
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding and controlling the covalent functionalisation of graphene†
Adam J.
Clancy
 *a,
Heather
Au
b,
Noelia
Rubio
c,
Gabriel O.
Coulter
c and
Milo S. P.
Shaffer
*c
*a,
Heather
Au
b,
Noelia
Rubio
c,
Gabriel O.
Coulter
c and
Milo S. P.
Shaffer
*c
aDept. Chemistry, UCL, Gower Street, London, WC1H 0AJ, UK. E-mail: a.clancy@ucl.ac.uk
bDept. Chemical Engineering, Imperial College London, London, SW7 2AZ, UK
cDept. Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk
First published on 30th June 2020
Abstract
Chemical functionalisation is one of the most active areas of graphene research, motivated by fundamental science, the opportunities to adjust or supplement intrinsic properties, and the need to assemble materials for a broad array of applications. Historically, the primary consideration has been the degree of functionalisation but there is growing interest in understanding how and where modification occurs. Reactions may proceed preferentially at edges, defects, or on graphitic faces; they may be correlated, uncorrelated, or anti-correlated with previously grafted sites. A detailed collation of existing literature data indicates that steric effects play a strong role in limiting the extent of reaction. However, the pattern of functionalisation may have important effects on the resulting properties. This article addresses the unifying principles of current graphene functionalisation technologies, with emphasis on understanding and controlling the locus of functionalisation.
Introduction
Graphene, a framework of sp2 hybridised carbons arranged as a 2D hexagonal lattice, is widely considered as one of the most important materials of the current generation. Ideal, perfect samples exhibit excellent mechanical properties1–3 (tensile strength of >100 GPa, Young's modulus 1 TPa, toughness of 3.9 MPa m1/2), thermal conductivity4,5 (5000 W mK−1) and electron mobility6–8 (>200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1). Whilst it lacks a true bandgap, the linear dispersion relation, and vanishing density of states at the undoped Fermi level, give rise to a range of important ambipolar optoelectronic phenomena. Graphene-derived materials have been applied to a vast range of applications, as diverse as structural composites, photonics, transistors, drug delivery, superconductors, sensors, nanofiltration, and isotopic enrichment.9–12 To adapt graphene to these opportunities, the intrinsic properties are often manipulated using chemical functionalisation, for example, to open a band gap, or increase solubility, electronic conductivity, biocompatibility, or wetting/interfacial adhesion. In addition, a range of specific functions may be introduced, for example, by grafting a biomarker, a fluorophore, an (electro)catalyst, or sorbent. While the desired outcomes vary significantly, much of the underlying graphene functionalisation chemistry is shared.
000 cm2 V−1 s−1). Whilst it lacks a true bandgap, the linear dispersion relation, and vanishing density of states at the undoped Fermi level, give rise to a range of important ambipolar optoelectronic phenomena. Graphene-derived materials have been applied to a vast range of applications, as diverse as structural composites, photonics, transistors, drug delivery, superconductors, sensors, nanofiltration, and isotopic enrichment.9–12 To adapt graphene to these opportunities, the intrinsic properties are often manipulated using chemical functionalisation, for example, to open a band gap, or increase solubility, electronic conductivity, biocompatibility, or wetting/interfacial adhesion. In addition, a range of specific functions may be introduced, for example, by grafting a biomarker, a fluorophore, an (electro)catalyst, or sorbent. While the desired outcomes vary significantly, much of the underlying graphene functionalisation chemistry is shared.
Real graphene samples vary enormously in quality and effective properties, depending on the synthetic strategy; typically, mechanically exfoliated samples are most crystalline, and chemically exfoliated the least.13,14 Whilst concentrations vary by many orders of magnitude, all samples contain a variety of possible defects (Fig. 1), including carbon vacancies, heteroatom substitutions, in-plane rearrangements, and sp3 point defects where another species is covalently bound to a basal plane carbon. These basal plane defects are known to diminish the mechanical properties of graphene,15–17 and alter the optoelectronic properties, even at low concentrations.18 Graphene edges may also be described as a defect, with a variety of characteristic properties depending on orientation. They are particularly prevalent in bulk graphene-related powders/dispersions and laterally-confined graphene nanoribbons and nanoplatelets.
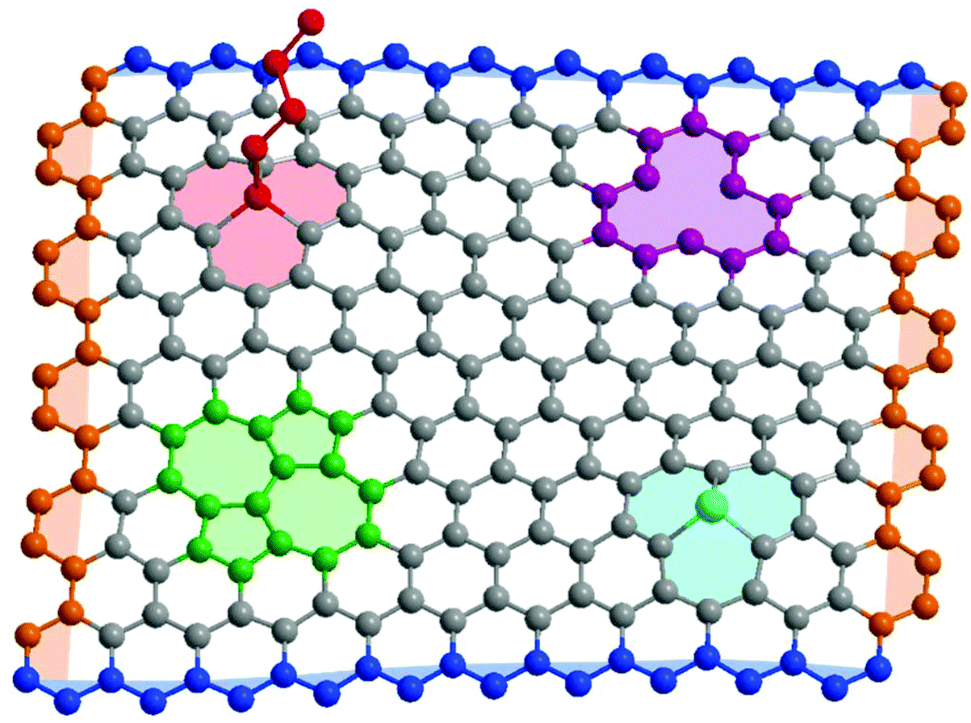 | ||
| Fig. 1 Schematic structure of typical graphene defects. [Red] sp3 functionality, [green] Stone–Wales rotation, [purple] vacancy, [light blue] heteroatom substitution, [dark blue] zig-zag edge, [orange] armchair edge. | ||
In part due to the well-established difficulty in creating high quality graphene in large quantities, there has been a tendency to describe a host of high aspect ratio graphitic materials as “graphene”, irrespective of crystalline quality, or indeed number of layers. Thicker materials with >1 layer are bi-/tri/few-layered graphenes, graphite nanoplatelets, or indeed, simply graphite. Strong oxidation produces exfoliated graphene oxide (GO) which can be treated to form reduced-GO (rGO), but not to regenerate graphene. While each of these species have distinct applications and interesting properties, they are not graphene. Here, we attempt to use the nomenclature set out by the editorial board of Carbon,19 and encourage other researchers to do the same.
The properties of graphene may be modified non-covalently by materials adsorbed on the basal plane, for example, doping with oxygen, coating with a polymer, or deposition on a substrate.20–22 Alternatively, graphene may be functionalised through covalently bonding a moiety to the carbon framework.23,24 Covalent modification is a more robust and typically irreversible approach to modifying the graphene, and is both of practical importance and fundamental scientific interest. This review summarises the typical reaction types and focuses on the increasing efforts to control the locus of functionalisation.
Functionalisation reactions
The functionalisation chemistry of graphene is similar to that of other sp2 hybridised carbon allotropes: carbon nanotubes and fullerenes.6,25,26 However, the curvature of the 1D/0D analogues creates strain in the sp2 carbon bonds which is alleviated by introduction of sp3 sites. The relaxation of the strained nanocarbons lowers the energy barrier for functionalisation, causing higher reactivity than for (ideally) non-curved/non-strained graphene.27–29 The overall reactivity of graphene is a function of the type, particularly the degree of perfection and number of layers; single layer graphene is significantly more reactive than bilayer and higher layer graphenes.30–33The simplest reactions occur at a pre-existing defect within the graphene layer. Whilst avoiding the introduction of any new defects and associated property degradation, this approach offers only very limited control of the degree of functionalisation which may be very low for high quality graphene. Adventitious defect groups (usually oxygen-based) remaining from graphene synthesis or exfoliation may be desorbed thermally, generating radicals that can trap an alternative functionality.34,35 Alternatively, specific defect groups may be coupled to further reagents. Most commonly, carboxylic acids, which may be adventitious or deliberate by-products of oxidation (see below), are subjected to esterification or amidification reactions with alcohols and amines, respectively.36
The edges of graphene flakes, or the grain boundaries within polycrystalline layers, which may be viewed as 1D defects within the 2D material, provide many correlated sites for reaction. The carbon framework of the core of the flake retains its sp2 hybridisation, so properties are not compromised, although doping and spin effects are under investigation.37 For large diameter graphene flakes, the proportion of carbons at edge sites is low, limiting the extent and influence of grafting. Edge functionalisation is particularly pertinent for graphitic species with lateral confinement, such as graphene nanoribbons or graphene nanoplatelets. They have a large quantity of edge sites and display bulk optoelectronic properties that can be tuned via the edge chemistry.38 The functionalisation options are broadly dependent on the existing chemistry of the edge termination site; for example, carboxylic acid terminations may be transformed into anhydrides, which subsequently may be transformed into amides or metal-ion complexes.39
A variety of direct reactions on the graphene basal plane are possible, although relatively reactive species are required to attack the π-bonded network. The most simple approach uses plasmas (e.g. H, F, Cl) for activation, although this approach can etch away the graphene framework.40 In the absence of plasma enhancement, perhalogenated graphene can be prepared using molecular F2 (or other strong fluorinating agents) via a radical mechanism,41 or using photochemically-generated chlorine radicals.40
In the liquid phase, graphene may be functionalised through chemical oxidation, usually with nitric acid, introducing oxygen-containing groups which may subsequently be used for the defect modifications mentioned above, and discussed in more detail elsewhere.42 With extreme oxidation, in the case of GO, the intrinsic properties are irreversibly affected,43 although damage may be moderated by use of lower temperatures or milder oxidants44 (e.g. S2O82−). rGO still contains a significant fraction of oxygen atoms, and the ‘reduction’ process typically removes some framework carbon atoms, for example due to the evolution of CO or CO2 on heating, leaving holes or pores in the layers. Graphene which is mildly oxidised to give only hydroxyls on the basal plane has been termed ‘oxo-graphene’ to distinguish it from typical GO, as it may be reduced without significant carbon loss.44
By far the most common non-oxidative functionalisations rely on reactions with organo radicals, typically using aryl diazonium salts. Here, the graphene reduces the aryl through transfer of a π-electron to the diazonium cation to form an ArN2˙ radical, which decomposes to release N2. The resultant aryl radical may react directly with the graphene carbon framework forming a covalent bond. Other radical sources are known (including nitrenes14 and aliphatic carbenes11), but aryl diazoniums are the most well established due to their relative stability, flexibility of substituents, and availability (either of the diazonium salt itself, or the parent aromatic amine which may be trivially converted).
The unsaturated nature of graphene has also been exploited by co-opting several classic alkene reactions. All the graphitic nanomaterials are susceptible to 1,3-dipolar cycloadditions, using azomethine ylides45 (the Prato reaction), ozone, or oxazolones.46 Graphene may also undergo Diels–Alder (DA) cycloadditions;47 interestingly, these reactions show the classic reversibility of DA reactions, and the graphene may react as either diene or dienophile.48
As an alternative to these neutral-graphene reactions, anionic graphene sheets can be prepared by doping with group 1 metals, either directly or via charge transfer agents. These ‘graphenides’ can initiate reductive functionalisation reactions with a host of functional groups, most commonly organohalides, but also disulphides, peroxides, silyl halides, and protons.6 The charge may also be used to initiate anionic polymerisation to graft polymers ‘from’ the surface of the graphene.25 Reductive functionalisation offers a greater variety of potential graphene-addend bonds than the C/N/O bonds typically formed in other reactions, including with bromine,32 hydrogen,33 and silicon.24
The greatest benefit of anionic functionalisation, however, lies in the properties of the initial graphenide itself, which forms thermodynamically-stable solutions of monolayers.6 In principle, the dissolution provides access to the entire graphene surface for homogeneous functionalisation, without the steric occlusion associated with incomplete exfoliation49 and/or adsorbed surfactants common in other shear-based exfoliation processes (e.g. bath sonication, shear mixing, etc.). The intrinsic damage associated with GO formation or intense sonication is also avoided.6 Historically, reductive functionalisation has been assumed to proceed via single electron transfer (SET) from the graphenide to reduce the reagent to a radical anion, which degrades to an anionic leaving group and a radical (e.g. RBr to R˙ and Br−). The resulting radical subsequently grafts to the graphene similarly to other radical routes such as aryl diazoniums. However, the analogous reaction on anionic carbon nanotubes was recently found to proceed via complexation between nanocarbon anion and reagent, followed by a two-electron transfer.50 Further studies of the mechanism of graphenide reactions are needed.
In addition to chemical doping methods, graphene may be charged electrochemically. This approach may be used to facilitate the exfoliation of graphite, through both Coulombic repulsion and chemical modification reactions. At the potentials required for exfoliation, accidental or deliberate reactions with the electrolyte or other additives are to be expected, although not always discussed. More modest potentials can be used to accelerate SET reactions, such as the diazonium chemistry, by providing higher energy electrons to accelerate aryl formation.51
While covalent modification of graphene is typically an addition to the hexagonal 2D framework (summarised in Fig. 2c), it is possible to modify the carbon lattice itself through heteroatom substitutions (often referred to as ‘heteroatom doping’). Introducing nitrogen or boron during chemical vapour deposition synthesis of graphene is by far the most common approach,52 although post-synthetic substitution through ion implantation has been used for phosphorus,53 germanium,54 and silicon.55 Difficulties in scalability and control makes this approach currently more suitable for fundamental studies than devices.
 | ||
| Fig. 2 Degree of functionalisation for reactions with the graphene carbon framework: summary of available literature. (a) Comparison of grafting stoichiometries between XPS and TGA, based on reports in which both techniques are used on the same sample,7,8,14,16,25,32,56 (red line, linear fit; black dashed line, equivalence of TGA and XPS measurements). (b) Molecular weight of grafted moiety versus degree of functionalisation (R/C, left) and grafting stoichiometry (C/R, right) and, categorised by reaction type: organo-radical additions5,7,8,11,12,14,16,17,57,58 (black, diazonium/carbene/nitrene), halogenation40,59–62 (green), plasma-hydrogenation63 (puce), cycloadditions45,46,56,64–66 (red), reductive functionalisation21,22,24–26,28,29,32,33,35,67 (blue), thermochemical functionalisation34 (magenta), and oxidation (orange). Limits for oxidation estimated from typical maximum limiting stoichiometry of C8O4H5 from Pei et al.68 between 16 Da (epoxide) and 33 Da (carboxyl), with the lower limit for degree of oxidation from hydroxylation via oxo-graphene.44 Dashed lines illustrate steric grafting limits69 for a freely jointed –CH2– chain treated with de Gennes model70 for 1, 2, 5, 20 and 50-layer graphene stacks. The underlying literature data is tabulated in the ESI,† for reference. (c) Schematic showing classic examples of the different classes of functionalisation reactions on graphene (same colour scheme as (b)). | ||
Degree of functionalisation
The extent of grafting reactions is variously reported as ‘grafting ratio’, the mass of grafted moieties relative to the graphitic framework, ‘grafting stoichiometry’ (C/R), the number of graphitic carbons per grafted group, or degree of functionalisation (R/C), the proportion of reacted framework carbons. The values are usually determined either through thermogravimetric analysis (TGA) or X-ray photoelectron spectroscopy (XPS). TGA measures the weight loss as the more volatile grafted groups decompose, typically between 200–400 °C; ideally, the grafted species is identified by mass spectroscopy to avoid confounding effects of adsorbed/intercalated solvent.71 XPS measures the ratio of carbon to a distinctive non-carbon element in the grafted species. In general, the agreement between the two methods, where both are reported, is excellent (the correlation from the available literature in summarised in Fig. 2a). There is a possible weak tendency, overall, for XPS to give a lower estimate of functionalisation degree (i.e. higher grafting stoichiometry) than TGA (∼95% of TGA values, Fig. 2a), but values are within error. An overestimation in TGA measurements for lighter grafted species might relate to the sacrificial loss of graphene framework carbons during thermal decomposition. However, a number of factors may play a role in different circumstances, including the identity of the atom tracked by XPS, its location in the grafted layer, and the possibility of addend or excess solvent intercalation.71 Since XPS is a surface sensitive technique, results for thicker, multilayer flakes may diverge.The overall degree of graphene functionalisation may be limited by the reaction conditions chosen, the reaction type, or the steric bulk of the grafted species. A summary of the available data (Fig. 2b) on the functionalisation of graphitic layers (excluding defect-modifying functionalisations such as via GO esterification) shows that the size of the addend is a fundamental limiting factor. As the attached species become larger, they occlude the graphene region adjacent to the existing functionalisation, on the same side of the graphene sheet, decreasing the degree of functionalisation (R/C).67 For (bulky) polymeric species, two regimes exist due to steric effects: at low grafting densities, the polymer will act as a random coil and is described as a de Gennes ‘mushroom’ conformation,70 while at higher densities, the polymer is extended away from the surface as a ‘brush’. To achieve the brush conformation, and the associated high grafting stoichiometry, the polymer must be grown from the surface of the graphene, for example by in situ anionic polymerisation. Pre-synthesised polymers, “grafted-to” graphene are limited to the mushroom regime.25 The majority of graphene functionalisations can be considered as “graft-to” reactions. As a guide, a prediction of the maximum degree of functionalisation allowed, as a function of molecular weight, can be made by considering the area of the surface occluded by a freely-jointed (CH2)n chain. This model has been used to plot limits on the summary of available data (Fig. 2b); the slope of the limit arises from de Gennes’ expression69,70 relating degree of functionalisation to the chain length to the power −6/5. The exact position depends on assumptions about packing, the molecular weight of the repeat unit, and the chain stiffness. However, since the effects of increasing monomer weight and increasing chain stiffness tend to counteract each other, the sensitivity to the specific identity of the grafted species is relatively weak on the log–log plot. A stronger effect arises depending on the nature of the graphitic material: true graphene in solution offers two surfaces for reaction, but as the number of stacked layers increases, the available specific area and hence the maximum degree of functionalisation falls. The set of trendlines in Fig. 2b illustrate that differences in the extent of exfoliation could account for most of the observed range in grafting stoichiometry. Unfortunately, relatively few papers report the stack height after functionalisation, so the strength of this hypothesis is currently hard to assess. The degree of functionalisation for some reagents, especially diazonium species, and other organo-radical additions, may be complicated by the tendency for self-condensation, leading to the deposition of gradually thicker oligomer layers, in principle exceeding the steric limit.72 However, for most radical reactions (including aryl diazoniums) performed in the liquid phase, the most common limiting feature is the degree of exfoliation, as poor exfoliation leads to inaccessible carbons in the non-surface layers of the suspended, few-layer graphite flakes.6
Of course, the chain approximation becomes increasingly unreliable at lower molecular weights, most obviously in the limit of single atom additions. Indeed, plasma-based reactions may eventually proceed to completion, giving a purely sp3 structure with 1:1 stoichiometry, courtesy of the small size of the atomic grafting species, combined with their high intrinsic reactivity.41 In reality, lower stoichiometry is more typical, with the extent of reaction determined by exposure time and conditions. For example, for hydrogenation,63 C/R ∼ 10 is most common, reaching ∼2 in some cases; even greater hydrogenation is possible through reductive processing.33 In contrast, the degree of fluorination can exceed 1 due to formation of CF2/CF3 from etching effects.73 Both sides of graphene must be reacted to attain stoichiometric graphene analogues; interestingly, despite the possibly limited access, plasma reactions have been shown to react between layers in non-monolayer stacks.74 Graphenide reactions may fundamentally be limited by the stoichiometry of the added charge. As the reaction proceeds, the anionic charge is consumed, lowering the Fermi level of the graphene and consequently the reactivity, towards the unreactive neutral state.75 At most, the grafting stoichiometry is limited to the initial carbon/metal stoichiometry. In reality, significantly lower stoichiometries are seen, particularly for poorly reacting species29,67 and unoptimized metal stoichiometries/concentrations.67
Locus of functionalisation
Most graphene functionalisations focus on the degree of functionalisation, as measured directly by TGA and XPS, or indirectly via Raman spectroscopy. Deducing the spatial distribution of functional groups is more challenging, though it has been explored using high resolution electron and probe microscopies, as well as by optical and Raman techniques at lower resolutions. It should be noted that the distribution of defects/functionalisations will alter the Raman spectra, even for an unchanged total degree of functionalisation; the classic Tuinstra–Koenig relation for the D/G band intensities was derived with a stochastic distribution of defects and may be unreliable for other functionalisation patterns.25,32,76 Most fundamentally, it is necessary to understand the sequence of addition to the graphene basal plane, which is dictated by the mechanism of the given reaction, the steric bulk of the reagent, and importantly, the changes in the local electronic structure of the graphene. Possible distributions of the grafted moieties on the basal plane (Fig. 3) include entirely random, stochastic arrangements of individual groups,25,67 pairwise additions across a graphene bond,48,77 additions on one of the two graphene sub lattices,78 or progressive bands of functionalisation spreading from some initiation site.79 Additions may also occur on one, both, or alternating sides of the graphene sheet, or in regions with specific curvature, or in contact with other phases.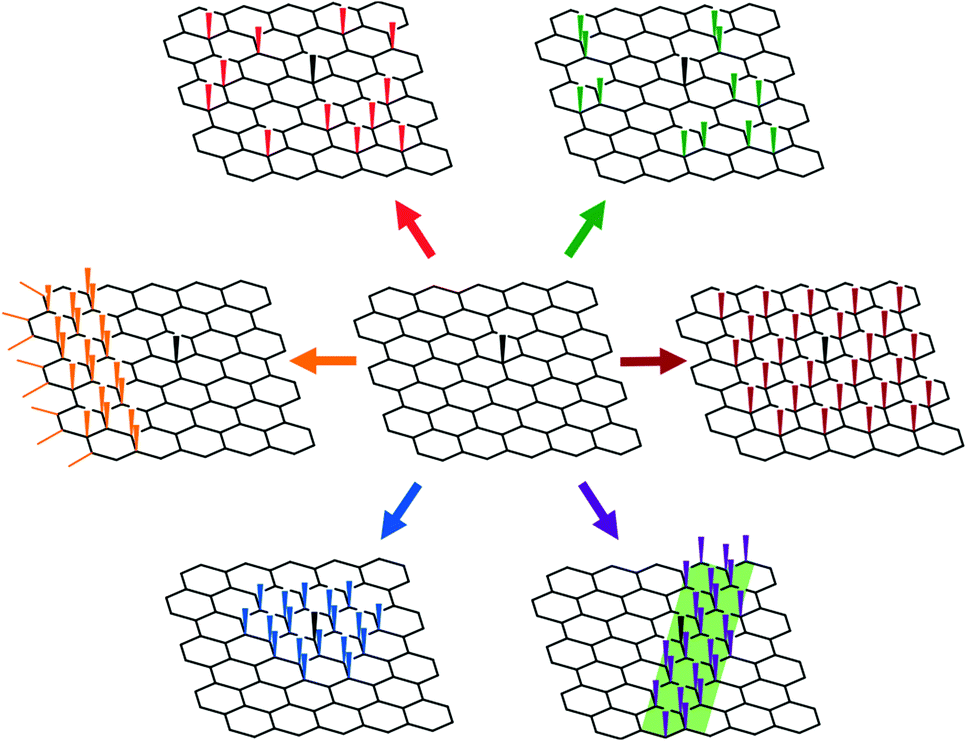 | ||
| Fig. 3 Schematic showing possible functionalisation patterns on graphene initially containing one defect (black). Clockwise from top left: stochastic random (red); pairwise addition (green); sub-lattice controlled (same as existing basal defect, maroon); substrate patterning (purple); basal-defect propagation (blue); edge propagation (orange). | ||
Pre-formed radical and plasma species have high reactivity and add irreversibly, typically forming homogeneous stochastic distributions, although in some systems, there may be higher selectivity towards edge sites.79 Most other reactions on graphene involve electron transfer from the graphene to the reagent; thus, the graphene carbons with higher local electron density and/or higher electron energies are more susceptible to reaction. For an infinite sheet of pristine graphene, all carbons have identical local electronic structure, however perturbations will bias the locus of functionalisation. Most simply, deposition of single layer graphene on a substrate will lead to fluctuations in the adjacent graphene, forming electron–hole puddles which can bias the reactivity towards the electron rich regions.30 Such fluctuations vary with the nature of the substrate and are dampened by additional layers in bi-/multi-layered stacks, leading to dramatically decreased reactivity of the top layer. Alternatively, the spontaneous wrinkles of the graphene may break the symmetry of the plane, providing sites of higher reactivity.80 In principle, the functionalisation of graphene could be manipulated using established methods of wrinkle engineering.81 Defects break the symmetry and local electronic structure of graphene, and therefore bias the locus of functionalisation. Edge sites are found in all real graphene samples, and have higher reactivities than sp2 carbons in the basal plane.30,82 The presence of sp3 or other in-plane defects (Fig. 1) leads to local fluctuations in electron density (Fig. 4) and buckling of the sheet, with both local strain and higher electron density increasing reactivity.77,83 This preference for reaction near an existing point defect can lead to ordering at greater length scales, as each new functionalisation introduces a new sp3 defect. As reactions proceed iteratively, the thermodynamic preference is for radial growth of functionalised islands, with subsequent addition preferred at the perimeter. This defect propagation process has been reported previously on CNTs.84 On 2D systems, the propagation may initiate at graphitic edges, with growth proceeding preferentially initially parallel to the edge, forming a row of sp3 defects, subsequently progressing inwards towards the flake centre85 (Fig. 5). This hypothesis has been explored using monodispersed molecular analogues of graphene (large aromatics) which can be explored by conventional chemical characterisation techniques (HPLC/NMR).86 The presence of an existing grafted species also tends to direct subsequent additions to the opposite face of the graphene (if accessible) to limit steric effects, and minimise strain.87
 | ||
| Fig. 4 The disruption of local electronic structure adjacent to a grafted species may influence subsequent reactions. Scanning tunnelling microscopy of graphene Diels–Alder functionalised with 3,5-bis(trifluoromethyl)phenyl maleimide shows standing waves forming in the surrounding region. Insert shows the Fourier transform of the image. Adapted with permission from Daukiya et al.77 Copyright 2017 American Chemical Society. | ||
 | ||
| Fig. 5 Evidence for progressive functionalisation from graphene sheet edges. Optical micrographs (a and c) and Raman spectra (b and d) of graphitic steps before (a and b) and after (c and d) reductive functionalisation illustrate higher degrees of functionalisation at edge sites. Adapted with permission from Feng et al.85 Copyright 2013 American Chemical Society. | ||
These coordinated sequential processes may be suppressed in real systems, due to both kinetic and steric effects. However, the limiting influence of steric occlusion is observed across a broad range of functionalising species, as noted above (Fig. 2b).67 The first grafted species occlude the local, electronically-preferable, defect-adjacent sites from subsequent functionalisation, likely preventing ordered additions, particularly at higher molecular weights.25 In the case of reactions that occur at a low concentration of intrinsic defects, without propagation (such as the thermochemical method), the degree of functionalisation depends only on the quality of the graphene, not the size of the grafted species34 (Fig. 2b).
For reactions initiated by donation of a π electron from the graphene (ultimately to form a radical), the loss of an electron leads to a localised unpaired electron on a carbon of the opposite sublattice to the sp3 aryl addend,88 which may direct an alternating pattern of functionalisation.78 Given the electron mobility in the plane, the fate of these unpaired electrons is unclear, and may depend on reaction density. The influence of functionalisation on the nature of the remaining conjugated π cloud is hard to predict, and it is indeed difficult to draw conventional resonance structures (Fig. 2c). While less studied than sp3 defects, in-plane vacancies, heteroatom substitutions,89 and Stone–Wales rotations will significantly affect the local electronic character. Carbons local to the in-plane defects with higher energy/modified electron density, or higher local strain, may preferentially functionalise over a non-defect-adjacent basal plane carbon.90,91 In principle, the electron-donating or withdrawing character of substituent atoms (such as boron or nitrogen) should direct the nature of the local reactions; however, current evidence is limited or even contradictory.92,93
Directed site functionalisation
As graphene chemistry matures, there are increasing efforts to dictate the location of additions. As a most simple example, one side of a graphene sheet can be functionalised selectively by masking the other face, as naturally occurs on a substrate.94 By ‘flipping’ the graphene onto a new substrate to expose the unfunctionalized side, a second reaction is possible,94,95 leading to separate functionalities on each side, creating a so-called Janus graphene. Reagents may also be embedded in photolithographically-patterned trenches to functionalise the underside of a graphene sheet locally while the topside is functionalised with more classic fluid-based reactions96 (Fig. 6). In principle, solid–liquid interface reactions may be scaled up by performing the reactions at liquid–liquid interfaces, as seen for other non-graphene Janus materials.97 The interfacial route has been applied to pre-functionalised graphene98 and GO99 to synthesise Janus species, but this approach has yet to be applied to pristine graphene, even though graphene can be assembled at liquid–liquid interfaces.100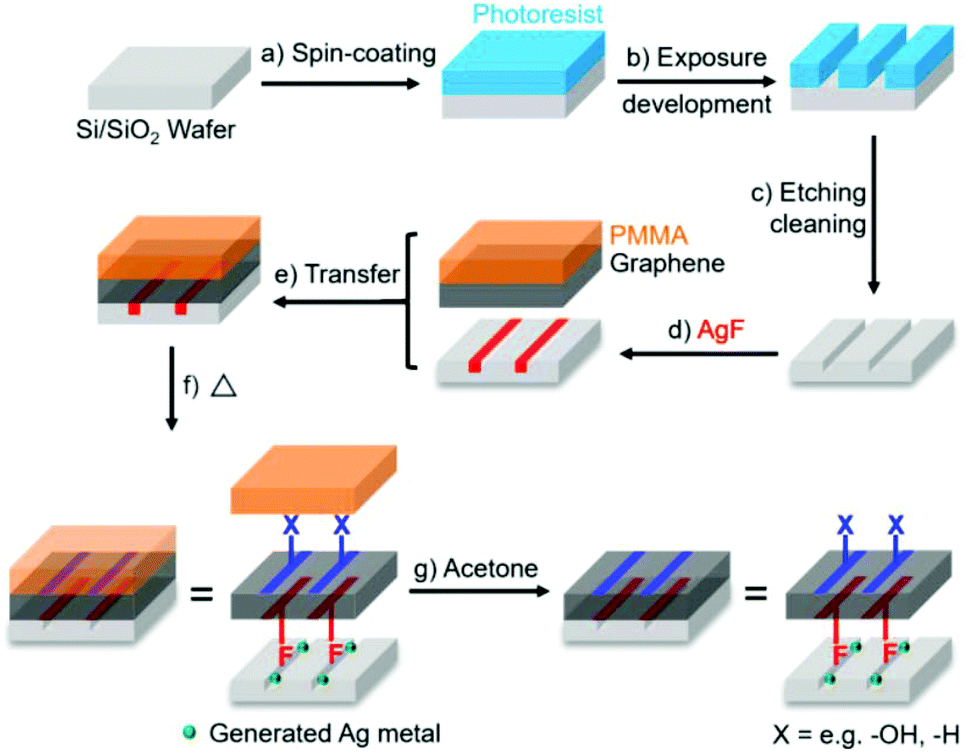 | ||
| Fig. 6 Process of Janus functionalisation of graphene using AgF embedded in substrate trenches for bottom-side fluorination. Reproduced with permission from Bao et al.96 Licenced under CC BY-NC-ND 4.0. | ||
Patterns in the substrate may also influence the chemistry on the upper surface through electronic effects,30 particularly for materials with large fluctuations in dipole.101 Lithographically altering the substrate allows manipulation of reactivity on the same scale as the lithographic patterning, typically tens of microns using optical methods. Lithographic patterning may also be performed on top of the graphene itself, through either patterning of a removable mask to prevent functionalisations at covered sites102,103 or deliberate introduction of defects to increase local reactivities of irradiated regions.82 Control using substrate patterning/masking down to the nanometre scale is likely possible with more advanced lithography; other direct write and self-assembly strategies are discussed below.104
Asymmetric lateral bands of functionalisation may be initiated by bipolar electrochemistry.105 When held in a static electric field, the electron density of a graphene sheet may be biased towards the edge closest to the cathode (complemented by an electron-poor edge near the anode). The cathodic edge is a better electron donor, accelerating functionalisation with aryl diazoniums. To balance charge, the graphene is reduced at the anodic side using a non-functionalising reaction. As functionalisation proceeds, the modified area is no longer conductive, and the polarised ‘edge’ of conjugated carbons moves inwards, with these graphitic carbons becoming more reactive. The result is a band of functionalisation which proceeds away from the graphene edge. Notably, this effect depends on the strength of the electric field and the distance from the graphene edges, not the distance from the field-generating electrode, allowing multiple graphene sheets in an array to be banded simultaneously (Fig. 7).
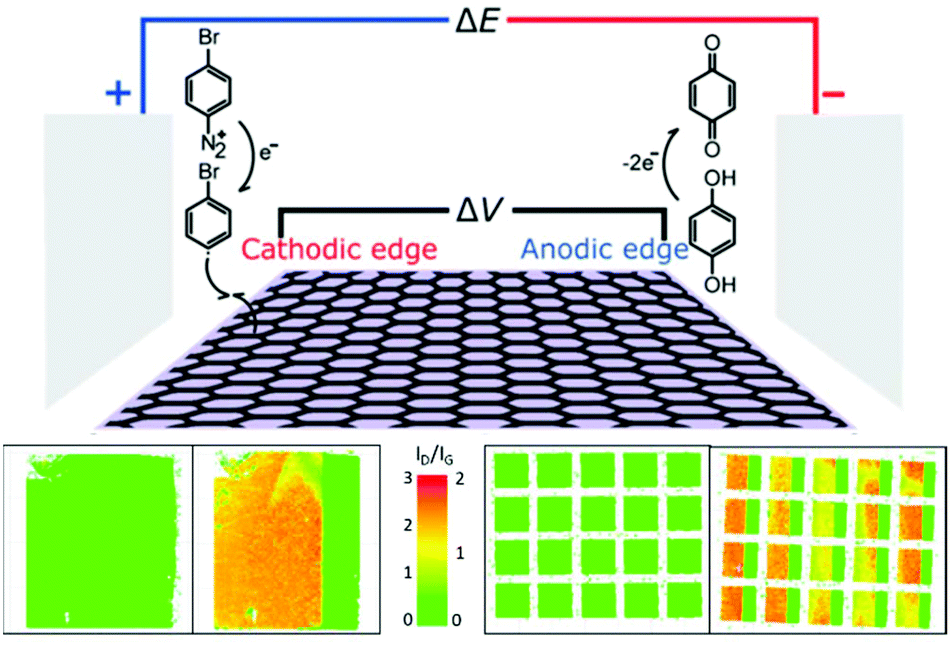 | ||
| Fig. 7 Progressive grafting of graphene using a bipolar electrochemical method. (Top) Schematic of setup. (Bottom) Raman D/G mode intensity ratio maps of graphene sheets before and after functionalisation for (left) single 10 mm2 sheet (right) 5 × 4 array of 1 mm2 graphene sheets. Adapted with permission from Koefoed et al.105 Copyright 2016 American Chemical Society. | ||
The functionalisation can be patterned at the nanoscale through probe microscopy. Scanning electrochemical cell microscopy allows for electrochemical reactions to be performed within sub-micron droplets on the graphene surface.106 At even higher resolution, the probe tip can drive local hydrogenation/oxidation by decomposing water with negative/positive tip bias, respectively.107 Higher oxidative voltages lead to local degradation of the graphene, facilitating cutting of the graphene for example into nanoribbons.108 Single-molecule control over functionalisation is also possible using the electron beam in a transmission electron microscope to decompose organochlorides to radicals which covalently adhere to the graphene layer.109 In principle, patterns of defects introduced by top-down methods, might be functionalised and expanded with sequential chemical reactions.
Fine nanoscale control may be achieved over larger areas of graphene through molecular self-assembly. Patterns of inert adsorbed molecules, such as linear alkanes, may be used as a mask to permit (radical) reactions only at the unmasked sites (Fig. 8a–b).110 Alternatively, the adsorbed species may be the reagent (typically a self-organising moiety terminated with diazonium),72 or even a side product from the reaction; as an example, surface-adhered bubbles of nitrogen from diazonium decomposition lead to circular patterns at the ∼100 nm scale (Fig. 8c–d).111
 | ||
| Fig. 8 AFM micrographs of graphene functionalised in the presence of self-assembled molecular masks. (a) Graphene functionalised after masking with C40H82, inset image Fourier transforms (b) schematic of masking and subsequently banded functionalisaiton. Reproduced from Tahara et al.110,58 Copyright 2018 American Chemical Society. (c and d) Graphene electrochemically functionalised with 1:1 TBD/4-nitrobenzenediazonium at different voltages (vs. Ag/AgCl) leading to formation of functionalisation-occluding nitrogen bubbles from diazonium decomposition. Higher magnification insets. Adapted with permission from Phan et al.111 Copyright 2019 American Chemical Society. | ||
Conclusions and perspective
Graphene functionalisation is essential for a range of practical processes and applications. However, as an archetypical nanomaterial, graphene is poised between molecular and continuum behaviour, and a range of fundamental chemistry questions remain to be addressed. The extent of charge delocalisation may be expected to vary as reactions proceed, and the π-bonded network becomes disrupted in ways that depend on the locus of functionalisation, the influence of substrates, and the presence of wrinkles and other defect features. The emergence of conjugated domains will effect properties (as already observed in the fluorescence of GO112) and subsequent reactivity. The nature of the radicals formed by SET and other radical additions to graphene remains to be clarified and is likely to vary with concentration and the remaining degree of conjugation; spin effects may also be generated by selective sub-lattice reactions or edge specific chemistries; in situ EPR experiments would be very insightful. Although SET mechanisms are widely invoked, complex formation50 and coordinated reaction paths may prove to have a larger role. Whilst many chemistries are considered to be irreversible, at least under the reaction conditions applied, the effects of backward reactions, and the stability of intermediates, deserve more attention. Currently, the evidence for propagation of functionalisation reactions from defects is limited, circumstantial, or indirect (i.e. based on graphene-like molecules or poorly exfoliated materials, rather the single layer graphene). That steric effects are so often dominant (Fig. 2b) suggests that any propagation effects tend not to limit the extent of reactions. However, further study of the development of functionalisation reactions at the atomic scale, on well-defined systems, is needed.Despite these fundamental issues, there are now a wide range of established practical chemistries for grafting useful moieties to single layer graphenes and other graphene-related materials. Their efficacy may be as much a function of graphene quality and type as the reaction conditions. The emphasis has largely been to show that a particular approach is ‘successful’, preferably with a high degree of functionalisation. Further work is needed to control the degree of functionalisation more appropriately for a range of different starting materials, particularly on more exfoliated/fewer layer feedstocks. The extent of functionalisation should be routinely reported relative to the degree of exfoliation. In general, there is an optimum extent of functionalisation balancing the benefits of the grafted groups with the loss of intrinsic properties; the use of macromolecular or branched addends can offer greater benefits for fewer disrupted graphene lattice sites. For systems with lower degrees of functionalisation (e.g. graphenide/DA), increasing degree of functionalisation may still be needed. More aggressive plasma/oxidation chemistry may require a lower degree of more controlled addition. In either case, whilst absolute grafting stoichiometry is important, the locus of functionalisation will alter the resulting graphene properties, influencing both final application and characterisation of the material. Further improvements in methods to characterise or observe the locus of functionalisation are needed.
Pure edge functionalisation whilst retaining undamaged basal plane properties, is attractive if sufficient. Most obviously, this approach applies to graphene-based ‘carbon-dots’ and graphene nanoribbons, which have optoelectronic properties modulated by the edge effects. For larger flakes, progressive functionalisation radially inwards allows a higher grafting stoichiometry without loss of core performance. However, avoiding parasitic islands forming in the core region will require a low concentration of initial basal plane defects. Still larger flakes, even wafer scale monolayers, could be compartmentalised into parallel ribbons or other confined architectures with patterned functionalisation, using lithography or leveraging more bottom-up processes such as wrinkle engineering, to access shorter length-scales across large areas. Further, full control over the locus of functionalisation will not only improve existing processing and tune properties, but open new avenues of research. For example, full functionalisation of a single sublattice is expected to generate magnetic carbon materials.113 Regular arrays of functional group sites will modulate optoelectronic properties, potentially introducing band gaps and useful plasmonic114 and magnetic115,116 features.
To realise these opportunities will require a significant expansion of our understanding of graphene chemistry, and the methods to control and characterise it. The knowledge acquired will extend beyond graphene, since many of the underpinning chemistries, challenges and opportunities are shared with other 2D nanomaterials including carbon nitrides, transition metal dichalcogenides, and other 2D allotropes (phosphorene, silicene, germanene, stanene). Many of these nanomaterials also have 1D embodiments, either as nanotubes6 or nanoribbons of the 2D species.117
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AJC would like to thank The Society of Chemical Industry (SCI) and the Ramsay Memorial Trust for funding. AJC, MSPS, and NR were supported by the EU Graphene Flagship under Horizon 2020 Research and Innovation programme grant agreement no. 785219 – GrapheneCore2. HA would like to acknowledge the Faraday Institution's LiSTAR project (EP/S003053/1, Grant FIRG014) for funding. GOC would like to thank EPSRC for funding through the Centre for Doctoral Training in Plastic Electronic Materials (EP/L016702/1) and Thomas Swan & Co. Ltd.References
- A. Zandiatashbar, G.-H. Lee, S. J. An, S. Lee, N. Mathew, M. Terrones, T. Hayashi, C. R. Picu, J. Hone and N. Koratkar, Nat. Commun., 2014, 5, 1–9 CrossRef PubMed.
- A. Shekhawat and R. O. Ritchie, Nat. Commun., 2016, 7, 10546 CrossRef CAS PubMed.
- P. Zhang, L. Ma, F. Fan, Z. Zeng, C. Peng, P. E. Loya, Z. Liu, Y. Gong, J. Zhang and X. Zhang, Nat. Commun., 2014, 5, 3782 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- S. P. Economopoulos, G. Rotas, Y. Miyata, H. Shinohara and N. Tagmatarchis, ACS Nano, 2010, 4, 7499–7507 CrossRef CAS PubMed.
- A. J. Clancy, M. K. Bayazit, S. A. Hodge, N. T. Skipper, C. A. Howard and M. S. Shaffer, Chem. Rev., 2018, 118, 7363–7408 CrossRef CAS PubMed.
- C. E. Hamilton, J. R. Lomeda, Z. Sun, J. M. Tour and A. R. Barron, Nano Res., 2010, 3, 138–145 CrossRef CAS.
- X. Zhang, M. Han, S. Chen, L. Bao, L. Li and W. Xu, RSC Adv., 2013, 3, 17689–17692 RSC.
- K. S. Novoselov, V. Fal, L. Colombo, P. Gellert, M. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- A. J. Clancy, D. B. Anthony and F. De Luca, ACS Appl. Mater. Interfaces, 2020, 12, 15955–15975 CrossRef CAS PubMed.
- T. Sainsbury, M. Passarelli, M. Naftaly, S. Gnaniah, S. J. Spencer and A. J. Pollard, ACS Appl. Mater. Interfaces, 2016, 8, 4870–4877 CrossRef CAS PubMed.
- J. Choi, K.-J. Kim, B. Kim, H. Lee and S. Kim, J. Phys. Chem. C, 2009, 113, 9433–9435 CrossRef CAS.
- M. Cai, D. Thorpe, D. H. Adamson and H. C. Schniepp, J. Mater. Chem. A, 2012, 22, 24992–25002 RSC.
- T. A. Strom, E. P. Dillon, C. E. Hamilton and A. R. Barron, Chem. Commun., 2010, 46, 4097–4099 RSC.
- A. Zandiatashbar, G.-H. Lee, S. J. An, S. Lee, N. Mathew, M. Terrones, T. Hayashi, C. R. Picu, J. Hone and N. Koratkar, Nat. Commun., 2014, 5, 3186 CrossRef PubMed.
- Y. Zhu, A. L. Higginbotham and J. M. Tour, Chem. Mater., 2009, 21, 5284–5291 CrossRef CAS.
- B. D. Ossonon and D. Bélanger, Carbon, 2017, 111, 83–93 CrossRef CAS.
- A. J. Marsden, P. Brommer, J. J. Mudd, M. A. Dyson, R. Cook, M. Asensio, J. Avila, A. Levy, J. Sloan and D. Quigley, Nano Res., 2015, 8, 2620–2635 CrossRef CAS.
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- J. Plšek, P. Kovaříček, V. Valeš and M. Kalbáč, Chem. – Eur. J., 2017, 23, 1839–1845 CrossRef PubMed.
- K. C. Knirsch, J. M. Englert, C. Dotzer, F. Hauke and A. Hirsch, Chem. Commun., 2013, 49, 10811–10813 RSC.
- J. M. Englert, P. Vecera, K. C. Knirsch, R. A. Schäfer, F. Hauke and A. Hirsch, ACS Nano, 2013, 7, 5472–5482 CrossRef CAS PubMed.
- G. Bottari, M. Á. Herranz, L. Wibmer, M. Volland, L. Rodríguez-Pérez, D. M. Guldi, A. Hirsch, N. Martín, F. D'Souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464–4500 RSC.
- K. C. Knirsch, F. Hof, V. Lloret, U. Mundloch, F. Hauke and A. Hirsch, J. Am. Chem. Soc., 2016, 138, 15642–15647 CrossRef CAS PubMed.
- N. Rubio, H. Au, H. S. Leese, S. Hu, A. J. Clancy and M. S. Shaffer, Macromolecules, 2017, 50, 7070–7079 CrossRef CAS.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinrück, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279 CrossRef CAS PubMed.
- S. Park, D. Srivastava and K. Cho, Nano Lett., 2003, 3, 1273–1277 CrossRef CAS.
- D. Dasler, R. A. Schäfer, M. B. Minameyer, J. F. Hitzenberger, F. Hauke, T. Drewello and A. Hirsch, J. Am. Chem. Soc., 2017, 139, 11760–11765 CrossRef CAS PubMed.
- F. Hof, R. A. Schäfer, C. Weiss, F. Hauke and A. Hirsch, Chem. – Eur. J., 2014, 20, 16644–16651 CrossRef CAS PubMed.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398–405 CrossRef CAS PubMed.
- F. M. Koehler, A. Jacobsen, K. Ensslin, C. Stampfer and W. J. Stark, Small, 2010, 6, 1125–1130 CrossRef CAS PubMed.
- H. Au, N. Rubio and M. S. Shaffer, Chem. Sci., 2018, 9, 209–217 RSC.
- R. A. Schäfer, D. Dasler, U. Mundloch, F. Hauke and A. Hirsch, J. Am. Chem. Soc., 2016, 138, 1647–1652 CrossRef PubMed.
- S. Hu, Z. P. Laker, H. S. Leese, N. Rubio, M. De Marco, H. Au, M. S. Skilbeck, N. R. Wilson and M. S. Shaffer, Chem. Sci., 2017, 8, 6149–6154 RSC.
- H. S. Leese, L. Govada, E. Saridakis, S. Khurshid, R. Menzel, T. Morishita, A. J. Clancy, E. R. White, N. E. Chayen and M. S. Shaffer, Chem. Sci., 2016, 7, 2916–2923 RSC.
- A. Kasprzak, A. Zuchowska and M. Poplawska, Beilstein J. Org. Chem., 2018, 14, 2018–2026 CrossRef CAS PubMed.
- V. V. Ivanovskaya, P. Wagner, A. Zobelli, I. Suarez-Martinez, A. Yaya and C. P. Ewels, in GraphITA, 2011, Springer, 2012, pp. 75–85 Search PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS PubMed.
- M. Rosillo-Lopez, T. J. Lee, M. Bella, M. Hart and C. G. Salzmann, RSC Adv., 2015, 5, 104198–104202 RSC.
- B. Li, L. Zhou, D. Wu, H. Peng, K. Yan, Y. Zhou and Z. Liu, ACS Nano, 2011, 5, 5957–5961 CrossRef CAS PubMed.
- W. Lai, X. Wang, J. Fu, T. Chen, K. Fan and X. Liu, Carbon, 2018, 137, 451–457 CrossRef CAS.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- S. Eigler, Chem. Commun., 2015, 51, 3162–3165 RSC.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS PubMed.
- G. Neri, A. Scala, E. Fazio, P. G. Mineo, A. Rescifina, A. Piperno and G. Grassi, Chem. Sci., 2015, 6, 6961–6970 RSC.
- V. Barbera, L. Brambilla, A. Milani, A. Palazzolo, C. Castiglioni, A. Vitale, R. Bongiovanni and M. Galimberti, Nanomaterials, 2019, 9, 44 CrossRef PubMed.
- S. Sarkar, E. Bekyarova, S. Niyogi and R. C. Haddon, J. Am. Chem. Soc., 2011, 133, 3324–3327 CrossRef CAS PubMed.
- A. J. Clancy, J. M. Serginson, J. L. Greenfield and M. S. Shaffer, Polymer, 2017, 133, 263–271 CrossRef CAS.
- A. J. Clancy, P. Sirisinudomkit, D. B. Anthony, A. Z. Thong, J. L. Greenfield, M. K. S. Singh and M. S. Shaffer, Chem. Sci., 2019, 10, 3300–3306 RSC.
- Z. Qiu, J. Yu, P. Yan, Z. Wang, Q. Wan and N. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 28291–28298 CrossRef CAS PubMed.
- H. Lee, K. Paeng and I. S. Kim, Synth. Met., 2018, 244, 36–47 CrossRef CAS.
- T. Susi, T. P. Hardcastle, H. Hofsäss, A. Mittelberger, T. J. Pennycook, C. Mangler, R. Drummond-Brydson, A. J. Scott, J. C. Meyer and J. Kotakoski, 2D Mater., 2017, 4, 021013 CrossRef.
- M. Tripathi, A. Markevich, R. Böttger, S. Facsko, E. Besley, J. Kotakoski and T. Susi, ACS Nano, 2018, 12, 4641–4647 CrossRef CAS PubMed.
- M. Tripathi, A. Mittelberger, N. A. Pike, C. Mangler, J. C. Meyer, M. J. Verstraete, J. Kotakoski and T. Susi, Nano Lett., 2018, 18, 5319–5323 CrossRef CAS PubMed.
- X. Zhong, J. Jin, S. Li, Z. Niu, W. Hu, R. Li and J. Ma, Chem. Commun., 2010, 46, 7340–7342 RSC.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- H. He and C. Gao, Chem. Mater., 2010, 22, 5054–5064 CrossRef CAS.
- K.-I. Ho, C.-H. Huang, J.-H. Liao, W. Zhang, L.-J. Li, C.-S. Lai and C.-Y. Su, Sci. Rep., 2014, 4, 5893 CrossRef CAS PubMed.
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson and P. E. Sheehan, Nano Lett., 2010, 10, 3001–3005 CrossRef CAS PubMed.
- J. Zheng, H.-T. Liu, B. Wu, C.-A. Di, Y.-L. Guo, T. Wu, G. Yu, Y.-Q. Liu and D.-B. Zhu, Sci. Rep., 2012, 2, 1–6 Search PubMed.
- K. Gopalakrishnan, K. Subrahmanyam, P. Kumar, A. Govindaraj and C. Rao, RSC Adv., 2012, 2, 1605–1608 RSC.
- M. Pumera and C. H. A. Wong, Chem. Soc. Rev., 2013, 42, 5987–5995 RSC.
- V. Georgakilas, A. B. Bourlinos, R. Zboril, T. A. Steriotis, P. Dallas, A. K. Stubos and C. Trapalis, Chem. Commun., 2010, 46, 1766–1768 RSC.
- X. Zhang, L. Hou, A. Cnossen, A. C. Coleman, O. Ivashenko, P. Rudolf, B. J. van Wees, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2011, 17, 8957–8964 CrossRef CAS PubMed.
- L. Yang and J. He, Chem. Commun., 2014, 50, 15722–15725 RSC.
- T. Morishita, A. J. Clancy and M. S. Shaffer, J. Mater. Chem. A, 2014, 2, 15022–15028 RSC.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- A. J. Clancy, H. S. Leese, N. Rubio, D. J. Buckley, J. L. Greenfield and M. S. Shaffer, Langmuir, 2018, 34, 15396–15402 CrossRef CAS PubMed.
- P. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- N. Rubio, H. Au, D. J. Buckley, C. Mattevi and M. Shaffer, Chem. – Eur. J., 2020, 26, 6545–6553 CrossRef PubMed.
- A. Kaplan, Z. Yuan, J. D. Benck, A. G. Rajan, X. S. Chu, Q. H. Wang and M. S. Strano, Chem. Soc. Rev., 2017, 46, 4530–4571 RSC.
- H. Touhara and F. Okino, Carbon, 2000, 38, 241–267 CrossRef CAS.
- S.-H. Cheng, K. Zou, F. Okino, H. R. Gutierrez, A. Gupta, N. Shen, P. Eklund, J. O. Sofo and J. Zhu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 205435 CrossRef.
- S. A. Hodge, D. J. Buckley, H. C. Yau, N. T. Skipper, C. A. Howard and M. S. Shaffer, Nanoscale, 2017, 9, 3150–3158 RSC.
- G. Bepete, A. Pénicaud, C. Drummond and E. Anglaret, J. Phys. Chem. C, 2016, 120, 28204–28214 CrossRef CAS.
- L. Daukiya, C. Mattioli, D. Aubel, S. Hajjar-Garreau, F. Vonau, E. Denys, G. N. Reiter, J. Fransson, E. Perrin and M.-L. Bocquet, ACS Nano, 2017, 11, 627–634 CrossRef CAS PubMed.
- R. I. Gearba, M. Kim, K. M. Mueller, P. A. Veneman, K. Lee, B. J. Holliday, C. K. Chan, J. R. Chelikowsky, E. Tutuc and K. J. Stevenson, Adv. Mater. Interfaces, 2016, 3, 1600196 CrossRef.
- L. Xie, L. Jiao and H. Dai, J. Am. Chem. Soc., 2010, 132, 14751–14753 CrossRef CAS PubMed.
- S. Deng and V. Berry, Mater. Today, 2016, 19, 197–212 CrossRef CAS.
- Y. Zhang, Q. Fu, Y. Cui, R. Mu, L. Jin and X. Bao, Phys. Chem. Chem. Phys., 2013, 15, 19042–19048 RSC.
- H. Li, L. Daukiya, S. Haldar, A. Lindblad, B. Sanyal, O. Eriksson, D. Aubel, S. Hajjar-Garreau, L. Simon and K. Leifer, Sci. Rep., 2016, 6, 1–7 CrossRef PubMed.
- G. Yang, L. Li, W. B. Lee and M. C. Ng, Sci. Technol. Adv. Mater., 2018, 19, 613–648 CrossRef CAS PubMed.
- S. Deng, Y. Zhang, A. H. Brozena, M. L. Mayes, P. Banerjee, W.-A. Chiou, G. W. Rubloff, G. C. Schatz and Y. Wang, Nat. Commun., 2011, 2, 382 CrossRef PubMed.
- L. Feng, Y.-W. Liu, X.-Y. Tang, Y. Piao, S.-F. Chen, S.-L. Deng, S.-Y. Xie, Y. Wang and L.-S. Zheng, Chem. Mater., 2013, 25, 4487–4496 CrossRef CAS.
- J. Holzwarth, K. Y. Amsharov, D. I. Sharapa, D. Reger, K. Roshchyna, D. Lungerich, N. Jux, F. Hauke, T. Clark and A. Hirsch, Angew. Chem., 2017, 129, 12352–12358 CrossRef.
- K. C. Knirsch, R. A. Schäfer, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2016, 55, 5861–5864 CrossRef CAS PubMed.
- D. Boukhvalov, Nanotechnology, 2010, 22, 055708 CrossRef PubMed.
- Y. Fujimoto and S. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245446 CrossRef.
- P. A. Denis and F. Iribarne, J. Phys. Chem. C, 2013, 117, 19048–19055 CrossRef CAS.
- M. Barrejón, A. Primo, M. J. Gómez-Escalonilla, J. L. G. Fierro, H. García and F. Langa, Chem. Commun., 2015, 51, 16916–16919 RSC.
- L. Ferrighi, M. Datteo and C. Di Valentin, J. Phys. Chem. C, 2014, 118, 223–230 CrossRef CAS.
- X. Ma, Q. Wang, L.-Q. Chen, W. Cermignani, H. Schobert and C. G. Pantano, Carbon, 1997, 35, 1517–1525 CrossRef CAS.
- M. A. Bissett, Y. Takesaki, M. Tsuji and H. Ago, RSC Adv., 2014, 4, 52215–52219 RSC.
- L. Zhang, J. Yu, M. Yang, Q. Xie, H. Peng and Z. Liu, Nat. Commun., 2013, 4, 1443 CrossRef PubMed.
- L. Bao, B. Zhao, V. Lloret, M. Halik, F. Hauke and A. Hirsch, Angew. Chem., 2020, 59, 6700–6705 CrossRef CAS PubMed.
- J. Hu, S. Zhou, Y. Sun, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 4356–4378 RSC.
- I. Jeon, M. D. Peeks, S. Savagatrup, L. Zeininger, S. Chang, G. Thomas, W. Wang and T. M. Swager, Adv. Mater., 2019, 31, 1900438 CrossRef PubMed.
- H. Wu, W. Yi, Z. Chen, H. Wang and Q. Du, Carbon, 2015, 93, 473–483 CrossRef CAS.
- J. Shim, J. M. Yun, T. Yun, P. Kim, K. E. Lee, W. J. Lee, R. Ryoo, D. J. Pine, G.-R. Yi and S. O. Kim, Nano Lett., 2014, 14, 1388–1393 CrossRef CAS PubMed.
- G. L. Paulus, Q. H. Wang and M. S. Strano, Acc. Chem. Res., 2013, 46, 160–170 CrossRef CAS PubMed.
- F. M. Koehler, N. A. Luechinger, D. Ziegler, E. K. Athanassiou, R. N. Grass, A. Rossi, C. Hierold, A. Stemmer and W. J. Stark, Angew. Chem., Int. Ed., 2009, 48, 224–227 CrossRef CAS PubMed.
- T. Wei, M. Kohring, M. Chen, S. Yang, H. B. Weber, F. Hauke and A. Hirsch, Angew. Chem., 2020, 59, 5602–5606 CrossRef CAS PubMed.
- Q. H. Wang, Z. Jin, K. K. Kim, A. J. Hilmer, G. L. Paulus, C.-J. Shih, M.-H. Ham, J. D. Sanchez-Yamagishi, K. Watanabe and T. Taniguchi, Nat. Chem., 2012, 4, 724 CrossRef CAS PubMed.
- L. Koefoed, E. B. Pedersen, L. Thyssen, J. Vinther, T. Kristiansen, S. U. Pedersen and K. Daasbjerg, Langmuir, 2016, 32, 6289–6296 CrossRef CAS PubMed.
- G. Zhang, P. M. Kirkman, A. N. Patel, A. S. Cuharuc, K. McKelvey and P. R. Unwin, J. Am. Chem. Soc., 2014, 136, 11444–11451 CrossRef CAS PubMed.
- I.-S. Byun, D. Yoon, J. S. Choi, I. Hwang, D. H. Lee, M. J. Lee, T. Kawai, Y.-W. Son, Q. Jia and H. Cheong, ACS Nano, 2011, 5, 6417–6424 CrossRef CAS PubMed.
- L. Tapasztó, G. Dobrik, P. Lambin and L. P. Biro, Nat. Nanotechnol., 2008, 3, 397–401 CrossRef PubMed.
- A. Markevich, S. Kurasch, O. Lehtinen, O. Reimer, X. Feng, K. Müllen, A. Turchanin, A. N. Khlobystov, U. Kaiser and E. Besley, Nanoscale, 2016, 8, 2711–2719 RSC.
- K. Tahara, T. Ishikawa, B. E. Hirsch, Y. Kubo, A. Brown, S. Eyley, L. Daukiya, W. Thielemans, Z. Li and P. Walke, ACS Nano, 2018, 12, 11520–11528 CrossRef CAS PubMed.
- T. H. Phan, H. Van Gorp, Z. Li, T. M. Trung Huynh, Y. Fujita, L. Verstraete, S. Eyley, W. Thielemans, H. Uji-i and B. E. Hirsch, ACS Nano, 2019, 13, 5559–5571 CrossRef CAS PubMed.
- H. R. Thomas, C. Vallés, R. J. Young, I. A. Kinloch, N. R. Wilson and J. P. Rourke, J. Mater. Chem. C, 2013, 1, 338–342 RSC.
- K. E. Whitener Jr., J. Vac. Sci. Technol., A, 2018, 36, 05G401 CrossRef.
- M. Dragoman and D. Dragoman, Prog. Quantum Electron., 2009, 33, 165–214 CrossRef CAS.
- N. García-Martínez, J. L. Lado, D. Jacob and J. Fernández-Rossier, Phys. Rev. B, 2017, 96, 024403 CrossRef.
- O. V. Yazyev and L. Helm, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125408 CrossRef.
- M. C. Watts, L. Picco, F. S. Russell-Pavier, P. L. Cullen, T. S. Miller, S. P. Bartuś, O. D. Payton, N. T. Skipper, V. Tileli and C. A. Howard, Nature, 2019, 568, 216–220 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0dt01589j |
| This journal is © The Royal Society of Chemistry 2020 |