
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrelations of acidity-basicity of solvent treated layered double hydroxides/oxides and their CO2 capture performance†
D. W. Justin
Leung
,
Chunping
Chen
,
Jean-Charles
Buffet
 and
Dermot
O'Hare
*
and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: dermot.ohare@chem.ox.ac.uk; Tel: +44 (0)1865 27268
First published on 18th June 2020
Abstract
The basicity and acidity of solvent-treated layered double hydroxide (ST-LDHs) and their layered double oxides (ST-LDOs) have been fully studied using Hammett titration, in situ FTIR, CO2-TPD and NH3-TPD. Five solvents (ethanol, acetone, isopropanol, ethyl acetate and 1-hexanol) were selected to treat [Mg0.72Al0.28(OH)2](CO3)0.14 (Mg2.5Al-CO3 LDH) and compared with traditional LDH co-precipitated from water. The Brønsted basicity strength of the ST-LDHs and ST-LDOs increased but was accompanied by a decrease in basic site density. In addition, the Lewis acidity of ST-LDOs also changes significantly, with medium strength Lewis acid sites dissapearing after solvent treatment. We found that the CO2 capture capacity of solvent treated LDOs is 50% higher than that of traditional co-precipitated LDO sample. The ethanol treated LDO exhibited the highest CO2 uptake of 1.01 mmol g−1. The observed CO2 capture performance of the ST-LDOs correlates linearly with the ratio of total acid sites to total basic sites.
Introduction
Layered double hydroxides (LDHs), also known as anionic clays, are a family of inorganic layered materials with a general composition [(M1−xz+M′xy+(OH)2)]w+(An−)w/n·mH2O, where Mz+ and M′y+ are one or more different metal cations, z can be 1 or 2 and y can be 3 or 4; overall the metal hydroxide layer charge (w) is determined by w = z(1 − x) + xy − 2 which is compensated by w/n An− interlayer anions.1 Most commonly, LDHs contain both M2+ and M′3+ cations, and x has been found to give crystalline phase pure LDHs when 0.35 ≥ x ≥ 0.16.2 Water molecules may also be incorporated into the interlayer region during synthesis, which form hydrogen bonds to the hydroxyl groups on the surface of the cationic layers as well as solvating the hydrophilic interlayer anions. This stabilises the layered structure while still maintaining anion mobility.3 Upon thermal treatment at low temperatures (100–200 °C), LDHs lose both surface-bound and interlamellar water. At 300–600 °C, LDOs are formed by de-hydroxylation and anion-decomposition.4–7 At higher temperatures, LDHs irreversibly decompose to form crystalline spinel phases that cannot be reconstructed back.8Due to their inherent basicity, both LDHs and LDOs have been widely used as heterogeneous catalysts.9–11 Efforts have been made to increase their basicity in order to amplify their catalytic behaviour by doping with various cations into the layer structure or by surface impregnation.12,13 They have also been attracting increasing interest for CO2 capture, many studies have been published on the effects of calcination temperatures, cation doping and intercalated anion on CO2 capture performance.14–19 However, due to the hydrophilic nature of LDHs, the primary platelet particles are usually highly aggregated by a strong hydrogen bonding network, resulting in low surface areas and formation of large platelet agglomerates, which can severely limit their use in catalysis and CO2 capture.20 O'Hare and co-workers have developed post synthesis solvent treatment (ST) methods to create highly dispersed LDHs (ST-LDHs). Initially, an aqueous miscible organic solvent treatment method (AMOST) was reported;21 more recently, an aqueous immiscible organic (AIM) solvent treatment has proved to be equally effective.22 It has been proposed that the solvent penetrates the interlayer region to varying extents, disrupting the hydrogen bonding network and displacing any surface bound water. This weakens the interlayer interactions, allowing the cationic layers to be delaminated, resulting in a highly dispersed platelets with high surface area and large pore volume.22,23 These newly discovered LDHs families have been found to be highly effective solid supports for single-site catalysts in slurry phase ethylene polymerisation.24,25 Very recent studies have been carried out on the impact of AMO-LDHs (using acetone as AMO solvent) on CO2 capture performance.26,27 However, there has been no systematic study of the changes in surface chemistry of LDHs following solvent treatment and in particular how this may relate to their use as CO2 adsorbents. Understanding the changes in surface chemistry of solvent treated LDHs will allow for optimisation of their catalytic and absorbent abilities.
Herein, we have investigated the acid and base properties of solvent treated LDHs (AMO- and AIM-LDHs) and their respective LDOs. The Brønsted basicity of both solvent treated LDHs and LDOs were evaluated using Hammett indicators in solution. The Lewis basicity and acidity of solvent treated LDOs were analysed using in situ FTIR and temperature programmed desorption (TPD). The CO2 capture performance of the ST-LDHs was evaluated using chemisorption experiments.
Results and discussion
Basicity studies of ST-LDHs
A conventional [Mg0.72Al0.28(OH)2](CO3)0.14 (Mg2.5Al-CO3) LDH (LDH-W) was synthesised using an adapted literature co-precipitation procedure in water.21 Solvent treated LDH samples (ST-LDHs) were prepared by re-dispersing a 30% solid content aqueous dispersion of Mg2.5Al CO3 LDH wet cake in the appropriate dispersing solvent for 4 h. The dispersing solvents used in this study were ethanol, acetone, isopropyl alcohol, ethyl acetate and 1-hexanol to yield LDH-E, LDH-A, LDH-IPA, LDH-EA and LDH-1H, respectively. The chemical composition of all the LDHs are provided in Table S1.†The powder X-ray diffraction (XRD) data for all the samples (Fig. S1†) are typical diffractions of an Mg2.5Al-CO3 LDH. As previously reported, we observe small variations in the lattice constants (Table S2†) and the peak widths of the Bragg reflections for ST-LDHs compared to the conventional sample (LDH-W).21,22 There is a large increase in the available surface area and pore volume of the ST-LDHs compared to LDH-W (Table S3†). LDH-E showed the highest surface area of 196 m2 g−1 while LDH-EA had the highest pore volume of 0.83 cm3 g−1.
Variation of Brønsted basicity of these materials was studied using Hammett indicators, three indicators were used: bromothymol blue (pKa = 7.1), phenolphthalein (pKa = 9.3) and clayton yellow (pKa = 12.7). For the purpose of this study, Brønsted basic sites pH > 12.7, 9.3 ≤ pH ≤ 12.7 and 7.1 ≤ pH ≤ 9.3 are assigned to the label ‘strong’, ‘medium’ and ‘weak’, respectively. All samples showed similar responses of having fewer medium Brønsted basic sites compared to the strong and weak ones (Fig. 1). LDH-IPA exhibited the highest number of medium basic sites (0.15 mmol g−1), double that of any other ST-LDHs. Analysis of the total number of Brønsted basic sites (Table 1), revealed that all ST-LDHs exhibited a higher total number compared to LDH-W (0.41 mmol g−1). LDH-IPA and LDH-1H showed the highest total basicity at 0.67 mmol g−1. However, once the surface area was taken into account, the AMO-LDHs showed lower basic site densities compared to LDH-W (4.52 μmol m−2). LDH-1H had the highest Brønsted basic site density of all ST-LDHs at 4.04 μmol m−2.
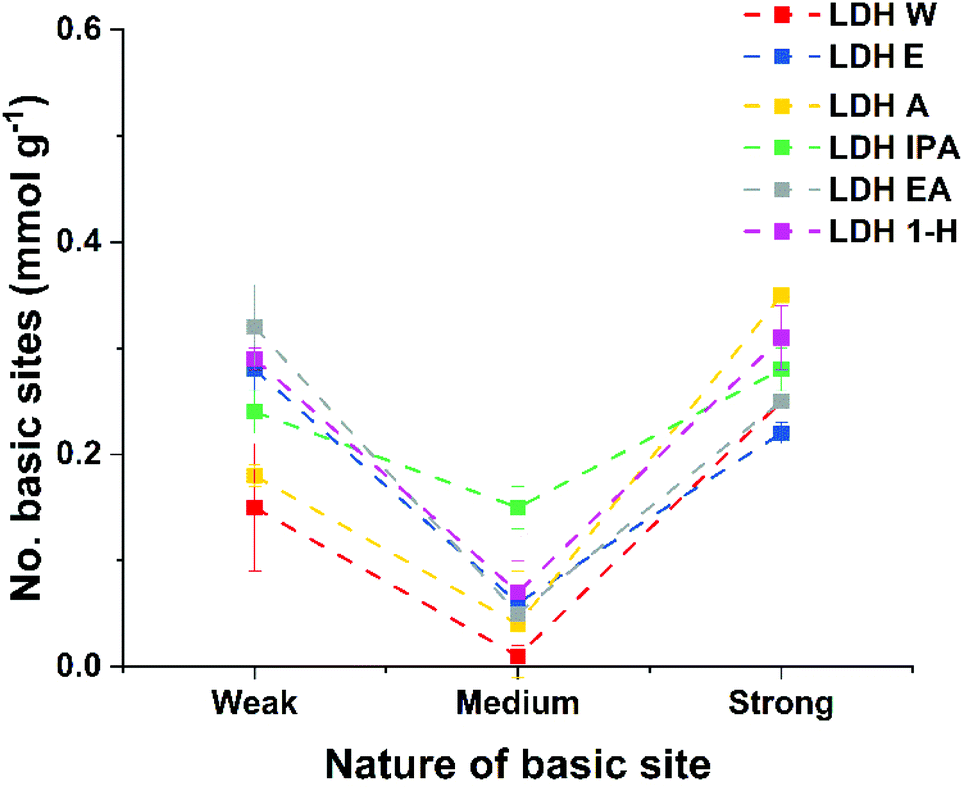 | ||
| Fig. 1 Plot of the total number of basic sites for ST-LDHs (dotted lines are just a guide to the eye). | ||
| Sample | Total basic sites (mmol g−1) | Basic site density (μmol m−2) |
|---|---|---|
| LDH-W | 0.41 ± 0.06 | 4.52 ± 0.65 |
| LDH-E | 0.57 ± 0.02 | 2.92 ± 0.10 |
| LDH-A | 0.57 ± 0.01 | 3.29 ± 0.06 |
| LDH-IPA | 0.67 ± 0.02 | 3.62 ± 0.01 |
| LDH-EA | 0.61 ± 0.04 | 3.48 ± 0.23 |
| LDH-1H | 0.67 ± 0.01 | 4.04 ± 0.06 |
The solvent treatment method increases LDH surface area without changing the structural arrangement of the cation within the layers. Although solvent treatment increases the available specific surface area, a priori, it would not be expected to change the basic site density of all samples. Especially as the previous reports suggest that the pre-treatment temperature of 200 °C only removes solvents leaving the underlying LDH structure intact.21,28,29 The decrease in the total basic site density for some solvent treated samples may be due to these solvents blocking basic sites. This site blocking could be a result of trace amounts of solvent absorbed on the surface. Perhaps treatment temperature of 200 °C not sufficiently high to desorb all surface sorbed solvents. It has been shown previously that hydroxyl-containing molecules may bind to the surface of an LDH, resulting in the surface hydroxyl groups not being accessible.30
In situ Fourier-transform infra-red (FTIR) spectroscopy was used to probe the LDHs’ Lewis basicity. As shown in Fig. S2,†LDH-W exhibits a strong absorbance between 1360–1400 cm−1 and a less intense absorbance between 1470–1580 cm−1, which are associated with strong Lewis basic sites due to monodentate carbonate surface coordination. A weak bidentate carbonate stretching vibration (1580–1670 cm−1) can be assigned to a medium strength basic site in LDH-W. These findings are consistent with previously reported literature results.31,32 Solvent treated LDH samples exhibit varying levels of both strong and medium strength Lewis basic sites. Solvent treatment appears to alter the overall Lewis basicity profile of ST-LDHs. In particular, both LDH-E and LDH-1H exhibited enhanced numbers of medium strength basic sites, while the medium basic sites are eliminated by treatment with acetone and isopropanol.
Basicity and acidity studies of ST-LDOs
Solvent treated LDOs (ST-LDOs) were prepared by calcining the appropriate ST-LDH at 450 °C for 12 h with a heating ramp rate of 5 °C min−1. The LDO products were named LDO-W, LDO-E, LDO-A, LDO-IPA, LDO-EA and LDO-1H by calcination of the appropriate ST-LDH respectively. As shown in Fig. 2, the general trend for ST-LDO samples (except LDO-EA) is that the number and strength of the Brønsted sites increases compared to the parent LDH. This increase in the number of strong Brønsted basic sites is due to the decomposition of hydroxide groups to unsaturated oxygen atoms and metal–oxygen pairs on the surface of the LDH nanosheets, which have higher basicity. Interestingly, both LDO-E and LDO-IPA exhibit a negligibly low number of weak basic sites and LDO-EA did not have any medium sites. LDO-1H showed the highest number of total Brønsted basic sites at 1.58 mmol g−1. In general, both the total number of basic sites and the basic site density of LDOs increases from that of their parent LDH. This is due to an increase in surface area as the LDH nanosheets start decomposing by dehydroxylation, decomposition of intercalated carbonate, and loss of interlayer water. Furthermore, some aluminium ions migrate from octahedral into tetrahedral sites33 (leading to exposure of more basic sites). Solvent treated LDOs (except LDO-1H) displayed a lower basic site density compared to LDO-W (4.83 μmol m−2), while LDO-1H has the highest total number of basic sites and highest basic site density at 1.58 and 5.79 μmol m−2 respectively (Table 2). Both CO2 and NH3 temperature programmed desorption (TPD) have been used to probe the Lewis basicity and acidity for the LDO samples.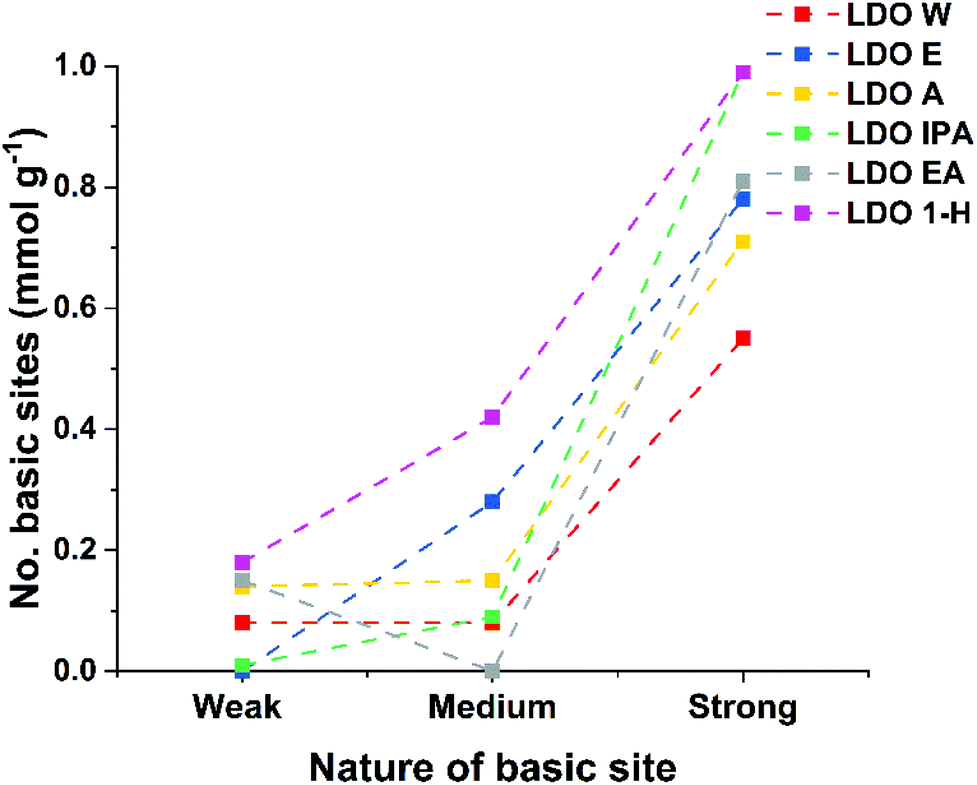 | ||
| Fig. 2 Titration plots of all basic sites of various ST-LDOs (dotted lines are just a guide to the eye). | ||
| Sample | Surface area (g m−2) | Total basic sites (mmol g−1) | Basic site density (μmol m−2) |
|---|---|---|---|
| LDO-W | 147 | 0.71 | 4.83 |
| LDO-E | 310 | 1.04 | 3.35 |
| LDO-A | 251 | 1.00 | 3.99 |
| LDO-IPA | 286 | 1.09 | 3.81 |
| LDO-EA | 243 | 0.89 | 3.67 |
| LDO-1H | 273 | 1.58 | 5.79 |
There is no noticeable difference in the general desorption profiles for the CO2 TPD (Fig. 3a), indicating that there is no change in the distribution profile of Lewis basic sites. The samples those treated with acetone, IPA and ethyl acetate had very similar number of basic sites ca. (1.96 mmol g−1). LDO-E and LDO-1H had Lewis basic sites in excess of 2.2 mmol g−1.
 | ||
| Fig. 3 (a) CO2 and (b) NH3 TPD profiles of ST-LDO samples. | ||
However, once the various surface areas of the LDOs are taken into account, all solvent treated samples present similar Lewis basic site density in the range of 7.22–8.10 μmol m−2 (Table 3), which are much lower than that of LDO-W (13.29 μmol m−2). The NH3 TPD experiments probe the Lewis acidity (Fig. 3b). LDO-W exhibited an intense, wide peak over the range of 100–450 °C. In contrast, ST-LDOs only exhibited two distinct individual peaks in the low temperature range (100–200 °C) and high temperature range (200–450 °C), which are assigned to the hydrogen bonding between NH3 and surface oxide/hydroxide groups and the strong strengths of Lewis acid sites, respectively. The medium strength Lewis acid sites disappeared after solvent treatment and their overall intensity decreased the compared to LDO-W. We believe this may arise because solvents strongly interact with acid sites during treatment, leading to blocking of some of the acid sites even after calcination. The total acidity of LDO-W is 0.38 mmol g−1 while the solvent treated LDOs had a significantly lower value in the range of 0.16–0.21 mmol g−1, where LDO-IPA has the lowest acidity value of 0.16 mmol g−1. Considering the Lewis acidic site density, all solvent treated LDO samples exhibit a Lewis acidic site density 3–5 times lower than that of LDO-W. It is interesting to find that the Lewis acid/base ratio of LDOs can be easily tuned from 0.08 to 0.19 by using different solvents. The Lewis acid/base ratio shows a correlation with the surface areas of the LDOs: the higher the surface area, the lower the ratio (Fig. S3†). This is due to both surface area and reduction in acid sites being a consequence of solvent penetration. A high surface area is the result of effective solvent replacement of surface and interlayer water molecules. This results in more solvent molecules being incorporated into the structure which will block more acid sites upon heat treatment.
| Sample | Total basic sites (mmol g−1) | Basic site density (μmol m−2) | Total acidic sites (mmol g−1) | Acidic site density (μmol m−2) | Total acidic sites/total basic sites |
|---|---|---|---|---|---|
| LDO-W | 1.95 | 13.29 | 0.38 | 2.59 | 0.19 |
| LDO-E | 2.24 | 7.22 | 0.19 | 0.61 | 0.08 |
| LDO-A | 1.97 | 7.86 | 0.21 | 0.84 | 0.11 |
| LDO-IPA | 1.97 | 6.88 | 0.16 | 0.57 | 0.08 |
| LDO-EA | 1.95 | 8.03 | 0.19 | 0.77 | 0.10 |
| LDO-1H | 2.21 | 8.10 | 0.21 | 0.77 | 0.09 |
Prinetto et al. have shown that the peak in the NH3 TPD at 150 °C is due to the hydrogen bonding between NH3 and surface oxide/hydroxide groups (Scheme S1(a)†).34 The resulting NH3 TPD features result from a combination of two different bonding modes involving the bonding between the nitrogen and the surface, and hydrogen bonding of an ammonia hydrogen to a neighbouring oxygen site (Scheme S1(b)†). These two bonding modes could not be resolved into separate features. However, it is important to note that both modes involve bonding to metal sites and both the medium and strong sites see a complete suppression and reduction in intensity respectively. While the NH3 bonding modes mainly occur through the metal sites, the CO2 bonding modes mainly concern surface oxygen sites. This indicates that the oxygen sites on the surface treated samples seem unaffected (Fig. 3a).
The change in the NH3 TPD curves, along with the reduction in basic site densities suggests that blocking of specific surface metal sites occurs upon solvent treatment. Our previous work has shown that the dispersing solvents remain on the surface of the AMO/AIM-LDH to variable degrees and these may result in the blocking of some active sites resulting in a decrease in basic and acidic site densities.
CO2 adsorption on LDOs
The CO2 adsorption capacity of the various ST-LDOs at 40 °C is shown in Fig. 4 and S4–S9.† CO2 adsorption by LDOs is a process involving both chemisorption and physisorption. For each sample, we observe that CO2 physisorption is around twice that of chemisorption. LDO-W showed a total CO2 adsorption of 0.64 mmol g−1 (0.44 and 0.20 mmol g−1 for physisorption and chemisorption respectively). Overall, ST-LDOs show a CO2 adsorption capacity increase of ca. 50% in both physisorption and chemisorption. | ||
| Fig. 4 CO2 capture of various ST-LDO samples at 40 °C and 100 kPa. | ||
LDO-E exhibited the highest total CO2 uptake of 1.01 mmol g−1, which represents a 58% increase compared with LDO-W. This is higher than previously reported figures for CO2 capture using LDOs under similar conditions. Sharma et al. have reported the highest to date of 0.38 mmol g−1 at 30 °C using a calcination temperature of 350 °C while Shang et al. reported a total CO2 capture value of 0.83 mmol g−1 for an acetone-treated Mg3Al CO3 LDH (pretreatment temperature of 180 °C).26,35LDO-EA exhibited a lowest CO2 uptake of 0.80 mmol g−1 amongst the ST-LDO samples tested. The chemisorption value of 0.30 mmol g−1 was comparable with the other solvent treated samples, but the physisorption figure has decreased due to the lower surface area.
It can be observed that the CO2 adsorption per g of all LDOs have a positive correlation with their surface area (Fig. S10†). Simply expressed, the higher the surface area, the more active adsorption sites are exposed, leading to a higher CO2 adsorption.
All solvent treated samples, with the exception of LDO-EA, exhibited a physisorption value similar to the total adsorption value of LDO-W. There seems to be a linear correlation (R = 0.94) relating the CO2 adsorption per surface area to the ratio total acidic to basic sites (mmol g−1) of the LDOs (Fig. 5). This atomic-level origin of this linear relationship to not clear at this time. However, it is worth noting that the acid sites as measured heavily involve the surface hydroxyl groups as shown in Scheme S1.† These hydroxyl groups can participate in CO2 adsorption via weak CO2 bidentate bonding. But this bonding mode is not present in the CO2 TPD due to the adsorption temperature of 100 °C.31,34 This observation suggests that solvent treatment not only disperses individual nanosheets increasing the accessible surface area but also the solvent interacts with the surface and selectively blocks acidic sites, leading to a slightly less active surface for CO2 adsorption.
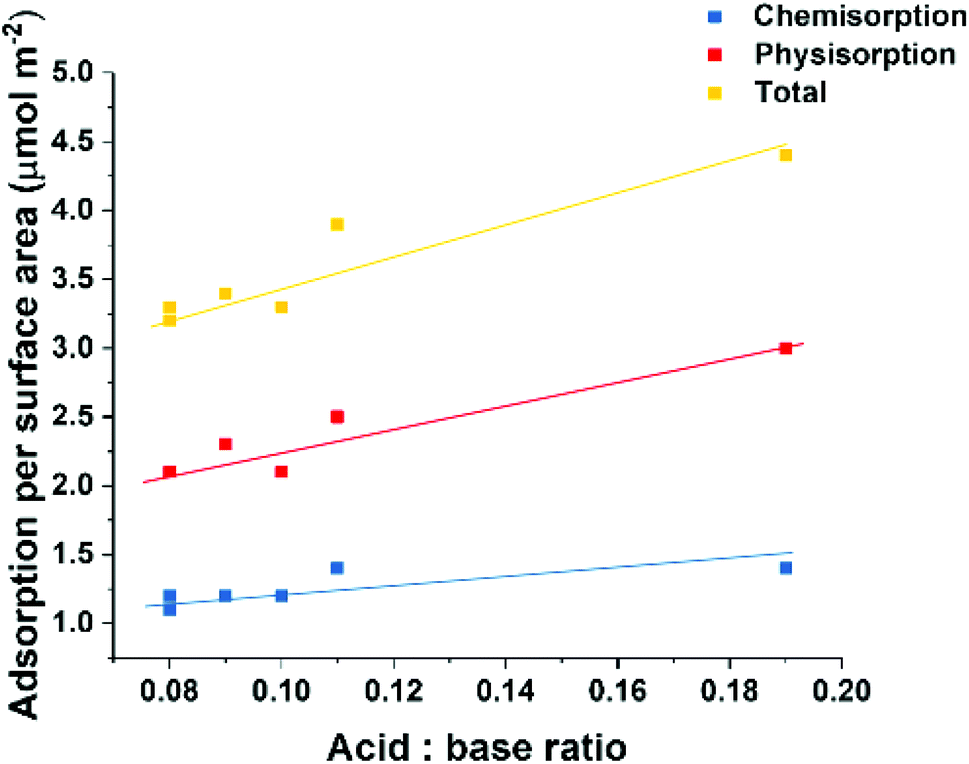 | ||
| Fig. 5 Molar CO2 adsorption per m2 as a function of the ratio of total acid sites to total basic sites for all ST-LDOs. | ||
Conclusions
A quantitative and comparative study of the basic and acidic features of conventional water washed and solvent treated LDHs and LDOs has been performed. Titration showed that solvent treatment leads to increase in Brønsted basicity in the LDHs while decreasing the basic site density. For Lewis basicity, all LDH samples showed evidence for monodentate carbonate binding while only LDH-E and LDH-1H exhibit stretching vibrations assigned to bidentate carbonate bonding for the solvent treated samples. These results show that changes in the Lewis basicity of LDHs can be achieved using the solvent treatment methods.Titration experiments on LDOs showed a general increase in total basic sites and basic site density compared to the parent LDH. All ST-LDOs exhibited lower basic site densities compared to LDO-W. All ST-LDOs showed no change in the distribution of basic strengths. The NH3 desorption profiles for ST-LDOs showed an elimination of the medium strength peak along with a reduction in intensity of the strong acidic peak, both of which involve NH3 bonding to the surface metal sites. This evidence, along with the reduction in basic site densities of solvent treated samples, strongly suggests that solvent molecules coordinate to the metal sites upon thermal treatment, which leads to blocking of active sites.
ST-LDOs displayed an increase in CO2 uptake capacity compared to conventional LDO (LDO-W; 0.64 mmol g−1). LDO-E exhibited the highest total CO2 uptake of 1.01 mmol g−1, which represents a 58% increase compared with LDO-W and we believe currently the highest value reported in the literature.
We found that the CO2 adsorption per surface area increases with an increasing acid/base ratio. Overall, study has found that the acid/basic properties of LDH can be fine-tuned by using (AMO or AIM) solvent treatments, which offers promising opportunities in selective catalysis and adsorption.
Experimental
Synthesis of ST-Mg2.5Al CO3-LDHs
[Mg0.72Al0.28(OH)2](CO3)0.14(H2O)0.66 (Mg2.5Al-CO3 LDH) (LDH-W) was synthesised using the co-precipitation method adapted from a literature procedure.21 Mg(NO3)2·6H2O (0.075 mol, 19.23 g) and Al(NO3)3·9H2O (0.025 mol, 9.37 g) were dissolved in 100 mL deionised (DI) water to make solution A. Na2CO3 (0.05 mol, 5.30 g) was then dissolved in another 100 mL DI water to make solution B. Solution A was added to solution B dropwise, while stirring, over the course of 1 h. The mixture was kept at pH 10 for the synthesis by the simultaneous addition of 4 M NaOH solution. When the addition of the solution A was completed and the final pH set to 10, the reaction was stirred for 24 h at room temperature. After the ageing process, the LDH product was washed with DI water until pH 7 and filtered. The product was dried overnight in a vacuum oven at 30 °C.For solvent treated samples, the wet cake was collected after the LDH was washed with water until the pH of the aqueous washing reached 7. The 30% solid content LDH wet cake was then re-dispersed in the appropriate solvent for 4 h then filtered and dried. The solvents used were ethanol, acetone, isopropyl alcohol, ethyl acetate and 1-hexanol to yield LDH-E, LDH-A, LDH-IPA, LDH-EA and LDH 1H, respectively. LDH samples were pre-treated to 200 °C for 1 h using a temperature ramping rate of 5 °C min−1 prior to characterisation and further studies.
Synthesis of ST-LDO samples
The various ST-LDH samples were calcined in a furnace at 450 °C for 12 h with a ramping rate of 5 °C min−1 and were used immediately after calcination to prevent rehydration and intercalation of impurities. The ST-LDOs were named LDO-W, LDO-E, LDO-A, LDO-IPA, LDO-EA and LDO-1H following from their parent ST-LDH respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank SCG Chemicals Co., Ltd. (Thailand) and the Balliol College Mehnert Scholarship for funding (DWJL).Notes and references
- A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191–3198 RSC.
- W. J. L. Pesic, S. Salipurovic, V. Markovic and D. Vucelic, J. Mater. Chem., 1992, 2, 1069–1073 RSC.
- G. Marcelin, N. J. Stockhausen, J. F. M. Post and A. Schutz, J. Phys. Chem., 1989, 93, 4646–4650 CrossRef CAS.
- M. Ogawa and K. Inomata, Chem. Lett., 2005, 34, 810–811 CrossRef CAS.
- K. L. Erickson, T. E. Bostrom and R. L. Frost, Mater. Lett., 2005, 59, 226–229 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- K. Chibwe and W. Jones, J. Chem. Soc., Chem. Commun., 1989, 926–927 RSC.
- J. Rocha, M. del Arco, V. Rives and M. A. Ulibarri, J. Mater. Chem., 1999, 9, 2499–2503 RSC.
- W. Kagunya, Z. Hassan and W. Jones, Inorg. Chem., 1996, 35, 5970–5974 CrossRef CAS.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 7040, 7040–7066 RSC.
- M. B. Gawande, R. K. Pandey and R. V. Jayaram, Catal. Sci. Technol., 2012, 2, 1113 RSC.
- S. J. Park, H. A. Ahn, I. J. Heo, I.-S. Nam, J. H. Lee, Y. K. Youn and H. J. Kim, Top. Catal., 2010, 53, 57–63 CrossRef CAS.
- J. Zhao, J. Xie, C.-T. Au and S.-F. Yin, Appl. Catal., A, 2013, 467, 33–37 CrossRef CAS.
- Q. Wang, Z. Wu, H. H. Tay, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, Catal. Today, 2011, 164, 198–203 CrossRef CAS.
- J. M. Silva, R. Trujillano, V. Rives, M. A. Soria and L. M. Madeira, Chem. Eng. J., 2017, 325, 25–34 CrossRef CAS.
- S. Kim, S. G. Jeon and K. B. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 5763–5767 CrossRef CAS PubMed.
- H. Du, A. D. Ebner and J. A. Ritter, Ind. Eng. Chem. Res., 2011, 50, 412–418 CrossRef CAS.
- K. Coenen, F. Gallucci, G. Pio, P. Cobden, E. van Dijk, E. Hensen and M. van Sint Annaland, Chem. Eng. J., 2017, 314, 554–569 CrossRef CAS.
- N. N. A. H. Meis, J. H. Bitter and K. P. de Jong, Ind. Eng. Chem. Res., 2010, 49, 1229–1235 CrossRef CAS.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301–6303 RSC.
- C. Chen, M. Yang, Q. Wang, J.-C. Buffet and D. O'Hare, J. Mater. Chem. A, 2014, 2, 15102–15110 RSC.
- K. Ruengkajorn, V. Erastova, J.-C. Buffet, H. C. Greenwell and D. O'Hare, Chem. Commun., 2018, 54, 4394–4397 RSC.
- V. Erastova, M. T. Degiacomi, D. O'Hare and H. C. Greenwell, RSC Adv., 2017, 7, 5076–5083 RSC.
- J.-C. Buffet, Z. R. Turner, R. T. Cooper and D. O'Hare, Polym. Chem., 2015, 6, 2493–2503 RSC.
- G. E. Hickman, C. M. R. Wright, A. F. R. Kilpatrick, Z. R. Turner, J.-C. Buffet and D. O'Hare, Mol. Catal., 2019, 468, 139–147 CrossRef CAS.
- S. Shang, A. Hanif, M. Sun, Y. Tian, Y. S. Ok, I. K. M. Yu, D. C. W. Tsang, Q. Gu and J. Shang, J. Hazard. Mater., 2019, 373, 285–293 CrossRef CAS PubMed.
- X. Zhu, C. Chen, H. Suo, Q. Wang, Y. Shi, D. O'Hare and N. Cai, Energy, 2019, 167, 960–969 CrossRef CAS.
- M. Yang, O. McDermott, J.-C. Buffet and D. O'Hare, RSC Adv., 2014, 4, 51676–51682 RSC.
- K. Ruengkajorn, C. M. R. Wright, N. H. Rees, J.-C. Buffet and D. O'Hare, Mater. Chem. Front., 2018, 2, 2277–2285 RSC.
- K. Muramatsu, Y. Kuroda, H. Wada, A. Shimojima and K. Kuroda, Dalton Trans., 2018, 47, 3074–3083 RSC.
- J. I. Di Cosimo, V. K. Díez, M. Xu, E. Iglesia and C. R. Apesteguía, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- R. W. Stevens, R. V. Siriwardane and J. Logan, Energy Fuels, 2008, 22, 3070–3079 CrossRef CAS.
- M. J. Hudson, S. Carlino and D. C. Apperley, J. Mater. Chem., 1995, 5, 323 RSC.
- F. Prinetto, G. Ghiotti, R. Durand and D. Tichit, J. Phys. Chem. B, 2000, 11117–11126 CrossRef CAS.
- U. Sharma, B. Tyagi and R. V. Jasra, Ind. Eng. Chem. Res., 2008, 47, 9588–9595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation technique details, XRD patterns and CO2 adsorption curves. See DOI: 10.1039/d0dt01587c |
| This journal is © The Royal Society of Chemistry 2020 |