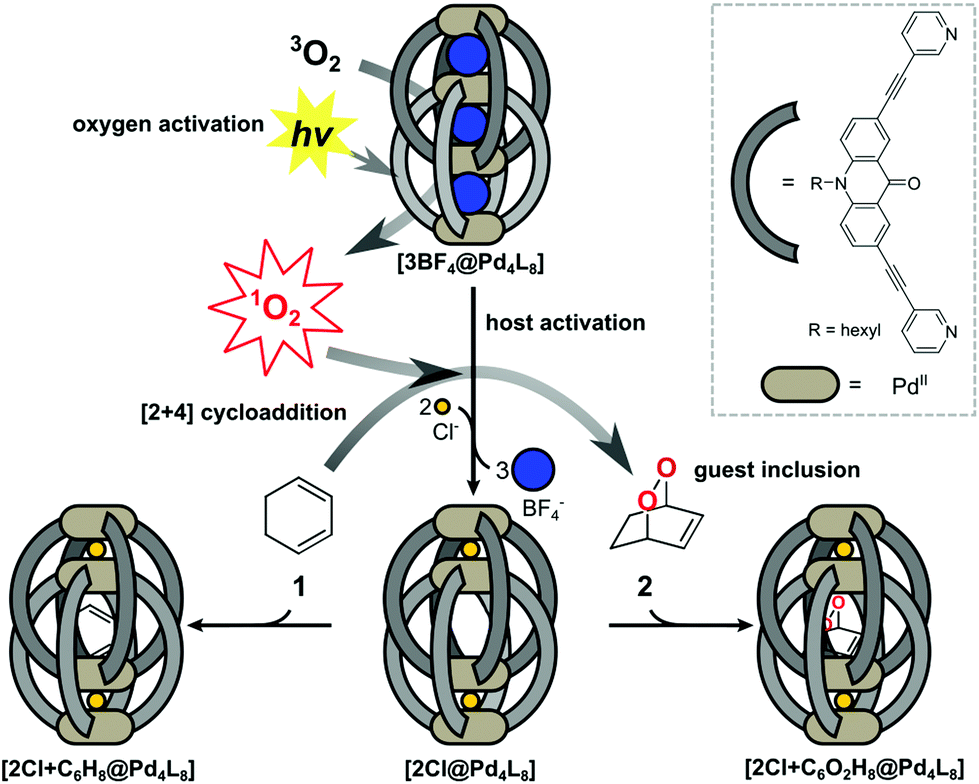 | ||
| Fig. 1 Catalytic function and guest inclusion behaviour of the acridone-based double cages [3BF4@Pd4L8] and [2Cl@Pd4L8]: both cages are able to generate singlet oxygen, reacting in a [2+4] cycloaddition reaction with 1,3-cyclohexadiene. Only [2Cl@Pd4L8] is able to encapsulate both substrate 1 and product 2. | ||
Six-membered rings, including bridged derivatives (i.e. norbornane- and bicyclooctane-type compounds), were found to be favourably accepted for encapsulation as compared to smaller or larger molecules.40 By comparing the results of systematic guest affinity experiments with high-level electron correlation calculations, London dispersion interactions were recognized as the main contributor to the driving force for neutral guest inclusion inside the central pocket of the charged host.41
Encouraged by these observations, we moved on to test the ability of [2Cl@Pd4L8] to take a role in catalytic reactions. Owing to the known dimensions of the central cavity and its preference for bicyclic organic compounds, we envisioned to conduct electrocyclic reactions inside the confined cavity. As a first example, we present the encapsulation of 1,3-cyclohexadiene 1, which is a common substrate for Diels–Alder reactions.42
Results and discussion
Synthesis of host–guest complexes
[3BF4@Pd4L8] and [2Cl@Pd4L8] were prepared according to our previously described procedure.39 The encapsulation of 1 by [2Cl@Pd4L8] was followed by 1H NMR spectroscopy and ESI mass spectrometry (Fig. 2a and c). After addition of 30 equivalents of 1 to an acetonitrile solution of the host and incubating the sample at room temperature for 24 h, a new set of signals were observed in the 1H NMR spectrum (Fig. 2a). A binding constant of 2.15(±0.04) × 103 L mol−1 for 1 was determined via1H-NMR signal integration of the equilibrated mixture of free host/guest and host–guest complex in slow exchange at 298 K (for details see ESI†). For the latter one, protons Hg′ and Ha′, which point inside the central pocket of the interpenetrated coordination cage, were found to be shifted significantly; Hg′ shifted upfield by Δδ = 0.25 ppm and Ha′ shifted downfield by Δδ = 0.16 ppm. Similar shifts were observed previously for the inclusion of other neutral guests.39,42 Based on these chemical shifts, we conclude that encapsulation of 1 takes place inside the central pocket of the chloride-activated coordination cage. In addition, high-resolution ESI mass spectrometry showed a series of species [2Cl+C6H8@Pd4L8+nBF4](6−n)+ (n = 0–3) with a variable number of tetrafluoroborate counter anions, confirming the formation of the host–guest complex (Fig. 2c). In contrast, the addition of 30 eq. 1 to the precursor cage [3BF4@Pd4L8] did not result in any change of the cage's 1H-NMR signals (ESI, Fig. S1†). This was expected as the neutral guest uptake only occurs in the presence of halide anions that trigger the enlargement of the central pocket.39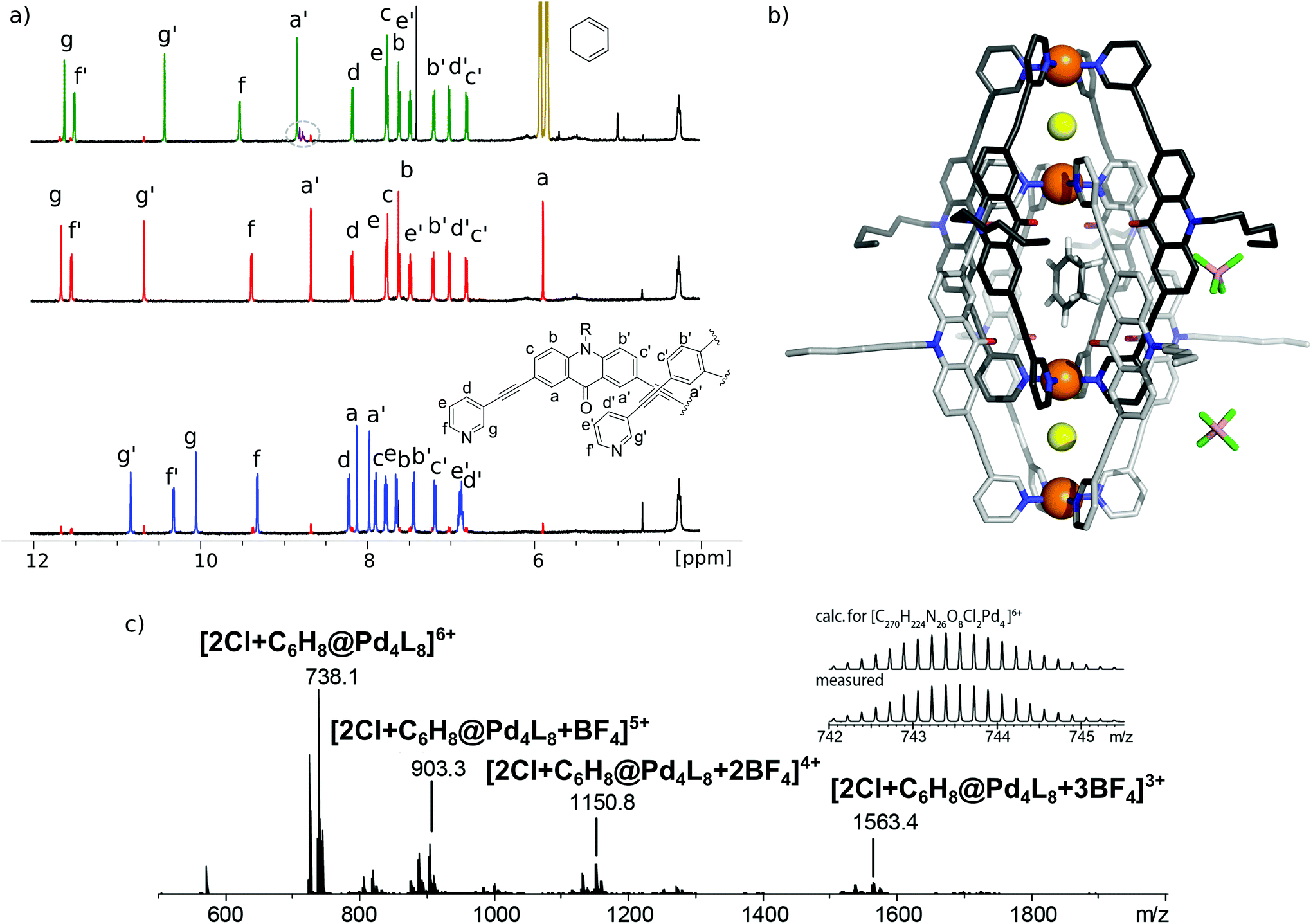 | ||
| Fig. 2 (a) 1H NMR spectra in CD3CN (0.35 mM) of [3BF4@Pd4L8] (blue), [2Cl@Pd4L8] (red) and [2Cl+C6H8@Pd4L8] (green); free guest (gold) and bound guest (purple; highlighted by a grey circle). (b) X-ray crystal structure of [2Cl+C6H8@Pd4L8] (colour scheme: C: light/dark grey; N: blue; O: red; Cl: yellow; F: green; B: salmon; Pd: orange, and H: white). The neutral guest molecule 1 was modelled in the central pockets of the cavity based on NMR and ESI-MS results and in agreement with the crystallographic data. It is depicted to adopt one of four symmetry-equivalent positions. For clarity, the hydrogen atoms of the coordination cage, co-crystallized solvent molecules and some of the counter anions were removed. (c) ESI-MS spectrum of the host–guest complex [2Cl+C6H8@Pd4L8]. | ||
Single crystal X-ray structure
Single crystals of [2Cl+C6H8@Pd4L8] were obtained via slow vapor diffusion of diethyl ether into an acetonitrile solution of the host–guest complex. X-ray data measured at a synchrotron radiation source reveal that the supramolecular structure consists of two monomeric cage units which interlace into the [Pd4L8] double cage architecture (Fig. 2b). The structure crystallized in space group P4/ncc with one fourth of the double cage in the asymmetric unit. Similar to results obtained in previous guest inclusion studies, the structure features three pockets, the outer two occupied by a chloride anion and the inner void occupied by the neutral guest molecule 1 (C6H8). The crystallographic analysis was complicated as the encapsulated guest molecule 1 is disordered over a special position (4-fold axis). Therefore, it is able to reside in four different, symmetry-equivalent orientations in the cavity, each of which shows an occupancy of 25%. Hence, the crystallographic data alone did not allow us to unequivocally interpret the exact co-conformation of host and guest in the complex. We solved this problem by modelling one cyclohexadiene guest molecule inside the cavity (in accordance with NMR spectroscopic and mass spectrometric data) and evaluated the crystallographic model using an omit map (see ESI, Fig. S21†). As a result, the guest in [2Cl+C6H8@Pd4L8] is slightly tilted off the vertical axis between the palladium cations flanking the inner void. The distances between the palladium cations for the outer pockets are 6.4 Å and for the inner cavity 10.6 Å. Modelling and refinement details can be found in the ESI.†Photocatalysis
1,3-Cyclohexadiene 1 is known to undergo a [4+2] cycloaddition with atmospheric oxygen in the presence of a photosensitizer such as Rose Bengal (RB)43 or tetraphenylporphyrin (TPP)44 under irradiation. The main product in this reaction is endoperoxide 2 with yields of 71% and 80%, respectively. Photosensitization produces singlet oxygen by triplet energy transfer from the dye molecules. Besides its biological relevance, for example in the oxidation of cholesterol,45 singlet oxygen is frequently used in synthetic organic photochemistry.46,47 A major problem with the typically employed dyes RB, TPP or methylene blue is their lack of long-term stability.48As acridone derivatives were reported before to serve as photosensitizers for singlet oxygen generation,48 we first tested ligand L for its ability to catalyse the photoreaction. However, ligand L turned out to be too light sensitive and even needs to be stored in the dark to prevent decomposition. Pleasingly, this lack of stability was completely abolished after cage assembly: both [3BF4@Pd4L8] and [2Cl@Pd4L8] did not show significant degradation after being exposed to light, even after several weeks. Encouraged by this observation, we anticipated that the acridone-based cages will be sufficiently stable as triplet photosensitizers.
Free acridone ligand L shows two absorption maxima at 262 and 357 nm with a shoulder at 319 nm. These bands are slightly blue shifted for the cages and the absorption of the shoulder is increased. [2Cl@Pd4L8] has three absorption maxima at 265, 231 and 360 nm and [3BF4@Pd4L8] has three absorption maxima at 265, 231 and 362 nm (ESI, Fig. S21†).
For testing photochemical oxygen activation, [2Cl@Pd4L8] was freshly prepared in CD3CN and 30 eq. of guest 1 was added. After an incubation time of 24 h, 1H NMR spectroscopy confirmed the formation of the host–guest complex [2Cl+C6H8@Pd4L8] in addition to a small amount of free host [2Cl@Pd4L8] (Fig. 3a and ESI, Fig. S4†). The sample was placed in a sealed screw cap vial (0.35 mM [2Cl@Pd4L8], sample volume 1 mL) and irradiated at room temperature in a photoreactor49 for 40 minutes with a 365 nm LED source (3.4 W). Subsequently, the 1H NMR signals of the cage protons Hg, Hg′ and Ha′ that are pointing inside the cavities shifted significantly, indicating a reaction of the encapsulated species (Fig. 3a). Furthermore, a new set of signals appeared at chemical shifts of δ = 6.63, 4.60, 2.22 and 1.43 ppm (Fig. 3b), matching with the reported literature values of endoperoxide 2 in CD3CN solution.50 Full conversion of 1 was observed, and endoperoxide product 2 had been formed as the main species. Hydroperoxide 3 was observed as a side product, which is the product of an ene reaction to singlet oxygen (light blue signals in Fig. 3).44
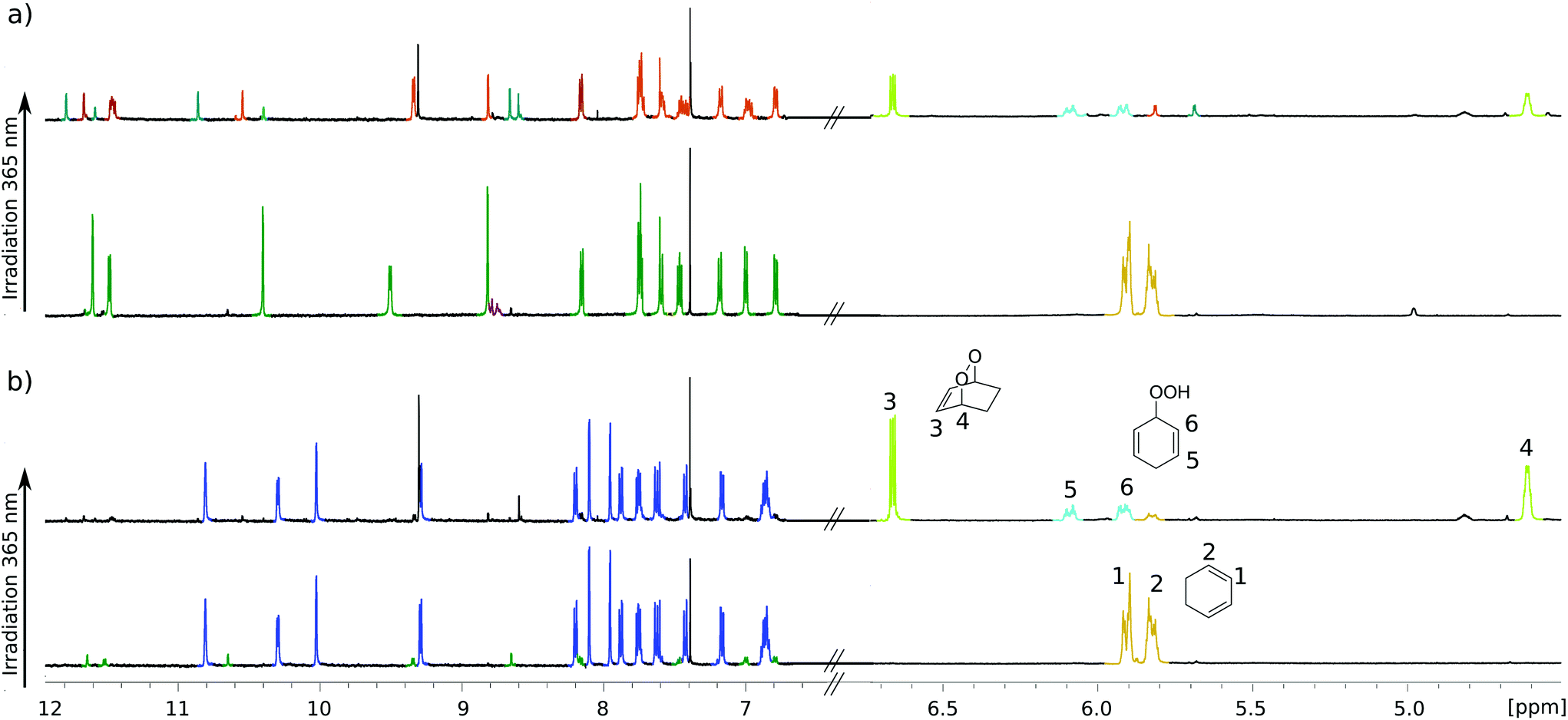 | ||
| Fig. 3 (a) 1H NMR spectra of [2Cl@Pd4L8] (0.35 mM) in the presence of 1,3-cyclohexadiene 1 (30 eq.) before (green) and after (orange and turquoise) irradiation with a 365 nm LED in the presence of air for 40 minutes. The transformation of 1 (gold) into products 2 (lime) and 3 (light blue) as observed in the same NMR spectrum (depicted in an expanded format in the right half). (b) 1H NMR spectra of [3BF4@Pd4L8] (0.35 mM) in the presence of 1 (30 eq.) (blue) before and after irradiation with 365 nm LED for 40 minutes. Right: conversion of 1 into 2 and 3 by [3BF4@Pd4L8]. | ||
Interestingly, proton signals for inward pointing Hg, Hg′ and Ha′ of the double cage were found to split into three sets, one assigned to [2Cl+2@Pd4L8], marked orange in Fig. 3a, and two further sets highlighted in turquoise. We assume that one of the latter signal sets stems from host including side product 3. The third group of signals may result from encapsulation of yet another product isomer or caused by 3 adopting a different, sterically locked position inside the host pocket. With the help of a DOSY experiment (ESI, Fig. S11†), we could confirm that all signal sets belong to species with the size of the double cage.
In order to confirm that species [2Cl@Pd4L8] served as a photocatalyst, we performed a series of control experiments, summarized in Table 1. First, addition of 1 to [2Cl@Pd4L8] under argon shows no formation of peroxides 2 or 3, which clearly confirms that a light induced [4+2] cycloaddition is taking place only in the presence of molecular oxygen (ESI, Fig. S12†). When only 1 in CD3CN was irradiated for 40 minutes, the solution turned brown with the formation of a sluggish precipitate and the 1H NMR spectrum shows neither substrate nor product signals (ESI, Fig. S13†). Likewise, a mixture of free acridone ligand L (2.8 mM in CD3CN) and 1 resulted in decomposition of both L and 1 after 40 minutes of irradiation as indicated by the formation of a brown precipitate and disappearance of 1H NMR signals corresponding to the ligand and substrate (ESI, Fig. S14†). In order to better mimic the electronic situation of a single acridone ligand as part of the cage, we prepared the complex [L(Pd-terpy)2] (terpy = 2,2′:6′,2′′-terpyridine), in which two terpy-capped palladium cations are coordinated to both pyridine nitrogen donors of the ligand (see the ESI† for synthesis conditions and characterization). Intriguingly, this species did not perform as a catalyst either, as demonstrated by 1H-NMR analysis (ESI, Fig. S15†). Altogether, these experiments point to an intrinsic photocatalytic activity of the coordination cage [2Cl@Pd4L8]. Previously, we have reported a [Pd2L4] cage with the same acridone backbone, but with isoquinoline instead of pyridine donor groups, which did not lead to cage dimerization.51 Under the same reaction conditions, this cage performed similarly to [2Cl@Pd4L8] (Table 1 and ESI, Fig. S19†), showing that cage interlocking is not necessary for photocatalytic activity.
|
|
||||
|---|---|---|---|---|
| No. | Condition | Conversion | 2 | 3 |
| a 0.35 mM (≙2.8 mM ligand) solution of the cage in CD3CN, 30 eq. of 1 under air. Irradiation with 365 nm LED. Yield determined by 1H NMR spectroscopy. Average of three samples. b 0.7 mM in CD3CN. c 2.8 mM in CD3CN. | ||||
| 1a | [2Cl@Pd4L8] | 100% | 58% | 42% |
| 2a | [3BF4@Pd4L8] | 88% | 70% | 30% |
| 3b | [Pd2L4] | 100% | 57% | 43% |
| 4c | [L(Pd-terpy)2] | — | 0 | 0 |
| 5 | only 1 in CD3CN | — | 0 | 0 |
| 6c | ligand L | — | 0 | 0 |
Next, we addressed the question whether the reaction takes place inside the central pocket of [2Cl@Pd4L8]. As stated above, the neutral substrate is taken up by the cage as a guest only in the presence of chloride anions. To probe whether substrate encapsulation was a prerequisite for a successful hetero-Diels–Alder reaction, 30 equivalents of 1 were added to a solution of [3BF4@Pd4L8] and the mixture was irradiated for 40 minutes in the presence of atmospheric oxygen. Interestingly, about 88% of 1 was converted into 2 as the major product (Fig. 3b). Further irradiation for 40 minutes resulted in the complete conversion of 1 (ESI, Fig. S1†). Importantly, cage [3BF4@Pd4L8] itself is not affected by irradiation as no change in its 1H-NMR spectrum was observed. Since the tetrafluoroborate-containing coordination cage [3BF4@Pd4L8] does not have any vacant pockets, it can be concluded that the hetero-Diels–Alder reaction is not dependent on encapsulation of 1.
In order to improve the selectivity of the reaction towards single product 2, we reduced the light intensity of the LED to 50% (ESI, Fig. S16†) or 20% (ESI, Fig. S17†) in order to slow down the reaction and yield only product 2, without success. Then, we performed the reaction at 5 °C, which was reported to be the optimal temperature with Rose Bengal as the photosensitizer.43 Also, under this condition, similar amounts of side product 3 were observed in the 1H NMR spectrum (ESI, Fig. S18†).
Finally, we were able to maximize the formation of product 2 by leaving the sample in ambient daylight for 94 days. Here, the formation of compound 2 and its host–guest complex [2Cl+2@Pd4L8] were clearly observed to be the only products. The 1H-NMR-signals of the host–guest complex correspond to the chemical shifts marked in orange in Fig. 3a (compare ESI, Fig. S20†). The hypothesis that the signal splitting described above is a result of different encapsulated products was thus confirmed. High resolution ESI mass spectrometry showed signals corresponding to [2Cl+C6H8O2@Pd4L8+nBF4](6−n)+ (n = 0–3) (Fig. 4), thus confirming the encapsulation of a species with a chemical formula of C6H8O2.
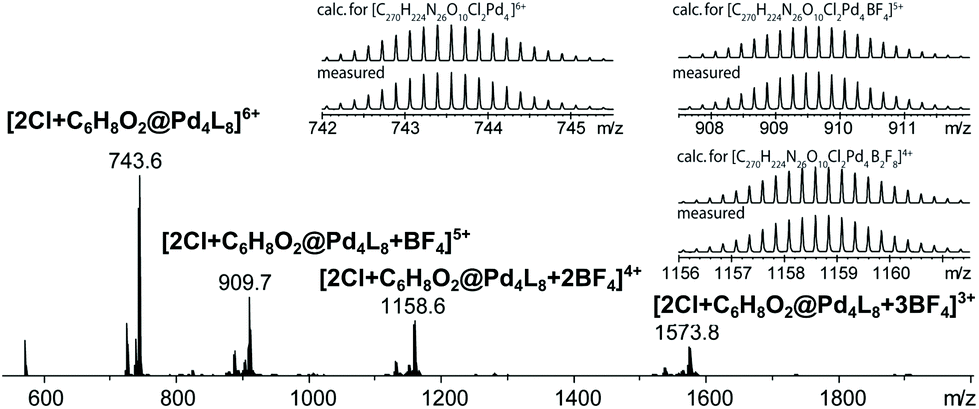 | ||
| Fig. 4 ESI-MS spectrum of the host–guest complex [2Cl+C6H8O2@Pd4L8] obtained by the slow photosensitization of a cage–substrate mixture in the presence of air after 94 days. | ||
To our delight, we were able to obtain single crystals of [2Cl+C6H8O2@Pd4L8]via slow vapor diffusion of diethyl ether into an acetonitrile solution of the host–guest complex. The crystal structure is similar to the structure of [2Cl+C6H8@Pd4L8] and also crystallized in the space group P4/ncc with one fourth of the double cage architecture in the asymmetric unit. Similar to the [2Cl+C6H8@Pd4L8] structure, the guest is positioned on the four-fold axis and was modelled in the inner cavity in accordance with the NMR spectroscopic and mass spectrometric data. The omit map (Fig. 5 and Fig. S22†) confirms the consistency of this position with the crystallographic data. The palladium–palladium distances in the [2Cl+C6H8O2@Pd4L8] complex are 6.4 Å for the outer pockets and 10.7 Å for the inner pockets. Guest 2 is slightly tilted off the vertical axis between the palladium cations in the central cavity of the interpenetrated coordination cage with the shortest palladium–guest distance measuring 3.0 Å. Close intermolecular contacts between the acridone ligands of the cage and neutral guest 2 were measured to range from 2.3 to 3.3 Å. The shortest intermolecular CH⋯O contact is 2.6 Å.
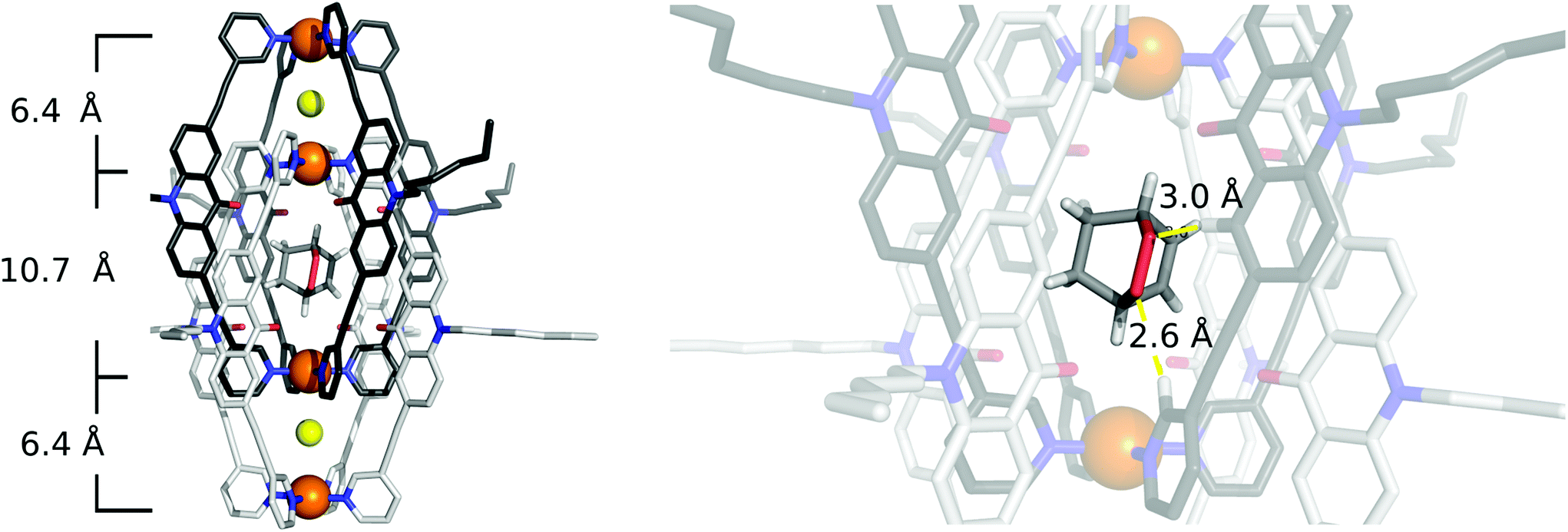 | ||
| Fig. 5 X-ray crystal structure of [2Cl+C6H8O2@Pd4L8] (colour scheme: C: light/dark gray; N: blue; O: red; Cl: yellow; F: green; B: salmon; Pd: orange, and H: white). Neutral guest molecule 2 in the central pocket of the cavity was modelled based on the NMR and ESI-MS results consistent with the crystallographic data. It is depicted to adopt one of four symmetry-equivalent positions. For clarity, hydrogen atoms of the coordination cage, co-crystallized solvent molecules and counter anions outside the cage structure were removed. | ||
Conclusions
We demonstrated the ability of interpenetrated coordination cages [2Cl@Pd4L8] and [3BF4@Pd4L8] to convert 1,3-cyclohexadiene 1 in the presence of atmospheric oxygen and light into the peroxides 2,3-dioxabicyclo[2.2.2]oct-5-ene 2 and 3-hydroperoxycyclohexa-1,4-diene 3. The metal-mediated supramolecular assembly stabilizes the acridone ligand toward photodegradation and thus enables its photochemical activity. As shown by 1H NMR spectroscopy, high-resolution mass spectrometry and X-ray structure analysis, [2Cl@Pd4L8] readily encapsulates both substrate and product of the examined reaction. Photocatalytic activity, however, is not restricted to the host–guest complex. Also, the unbound substrate in the presence of either one of the coordination cages is converted to the peroxide. In future studies, we will extend the substrate scope for this reaction and examine the system's capability to catalyse further electrocyclic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Training group ‘Confinement-controlled Chemistry’, funded by the Deutsche Forschungsgemeinschaft (DFG) under Grant GRK2376/331085229. Further support by the DFG through grants CL 489/2-2 and SPP1807 is acknowledged. S. P. and G. H. C. thank the European Research Council for supporting the project with a Marie-Sklodowska Curie Individual Fellowship (MOCCA Project No. 798103 ). Single crystal X-ray diffraction data of [2Cl+C6H8@Pd4L8] was collected at Petra III, DESY (a member of the Helmholtz Association, HGF), Hamburg, Germany. Anja Burkhardt is thanked for assistance in using macromolecular beamline P11 and travel grants from DESY are gratefully acknowledged (I-20160736).Notes and references
- R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813–1824 RSC.
- M. M. J. Smulders and J. R. Nitschke, Chem. Sci., 2012, 3, 785–788 RSC.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- O. Shyshov, R.-C. Brachvogel, T. Bachmann, R. Srikantharajah, D. Segets, F. Hampel, R. Puchta and M. von Delius, Angew. Chem., Int. Ed., 2017, 56, 776–781 CrossRef CAS PubMed.
- R. Sekiya, M. Fukuda and R. Kuroda, Org. Biomol. Chem., 2017, 15, 4328–4335 RSC.
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed.
- L. Yang, X. Jing, B. An, C. He, Y. Yang and C. Duan, Chem. Sci., 2018, 9, 1050–1057 RSC.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed.
- C. Tan, D. Chu, X. Tang, Y. Liu, W. Xuan and Y. Cui, Chem. – Eur. J., 2019, 25, 662–672 CrossRef CAS PubMed.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- T. A. Bender, M. Morimoto, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 1701–1706 CrossRef CAS PubMed.
- S. M. Bierschenk, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 733–737 CrossRef CAS PubMed.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS PubMed.
- W. Cullen, M. Misuraca, C. Hunter, N. Williams and M. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- V. Martí-Centelles, A. Lawrence and P. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef PubMed.
- S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef CAS.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- L.-J. Chen and H.-B. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS PubMed.
- S. Ganta and D. K. Chand, Inorg. Chem., 2018, 57, 5145–5158 CrossRef CAS PubMed.
- R. Lavendomme, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 12147–12158 CrossRef CAS PubMed.
- S. Chakraborty, K. J. Endres, R. Bera, L. Wojtas, C. N. Moorefield, M. J. Saunders, N. Das, C. Wesdemiotis and G. R. Newkome, Dalton Trans., 2018, 47, 14189–14194 RSC.
- R. Sekiya, M. Fukuda and R. Kuroda, J. Am. Chem. Soc., 2012, 134, 10987–10997 CrossRef CAS PubMed.
- M. Han, Y. Luo, B. Damaschke, L. Gómez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Angew. Chem., Int. Ed., 2016, 55, 445–449 CrossRef CAS PubMed.
- R.-J. Li, J. J. Holstein, W. G. Hiller, J. Andréasson and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 2097–2103 CrossRef CAS PubMed.
- A. J. Musser, P. Neelakandan, J. M. Richter, H. Mori, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 12050–12059 CrossRef CAS PubMed.
- Y. Furutani, H. Kandori, M. Kawano, K. Nakabayashi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2009, 131, 4764–4768 CrossRef CAS PubMed.
- W. Cullen, H. Takezawa and M. Fujita, Angew. Chem., Int. Ed., 2019, 58, 9171–9173 CrossRef CAS PubMed.
- S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed.
- M. Frank, M. D. Johnstone and G. H. Clever, Chem. – Eur. J., 2016, 22, 14104–14125 CrossRef CAS PubMed.
- M. Frank, J. Ahrens, I. Bejenke, M. Krick, D. Schwarzer and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 8279–8287 CrossRef CAS PubMed.
- M. Frank, J. Hey, I. Balcioglu, Y.-S. Chen, D. Stalke, T. Suenobu, S. Fukuzumi, H. Frauendorf and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 10102–10106 CrossRef CAS PubMed.
- R. Zhu, J. Lübben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796–2800 CrossRef CAS PubMed.
- J. M. Dieterich, G. H. Clever and R. A. Mata, Phys. Chem. Chem. Phys., 2012, 14, 12746–12749 RSC.
- S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060–1063 CrossRef PubMed.
- S. Löffler, A. Wuttke, B. Zhang, J. J. Holstein, R. A. Mata and G. H. Clever, Chem. Commun., 2017, 53, 11933–11936 RSC.
- J. Clayden, N. Greeves and S. G. Warren, Organic chemistry, Oxford University Press, Oxford, New YorK, 2nd edn, 2012 Search PubMed.
- T. Murase, Y. Nishijima and M. Fujita, J. Am. Chem. Soc., 2012, 134, 162–164 CrossRef CAS PubMed.
- R. Ziegert and S. Bräse, Synlett, 2006, 2119–2123 CAS.
- O. Dechy-Cabaret, F. Benoit-Vical, C. Loup, A. Robert, H. Gornitzka, A. Bonhoure, H. Vial, J.-F. Magnaval, J.-P. Séguéla and B. Meunier, Chem. – Eur. J., 2004, 10, 1625–1636 CrossRef CAS PubMed.
- R. C. Murphy and K. M. Johnson, J. Biol. Chem., 2008, 283, 15521–15525 CrossRef CAS PubMed.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- M. DeRosa, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- J. Eberhard, K. Peuntinger, S. Rath, B. Neumann, H.-G. Stammler, D. M. Guldi and J. A. Mattay, Photochem. Photobiol. Sci., 2014, 13, 380–396 RSC.
- C. C. Le, M. K. Wismer, Z.-C. Shi, R. Zhang, D. V. Conway, G. Li, P. Vachal, I. W. Davies and D. W. C. MacMillan, ACS Cent. Sci., 2017, 3, 647–653 CrossRef CAS PubMed.
- K. Feng, R.-Y. Zhang, L.-Z. Wu, B. Tu, M.-L. Peng, L.-P. Zhang, D. Zhao and C.-H. Tung, J. Am. Chem. Soc., 2006, 128, 14685–14690 CrossRef CAS PubMed.
- W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. CCDC 1952467 and 1952468. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/D0DT01674H |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |