
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative strategies for CO homologation
Richard Y.
Kong
 * and
Mark R.
Crimmin
*
* and
Mark R.
Crimmin
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, White City, Shepherds Bush, London, W12 0BZ, UK. E-mail: r.kong17@imperial.ac.uk
First published on 8th June 2020
Abstract
Recent approaches in which at least two metal or main-group centres are involved in the homologation of CO are reviewed. We have characterised the strategies into three broad areas: (i) the reductive homologation of atmospheric CO at a metal or main group centre (ii) the reductive homologation of metal–carbonyl CO units and (iii) reductive homologation of CO with M–M, B–Li, Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si, and B
Si, and B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B bonds.
B bonds.
Introduction
In response to concerns over anthropogenic CO2, interest in developing methods to produce liquid hydrocarbons from renewable resources has heightened in recent years. One established technology to achieve this is the Fischer–Tropsch (F–T) process. The F–T process converts syngas mixtures (H2/CO) into short-to-medium chain hydrocarbons using heterogeneous transition metal catalysts (eqn (1)). CO2 can be incorporated through the water–gas shift reaction (eqn (2)).| nCO + (2n + 1) H2 → CnH2n+2 + nH2O | (1) |
| CO2 + H2 ⇌ CO + H2O | (2) |
Although syngas is typically produced from coal or natural gas, biomass has also been identified as viable source.1 F–T catalysis produces hydrocarbons with a range of carbon chain-lengths determined by the Anderson–Schulz–Flory distribution.2
Carbon monoxide (CO) is a diatomic molecule with bond order of 3. The HOMO is a σ-orbital with a major contribution from the carbon atom, while the LUMO consists of two degenerate π* orbitals (Fig. 1). Accordingly, CO is capable of either acting as a Lewis base or Lewis acid through HOMO and LUMO based reactivity respectively. The reduction of CO is also possible through electron transfer to the LUMO from a suitable reductant.
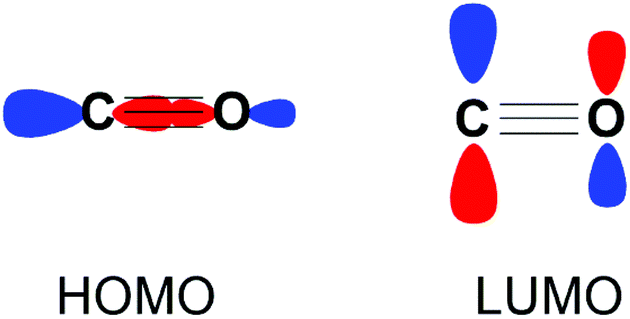 | ||
| Fig. 1 Frontier molecular orbitals of CO depicted as combinations of their constituent atomic orbitals. A second degenerate LUMO orthogonal to the plane of the page is not shown for clarity. | ||
While homogeneous F–T catalysis remains significantly underdeveloped with respect to the heterogeneous commercial process, there is growing interest in this area. Homogeneous models are amenable to study by solution techniques (NMR, IR, UV-vis spectroscopy) and can offer unique insight into fundamental reactivity that may be of relevance to heterogeneous F–T catalysis. In the longer term, using homogeneous catalysis to direct the selectivity of F–T reactions to high-value products is an attractive target.
In this frontier article, we summarise contemporary research in CO homologation using cooperative strategies. That is reactions in which two or more components (typically transition metal or main group complexes) react with CO to form carbon chains by forming new C–C bonds. These reactions typically involve three stages: initiation, propagation, and termination. Cooperative effects have been observed in each of the separate stages of chain growth.
The initiation step often involves the reduction of CO with electrons from a transition metal or main group complex. Reduction forms a high energy intermediate. The propagation step involves further reaction of this high energy intermediate with CO to form C2 fragments or less commonly C3/C4 chains. Termination involves the sequestration of reactive intermediates into stable, isolable products which do not react with further equivalents of CO. The available mechanisms for CO homologation are diverse, as is the range of compounds used. There are, however, some general themes that emerge. We have classified the cooperative approaches to CO homologation into three categories:
(i) Reduction of CO at a metal or main-group centre.
(ii) Reduction of transition metal carbonyls (M–CO) with an external reductant
(iii) Reduction of CO with M–M, B–Li, SiSi and BB bonds.
We focus on defined reactions in which CO units react to form carbon chains. Most of these reactions do not involve H2 and in only a few cases can the carbon chain be liberated from the reagents. Others have summarised reactions of CO/H2 mixtures with transition metal complexes including reactions that involve the insertion of CO into M–H bonds.3
Results and discussion
(i) Reduction of CO at a metal or main-group centre
The reductive homologation of CO was first observed by Justus von Liebig in 1834 through reaction with molten potassium to form “potassium carbonyl” (KCO).4 In 1960, “potassium carbonyl” was shown to be a mixture of potassium ethynediolate (1) and potassium benzenehexolate (2) salts (Scheme 1a).5 In 1985 Evans, Atwood and co-workers reported a homogeneous metal complex that could perform a similar reaction with CO to form coupled products (Scheme 1b). Hence, the samarium(II) sandwich complex 3·THF2 reacted with CO at ambient temperatures and pressures to yield multiple products including 4. 4 contains a ketenecarboxylate motif derived from three CO units that bridges two samarium(III) centres.6 Similar reactivity is accessed through the lanthanum congener of 3, in this case resulting in a diamagnetic product that is amenable to characterisation by multinuclear NMR spectroscopy.7 Evans noted that the inability of f-block complexes to form stable CO adducts alongside their high oxophilicity affords this unique chain growth reactivity. Both reactions occur with oxidation of Ln(II) to Ln(III) (Ln = Sm, La).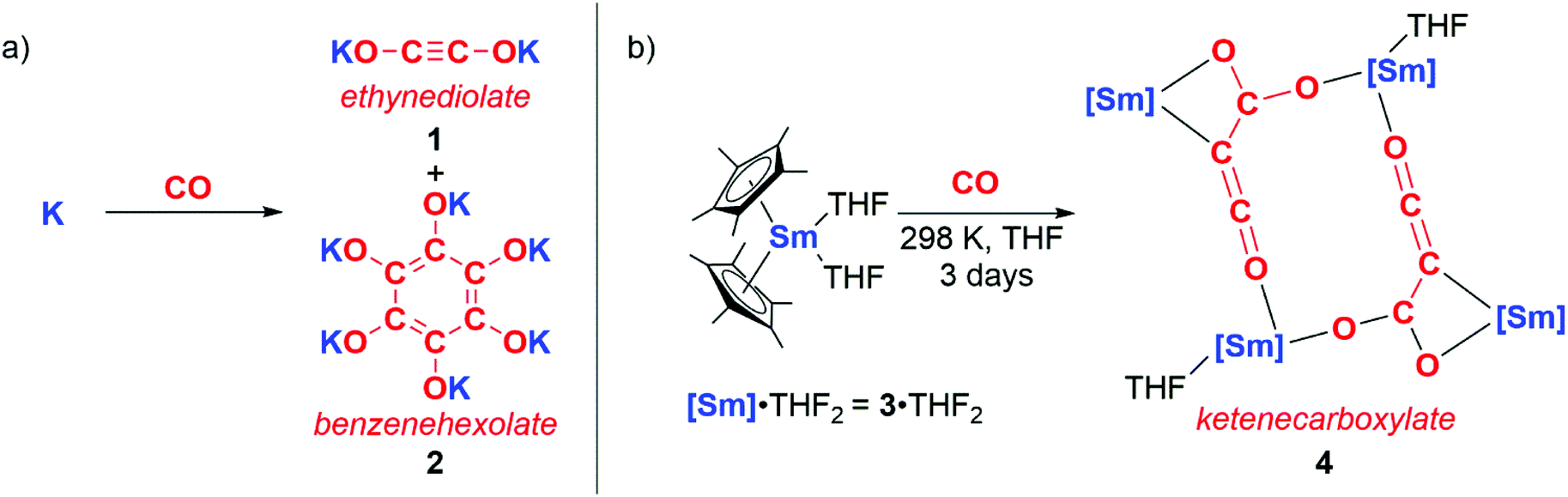 | ||
| Scheme 1 a) Formation of ‘potassium carbonyl’ reported by Liebig, confirmed by Büchner and Weiss. (b) Reductive homologation of CO by samarocene to form a ketenecarboxylate. | ||
In 2006, Cloke and co-workers reported the reductive cyclotrimerisation of CO with the uranocene(III) complex 5a to form a deltate anion (7).8 The reaction occurs with oxidation of U(III) to U(IV) and requires cooperative action of two equiv. of the actinide complex. Remarkably, further studies showed that acyclic and cyclic carbon chains of different lengths and shapes – ethynediolate (6) and squarate (8) – could be accessed by controlling either the stoichiometry of the reaction or the steric profile of the ligands on the uranium centre (Scheme 2a).9–11
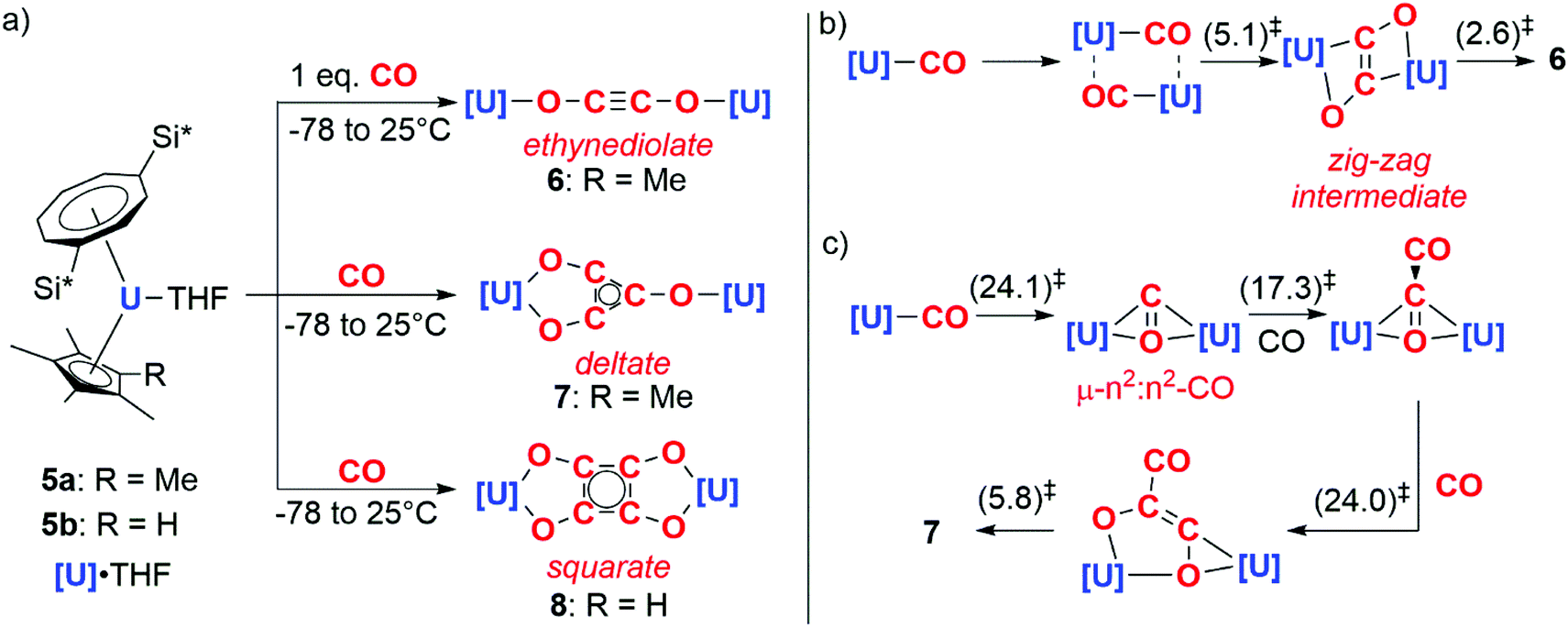 | ||
| Scheme 2 a) Experimental CO homologation. Si* = SiiPr3 (b) computational pathway towards the formation of ethynediolate. (c) Computational pathway towards the formation of deltate. Calculations performed using the B3PW91 functional. (Gibbs free energies in kcal mol−1). | ||
DFT calculations have been used to study how carbon chain growth occurs with this well-defined system. These calculations suggest that two distinct mechanisms are in operation, one leads to the formation of the ethynediolate and the other the deltate. In the case of ethynediolate, calculations by Green and co-workers suggest that an intermediate uranium(III) mono(carbonyl) is formed which can undergo sequential dimerisation and electron transfer steps, forming a ‘zig-zag’ intermediate which isomerises to the experimentally-observed linear ethynediolate (Scheme 2b).10 In the case of the deltate, Maron and co-workers propose that the same uranium(III) mono(carbonyl) is reduced by a second equivalent of the uranium(III) starting material resulting in the formation of a μ-η2:η2-CO ligand bridging two uranocene centres. A second CO equivalent reacts with this bridging CO ligand to directly form a new C–C bond and a {μ-η2:η2-C2O2}2− moiety that spans two uranium centres. A third equivalent of CO inserts into the U–C bond of this intermediate which, following a series of isomerisation steps, results in the formation of the deltate product (Scheme 2c).12 While the proposed uranium(III) carbonyl intermediates have not been isolated, closely related structurally characterised uranium(III) carbonyl complexes are known.13,14
The potential for f-block reductants to couple CO units together has been further elaborated by other groups, though exclusively towards the formation of ethynediolate products. Arnold and co-workers showed that simple uranium(III) alkoxide and amide complexes (9a–c) reduce CO under ambient conditions to ethynediolate containing products (10a–c). In the case of the amide (9a), further reactivity of the ethynediolate is observed; C–H activation of a methyl group of the ligand occurs by addition across the CC bond, forming metallocycle 11a (Scheme 3).15,16 A uranium(III) complex supported by a tripodal tris(amido)amine ligand (12) also reacts with CO to form an ethynediolate (13), as reported by Liddle and co-workers. Notably 12 can be regenerated from the ethynediolate uranium(IV) species through further silylation and reduction steps. This creates a closed chemical loop with respect to the uranium fragment (Scheme 4).17
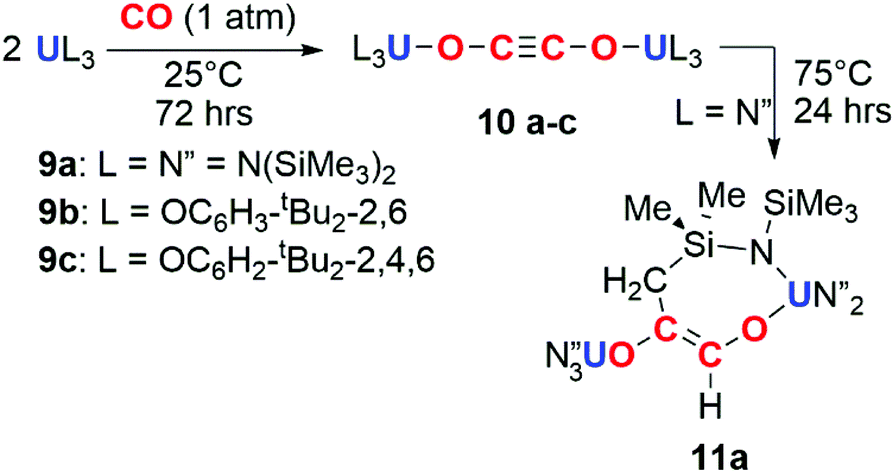 | ||
| Scheme 3 Reduction of CO by uranium tris(amide) (9a) and tris(aryloxide) (9b–c) complexes to the ethynediolate (10a–c). Further reactivity observed for 10a to 11a. | ||
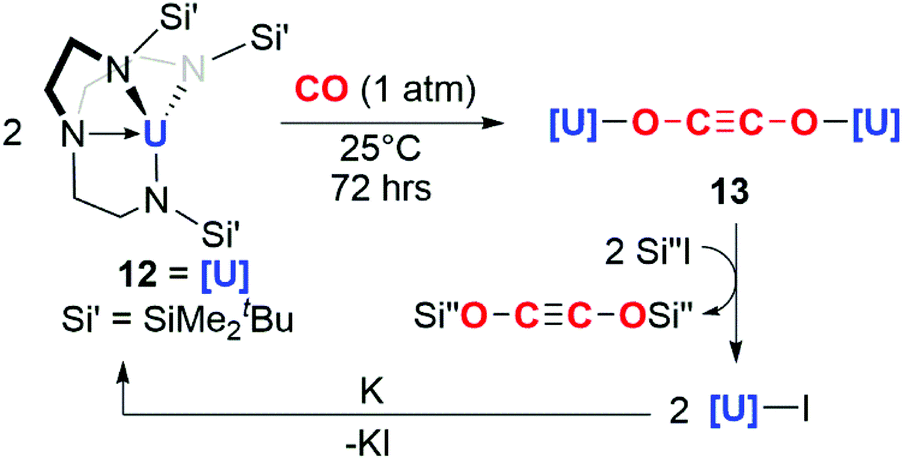 | ||
| Scheme 4 Reductive homologation of CO by a tris(amido)amine uranium(III) complex 12. Si′′ = SiMe3 or SiMe2Ph. | ||
Transition metal analogues of this type reactivity are far rarer, those that are known often involve direct participation of metal–ligand bonds. For example, in 2018, Kays and co-workers reported the deoxygenative coupling of CO using bis(terphenyl) iron(II) complex 14 to form squaraine 15, and iron carboxylate complex 16 along with concomitant formation of iron pentacarbonyl (Scheme 5).18 The reaction is proposed to proceed through the ketene intermediate 17, which derives from the fragmentation of CO into C and CO2. Dimeristion of 17 forms 18, which reacts with excess CO to yield the products 15 and 16, along with concomitant elimination of iron pentacarbonyl. This system is unusual as the organometallic reagent does not reduce CO but facilitates a disproportionation of CO. Cooperativity between the two iron centres is critical in formation of the squaraine.
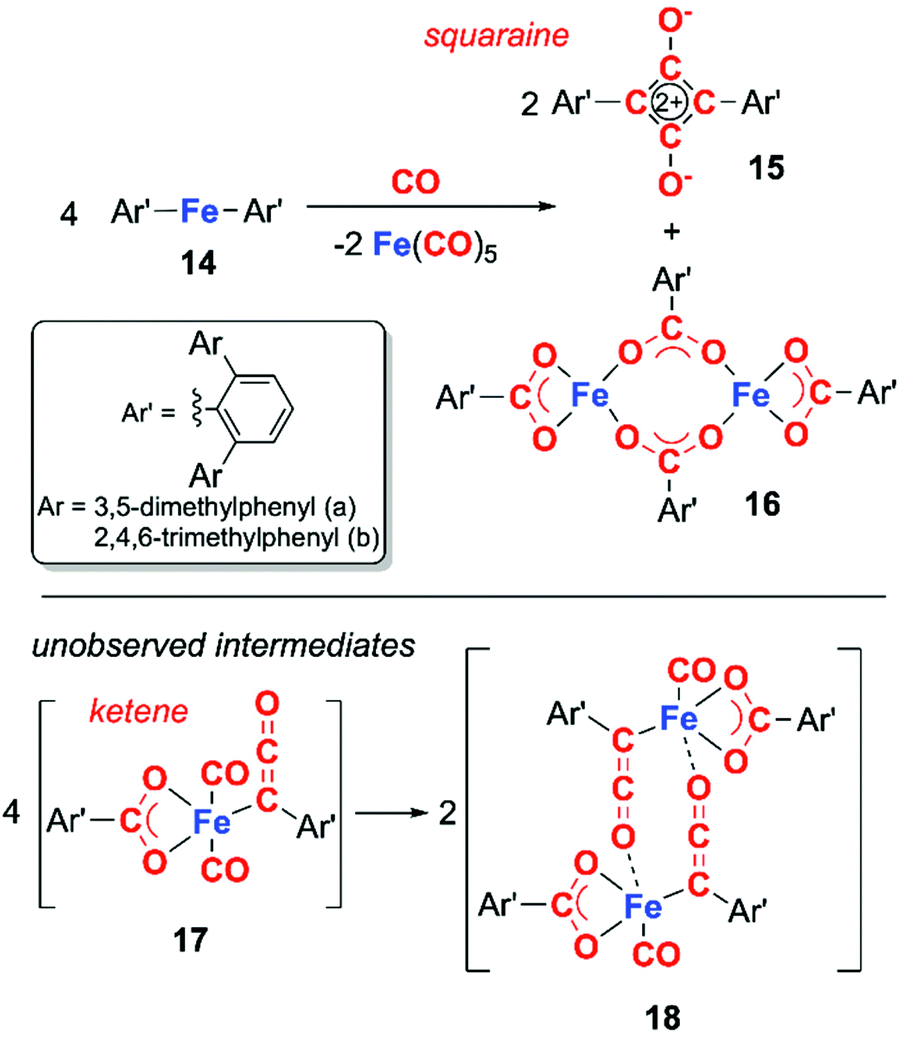 | ||
| Scheme 5 Deoxygenative coupling of CO to form a squaraine using bis(terphenyl) iron complex 14. | ||
While the reaction of low-valent f-block compounds with CO has been studied in detail in the last two decades, analogous chemistry with main-group reagents has only recently come into focus through the study of silylenes. In contrast to the 1e− reduction per metal centre of f-block complexes, silylenes function as 2e− reductants per silicon(II) centre. Early work in this area demonstrated that the addition of transient silylenes [R2Si:] (R = alkyl, aryl) to CO results in the formation of simple adducts of the form [R2Si:(CO)], stable only at −196 °C.19–21 The CO adduct of a gallium-substituted silylene has been isolated.22 No coupling products were observed. Only through the inclusion of cooperative strategies has CO coupling been observed with these reagents.
In 2019, Driess and co-workers reported the silicon-mediated coupling of CO with a bis(silylene) compounds supported by dinucleating ligands incorporating either a xanthenyl (18a) or ferrocenyl (18b) backbone (Scheme 6).23 The resultant products are 1,3-disilyloxetanes (19a–b) in which two silicon(IV) centres are bridged by a ketenylidene and an oxo ligand. The oxo ligand is derived from complete cleavage of a CO bond. A total of 4e− are transferred to two molecules of CO. Crucially, cooperativity between the silylene units is necessary for CO reduction: this type of reactivity is not observed using analogous amidinate or β-diketiminate mono(silylene) reagents or when the silicon sites of the bis(silylene) compound are separated by a distance of 6 Å.
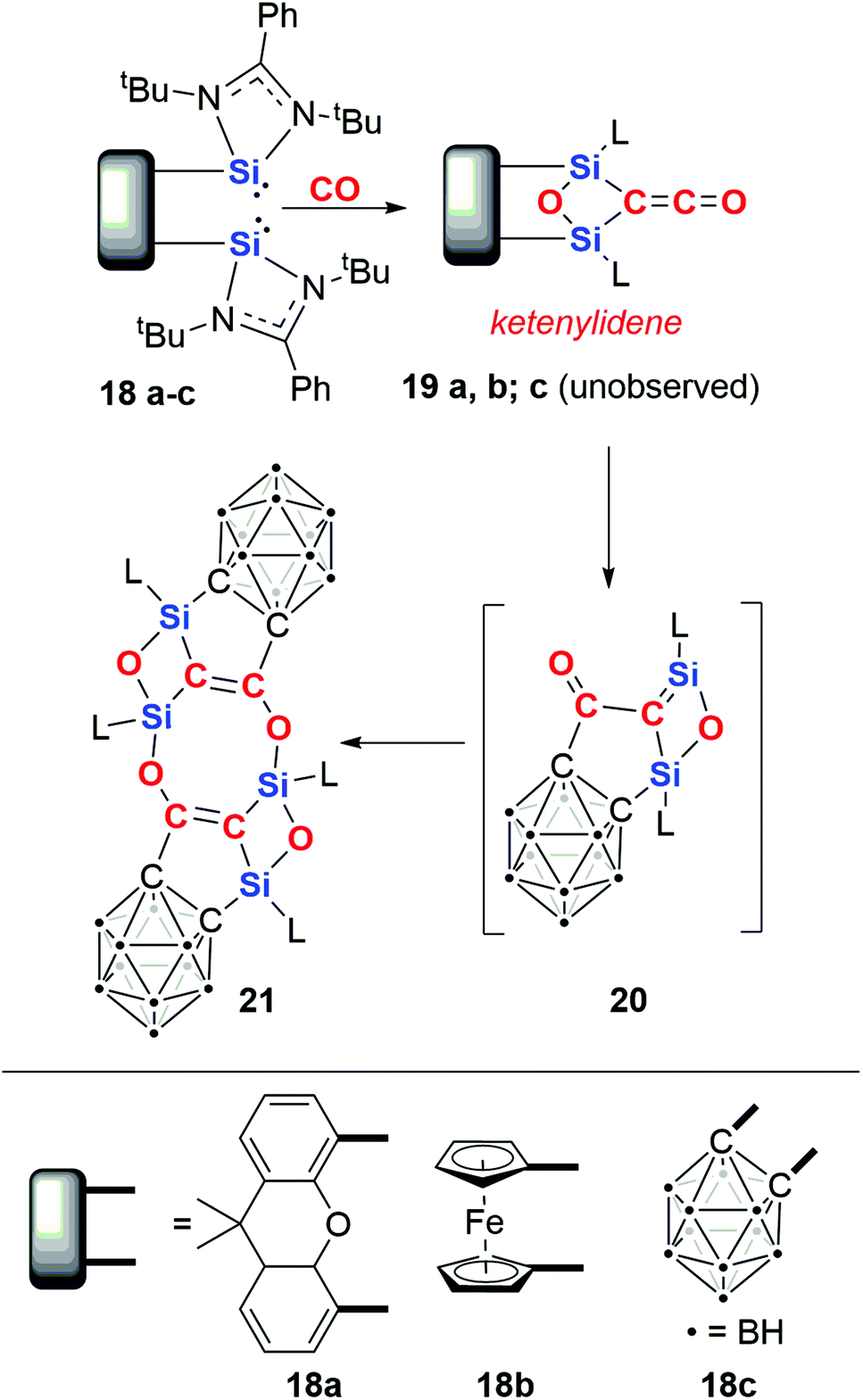 | ||
| Scheme 6 Reductive homologation of CO to a ketenylidene ligand by a (bis)silylene. | ||
Upon modifying the dinucleating ligand to include an ortho-carborane backbone, a different outcome was observed.24 Hence, 18c reacts to form 21via a multistep process (Scheme 6). The first step is proposed to form an identical intermediate to those observed with xanthenyl- and ferrocenyl-based ligands. In the case of the carborane-based ligand, this intermediate is unstable and reacts further with cleavage of the Si–Ccarborane bond and concomitant Cketene–Ccarborane bond formation, resulting in monomer 20. Dimerisation of 20 results in the crystallographically and spectroscopically characterised product, 21.
More recently, it has been shown that dinuclear systems are not required for CO homologation and that cooperative effects can be achieved through intermolecular rather than intramolecular assistance. Aldridge and co-workers reported the reductive coupling of CO using a boryl-substituted acyclic silylene 22 to form an ethenediolate species 23 (Scheme 7).25 The acyclic silylene 22 has previously been reported to activate H2; the high reactivity of 22 has been ascribed to the strong σ-donating ability of the boryl ligand and the resultant small HOMO–LUMO gap.26
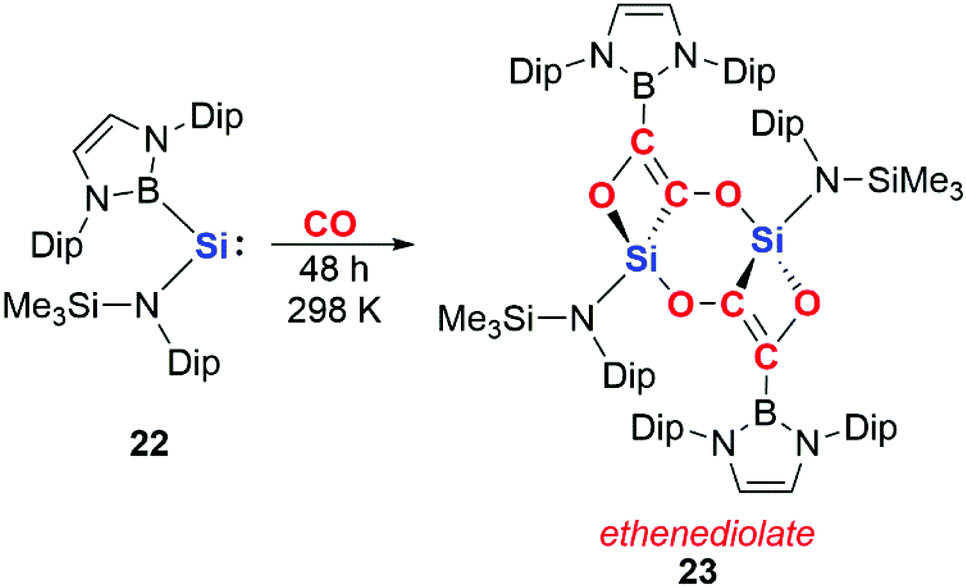 | ||
| Scheme 7 CO homologation by an acyclic silylene. Dip = 2,6-di-iso-propylphenyl. | ||
(ii) Reduction of M–CO with an external reductant
The previous section highlighted the capability of some redox-active metal centres to reduce atmospheric CO as a first step for carbon chain growth. An alternative cooperative strategy is to separate the CO binding event from the reduction and chain growth steps. To achieve this, transition metal carbonyls are typically used as the CO source, while a second low-oxidation state reagent is used as a reductant.In 1982 Bercaw, Mertes, and co-workers reported the reaction of a zirconocene(II) reductant (24) with an iron carbonyl dimer (Scheme 8a).27 In this reaction, 24 performs the 2e− reduction of two cis-oriented CO ligands in 25 to form the bis(alkylidene) moiety 26. Interestingly, the coupling of CO is reversible as reaction of 26 with CO regenerates 25 with concomitant formation of [(Cp*)2Zr(CO)2]. In 1986, Lippard and co-workers reported the reductive coupling of two cis-disposed carbonyl ligands of a group 5 metal complex (27) using sodium amalgam as the reducing agent (Scheme 8b). Subsequent addition of two equivalents a tri-alkylsilyl chloride (SiR3Cl, R = Me, iPr) resulted in a η2-coordinated silylated ethynediolate (28).28,29 Reduction of 27 followed by addition of one equivalent of tri-iso-propylsilyl chloride affords the siloxyalkylidyne complex 29. Addition of a further equivalent of tri-iso-propylsilyl chloride to 29 confirmed it to be an intermediate en route to 28.30,31 In 1996, Pampaloni and co-workers reported the reductive coupling of CO in which group 4 complexes played the role of both the transition metal carbonyl and the reductant (Scheme 8c). Hence, reaction of a group 4 metallocene dicarbonyl (30a–b) with the respective homoleptic group 4 N,N-dialkylcarbamate complex (31a–b) resulted in the formation of trinuclear metal clusters with ketenylidene ligands (32). The origin of the ketenylidene ligand is likely two cis-disposed CO units which are coupled with concomitant reductive cleavage of a CO triple bond to form a μ3-oxo ligand.32,33
 | ||
| Scheme 8 Reductive coupling of transition metal bound CO by (a) a zirconocene(II) reductant, (b) sodium/mercury amalgam, and (c) group 4 metallocenes. | ||
In 2013, Suess and Peters demonstrated that an iron bis(alkylidyne) complex derived from a metal carbonyl is capable of releasing an alkene upon hydrogenation (Scheme 9). The carbon atoms in the alkylidyne ligands originate from CO.34 Reduction of iron dicarbonyl complex 33 using potassium, followed by addition of trimethylsilyl triflate resulted in generation of the bis(alkylidyne) complex 34. Reaction of 34 with dihydrogen gas resulted in liberation of the C2 fragment 35, as exclusively the Z-alkene. Paramagnetic iron-containing products from the hydrogenation have eluded characterisation and are yet to be isolated. This example is notable as H2, a key component of syngas used in F–T catalysis, is used to achieve the dissociation of a CO derived hydrocarbon from the transition metal fragment.
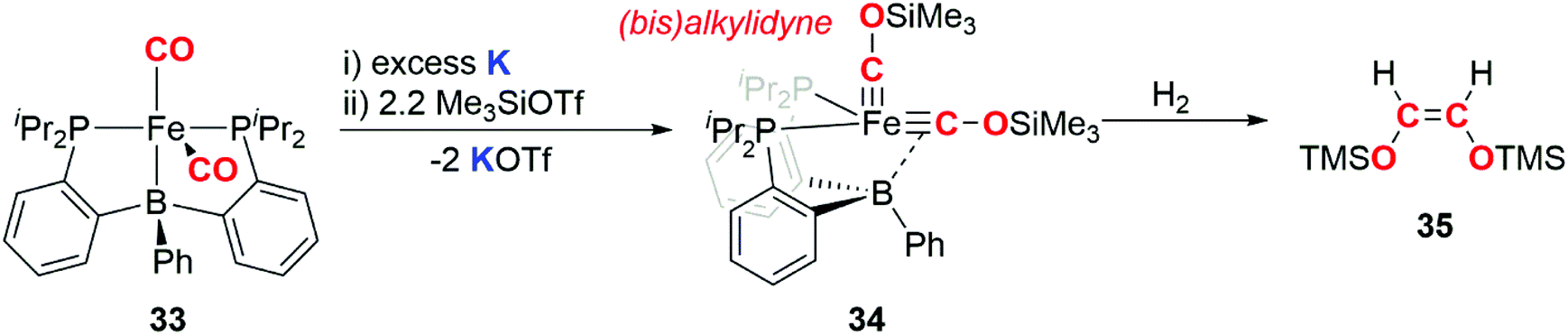 | ||
| Scheme 9 Reduction of CO to form a (bis)alkylidyne complex and reductive coupling of the (bis)alkylidyne to liberate a hydrocarbon fragment. | ||
In 2016, Buss and Agapie reported the 4e− deoxygenative coupling of CO using a molybdenum complex (Scheme 10).35 The Mo(II) dicarbonyl complex 36 can be successively reduced to the Mo(0) 37, Mo(-II) 38, and Mo(-III) 39 species with 2, 4, and 7 (excess) equiv. of KC8 respectively. The deoxygenative coupling of the CO ligands in the reduced complexes to form C2 fragments is effected by addition of trialkylsilyl chloride and further equivalents of reductant to form either disilaketene (42a, R = Me) or a disilaethynolate (42b, R = iPr) products and the molybdenum dinitrogen complex 41.
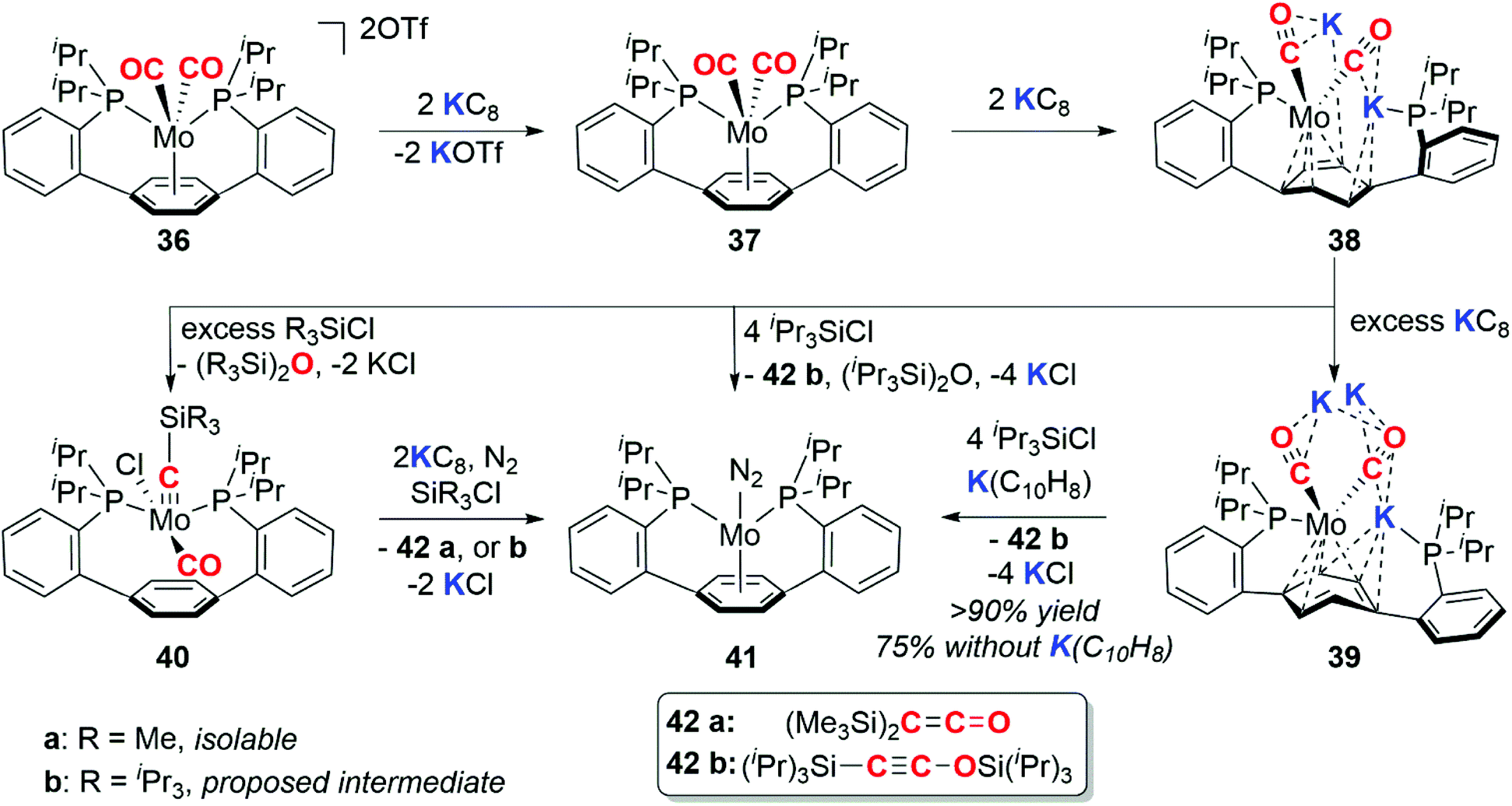 | ||
| Scheme 10 Four-electron deoxygenative coupling of CO at molybdenum. | ||
During the course of these experiments the siloxyalkylidyne intermediate 40a was isolated. Both the structure and reactivity of 40a parallel the reactivity of siloxyalkylidyne complexes explored by Lippard and co-workers (vide supra).30 The P–Ar–P pincer ligand is a key design motif in this chemistry as the arene functions as a ‘reservoir of electrons’ capable of stabilising the myriad of oxidation states of molybdenum through a range of coordination (η6-, η4-, η0-) modes.35 Subsequent computational and synthetic studies on the mechanism show that the C–C bond formation step proceeds via a (bis)alkylidyne-type complex.36
In 2018, our group reported the reductive homologation of CO by reaction of an aluminium(I) reductant 43 with [W(CO)6] (44), in the presence of CO gas (Scheme 11a). Three CO units are catenated from the transition-metal and chelated by the aluminium(III) ligands, forming a [4,6] ortho-fused bicyclic ring system in compound 45. Labelling experiments show that the chain contains CO units from both the tungsten hexacarbonyl reagent and atmospheric CO.37
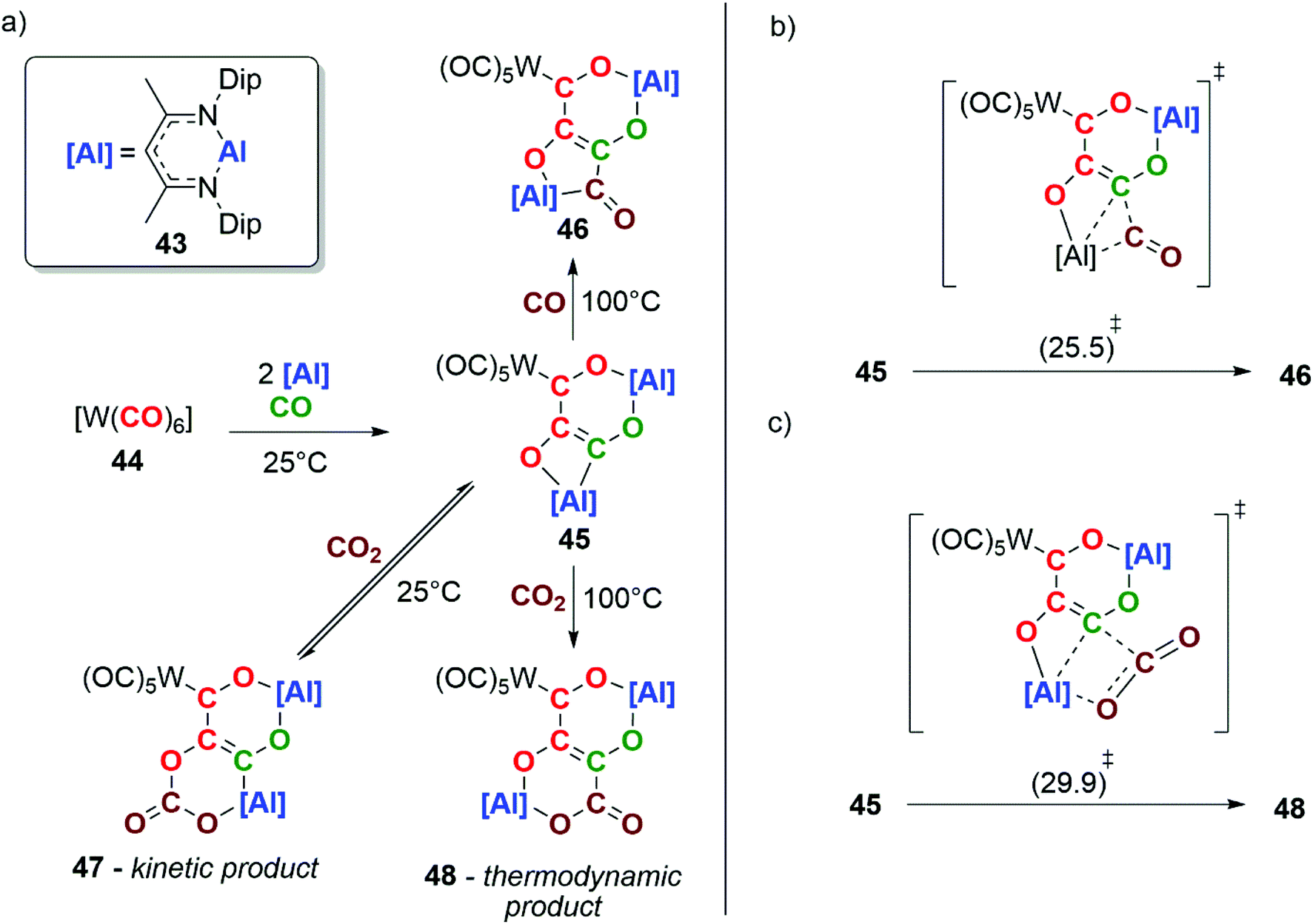 | ||
| Scheme 11 a) Experimentally determined CO coupling and chain-growth processes. Dip = 2,6-di-isopropylphenyl. DFT calculated transition states for (b) the insertion of CO into the Al–C bond of 45 and (c) the insertion of CO2 into the Al–C bond of 45. Calculations performed using the ωB97x functional (Gibbs free energies in kcal mol−1). | ||
Heating 45 under an atmosphere (1 atm) of CO gas at 100 °C for 18 h results in further chain-growth from the {C3O3}4− to the {C4O4}4− species 46. Alternatively, reaction of 45 with CO2 at room temperature results in the observation of the reversible insertion of CO2 into an Al–O bond of 45 forming the kinetic product 47. The same reaction at 100 °C yields the thermodynamic product 48, a four-carbon chain derived from the heterocoupling of CO and CO2. DFT calculations have been used to explore the chain-growth process. A single transition state was found to connect 45 and 46 (Scheme 11b). Likewise, the conversion of 45 to 48 involves a single step (Scheme 11c). Both can be conceptualised in terms of insertion of CO or CO2 into the Al–C bond of the carbon chain. The interaction of CO and CO2 with the Lewis acidic aluminium centre plays a key role in the chain-growth processes. This reaction is remarkable in that chain growth occurs with the formation of C3 and C4 intermediates that still have reactive sites in the form of reactive M–C bonds. This allowed the defined steps of chain-growth Cn to the Cn+1 homologue (n ≥ 2) in a homogeneous F–T model to be observed for the first time.37
In 2019, Roesky and co-workers documented carbon chain growth of the CO ligands of [Re2(CO)12] using a samarium(II) reductant, 3·2THF (Scheme 12). While 3·2THF has been shown to reductively couple two CO units to the ketenecarboxylate (vide supra),6 altering the source of CO to a rhenium carbonyl cluster 49 results in four CO units being coupled together. The coupled CO units form a bis(alkylidene) ligand bound to the rhenium centre in 50 (Scheme 12). Interestingly, CO coupling is only observed in the case of samarocene and the rhenium carbonyl: variation of the CO source to [Mn2(CO)10] or reductant to a bis(amidinate) supported lanthanide complex (Ln = Sm, Yb) yielded a suite of isocarbonyl bridged complexes but provided no evidence for carbon chain growth from CO.38
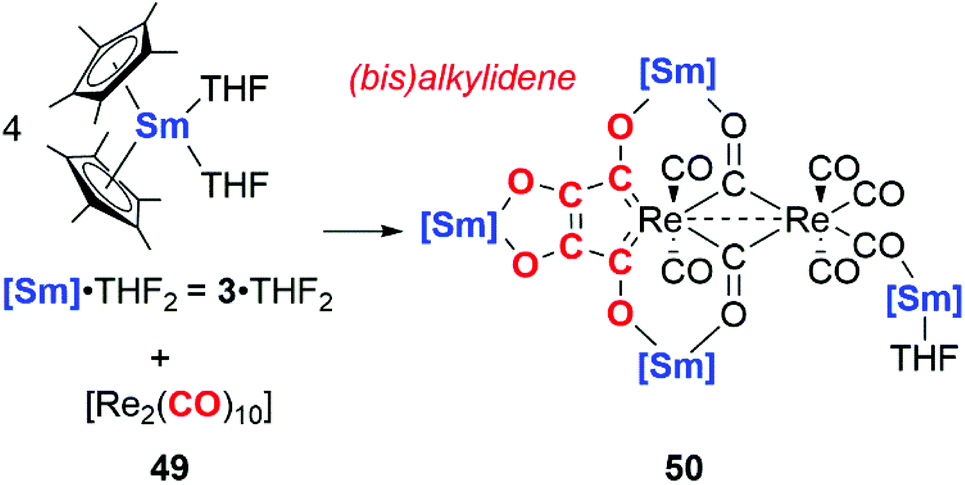 | ||
| Scheme 12 Homologation of rhenium bound CO units using a samarocene reductant. | ||
(iii) Reduction of CO with M–M, B–Li, Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) Si and B
Si and B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) B bonds
B bonds
The strategies for carbon chain growth from CO outlined above involve the use of low-valent metal centres which can transfer electrons to CO to initiate a chain growth sequence. An alternative approach to CO homologation involves the use of low-oxidation state compounds that possess a metal–metal or element–element bond. These bonds are potential reactive sites, and reduction of CO can occur by transfer of electrons in the bond to the substrate with concomitant oxidation of the metal centres. The cooperative action of two reactive sites, this time connected directly through a bond in the ground state of the starting material, is a key consideration for the reaction with CO.
Wayland and co-workers first utilised this strategy in 1989. They reported the selective dimerisation of CO using a rhodium porphyrin dimer (51) containing a Rh–Rh bond (Scheme 13). The product of reductive coupling is an ethanedionyl complex derived from the double insertion of CO into the Rh–Rh bond. The product (52) was not amenable to NMR or solid state characterisation and was inferred through IR studies on 12CO and 13CO isotopomers.39
 | ||
| Scheme 13 Formation of an ethanedionyl ligand from CO using rhodium porphyrin dimer 47. Ar = 3,5-dimethylphenyl. | ||
In 2016, Kinjo and co-workers reported the isolation of the boryllithium compound 53 (Scheme 14). Insertion of CO into the B–Li bond of this highly reactive species forms the bora-acyllithium 54, with tautomerises to 54′. Dimerisation of 54′ results in the lithium ethenediolate 55 which has been tentatively assigned using 11B NMR spectroscopy. 55 is unstable and can only be observed transiently, however reaction with trimethylsilyl chloride allows the isolation of 56, which has been structurally characterised. Overall the reaction results in the reductive coupling and functionalisation of two CO molecules. The high polarity of the B–Li bond is thought to be important in determining the reaction of 53 with CO, no reaction is observed for analogous [B]–M (M = Mg, Al, Au, Zn, Sb, Bi) complexes.40
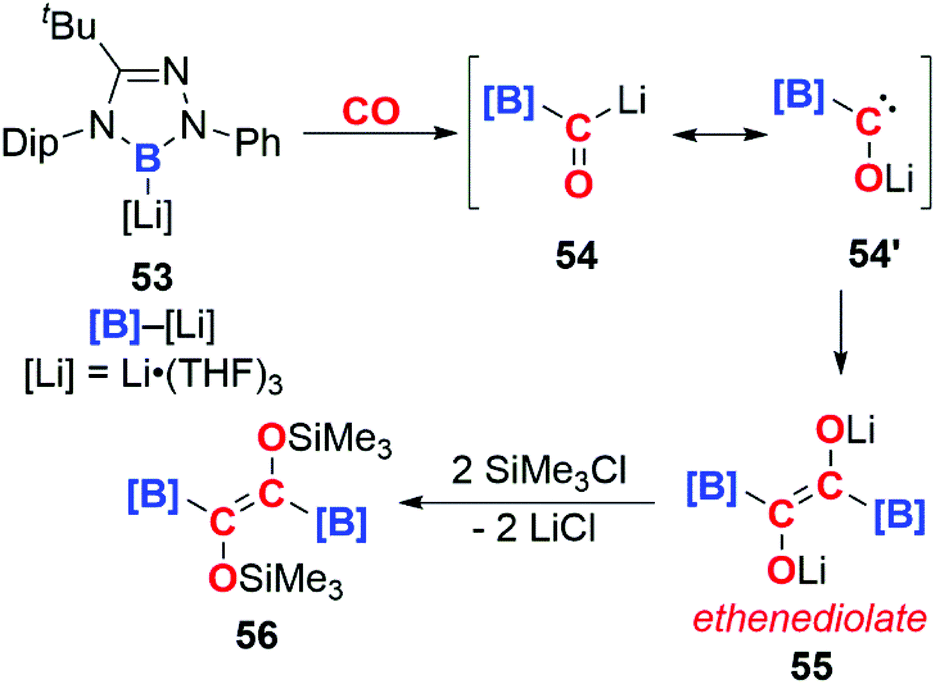 | ||
| Scheme 14 Formation of an ethenediolate from boryllithium 49. Dip = 2,6-di-iso-propyl-phenyl. | ||
In a similar manner, boryllithium 57 reacts with CO to form an unobserved ketene (58), which dimerises above −78 °C to form the isolable product 59 (Scheme 15).41
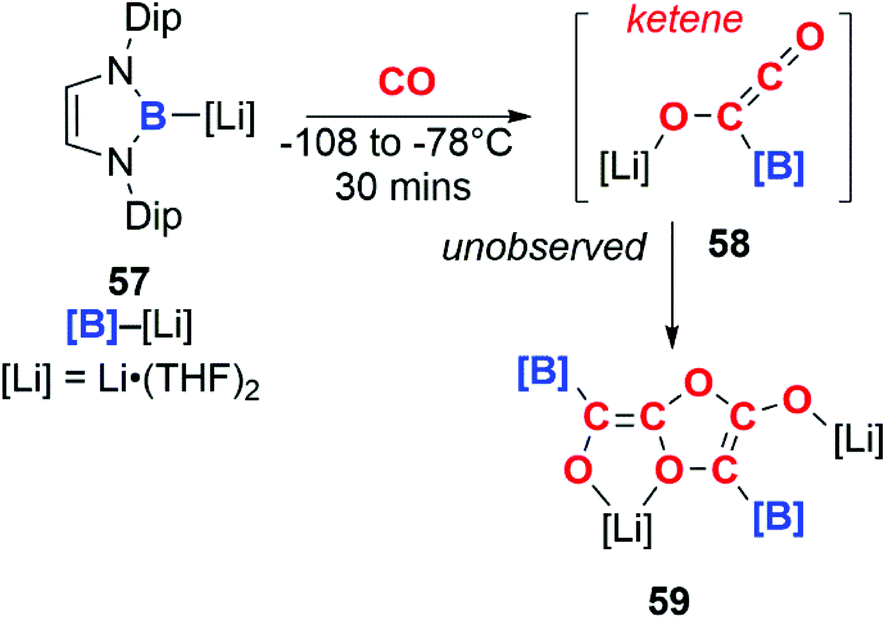 | ||
| Scheme 15 Tetramerisation of CO by a boryllithium. | ||
Incorporating SmCl3 into this reaction gave a completely different result (Scheme 16). Samarium(III) chloride (60) and boryllithium 57 react to yield samarium(III) boryl complex 61, which in the presence of CO can go on to form the bora-acyl samarium complex, 62. 62 is proposed to originate from the insertion of CO into the Sm–B bond of 61 and similar reactivity has been observed before.42 If the reaction with CO occurs in the presence of an excess of 57 both 58 and 62 can be generated in situ. These intermediates can combine to form a C3 carbon chain bearing both Sm and Li substituents, 63. Both isotopic labelling experiments and DFT calculations support the proposed mechanism. The tandem cooperative CO homologation strategy employed to access a C3 product (63), provides a novel approach towards the construction and elaboration of carbon chains.
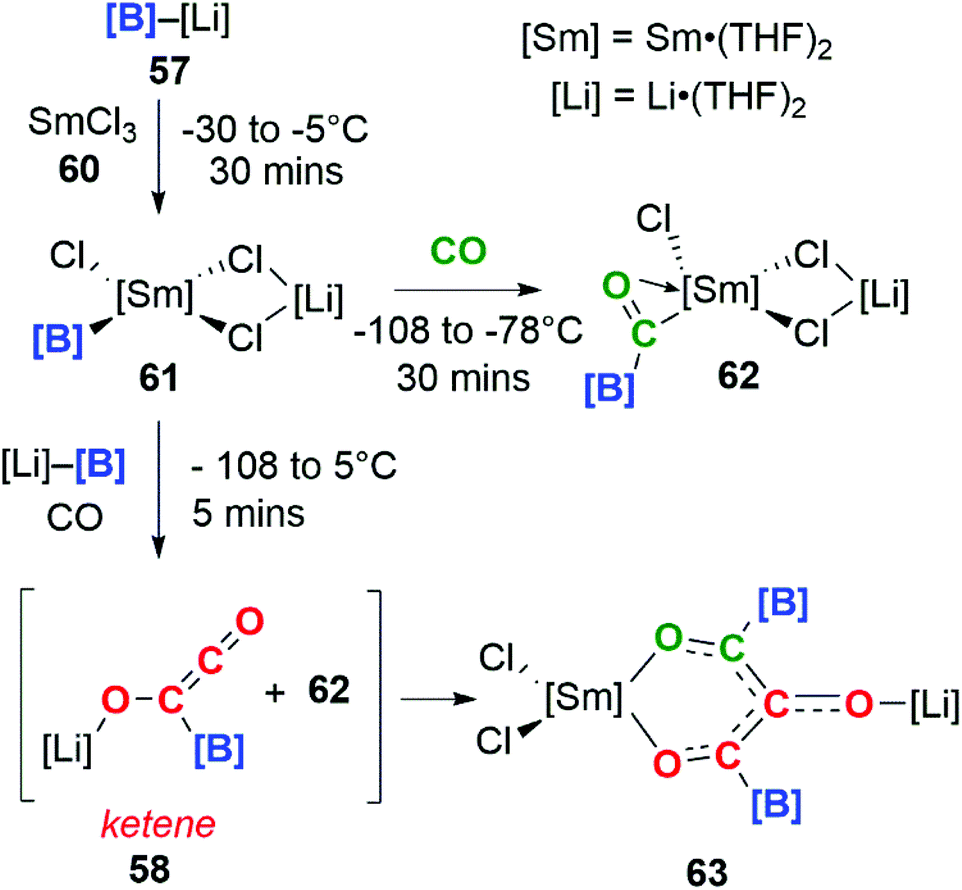 | ||
| Scheme 16 Trimerisation of CO by a boryllithium/SmCl3. | ||
The use of well-defined s-block metal complexes to reductively couple CO was first reported in 2019 by Jones, Maron and co-workers (Scheme 17a).43,44 Reaction of a magnesium(I) dimer 64a–d with one equivalent of a donor ligand (L = DMAP) yields an asymmetric derivative of the parent dimer 65a–d (Scheme 17a and b). Remarkably, while the parent dimer does not react with CO, the ligated dimer reduces CO and forms carbon chains. Sterically bulkier magnesium centres (64a–b) yield the deltate {C3O3}2−, while comparatively less crowded magnesium centres (64c–d) yield a bridging ethenediolate {C2O2}2. The steric profile of the magnesium centres controlling the degree of CO homologation parallels observations by Cloke and co-workers when studying uranocene reductants.8–12
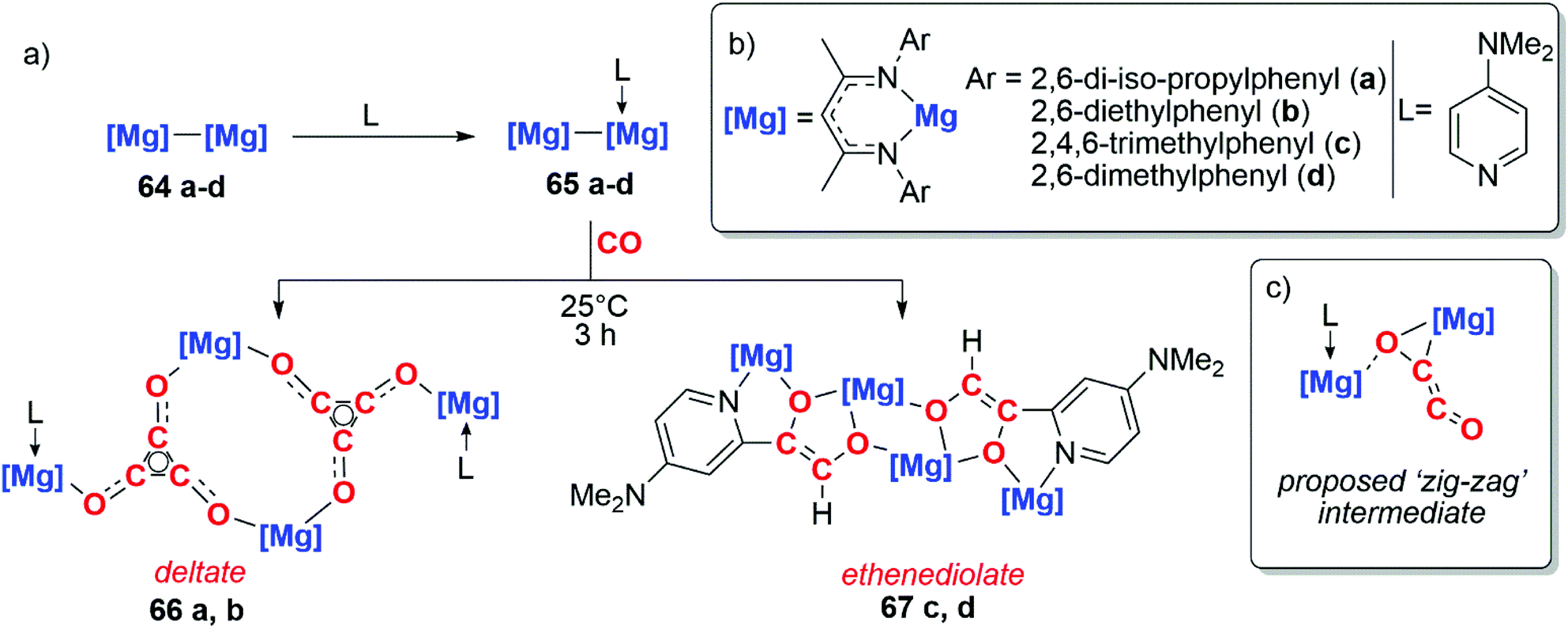 | ||
| Scheme 17 (a) Formation of the deltate and ethenediolate products from activated magnesium(I) dimers. (b) Structure of Mg–Mg compounds. (c) Proposed zig-zag intermediate. | ||
DFT calculations were employed to investigate the mechanism of chain growth. Insertion of CO into the Mg–Mg bond followed by insertion of a second equivalent of CO into the Mg–C bond results in formation of a ‘zig-zag’ intermediate (Scheme 17c).43,44 This ‘zig-zag’ intermediate bears a striking resemblance to the proposed intermediate in the ethynediolate formation with mixed uranocene complexes reported by Cloke and co-workers (vide supra). A barrierless insertion of a third equivalent of CO into the Mg–C bond results in formation of the deltate for compounds 66a–b. From the same intermediate, insertion into an ortho C–H bond of the DMAP ligand followed by isomerisation results in the ethenediolate products 67c–d.
In 2015, Scheschkewitz and co-workers reported the reductive coupling of CO by the disilenide compound 68. 68 contains a SiSi bond and the Si centres act in a cooperative manner in the reduction of CO (Scheme 18a).45 The complete cleavage of the CO triple bond is achieved resulting in the formation of a silanone dimer with pendant alkynylsilyl and silirene groups, 69. Two CO units are coupled per SiSi, with the SiSi bond providing 4e− in the reaction. The mechanism is proposed to proceed through a key intermediate 70 in which two CO units form a ketenylidene moiety along with concomitant cleavage of a CO triple bond to form a bridging oxo ligand. 70 is proposed to undergo a ligand metathesis to form two equiv. of silylene 71 and ethynolate 72. The extruded silylene units then recombine with 72 by (2 + 1) cycloaddition to the CC bond and oxidative addition to the C–OLi bond resulting in formation of the final product 69.
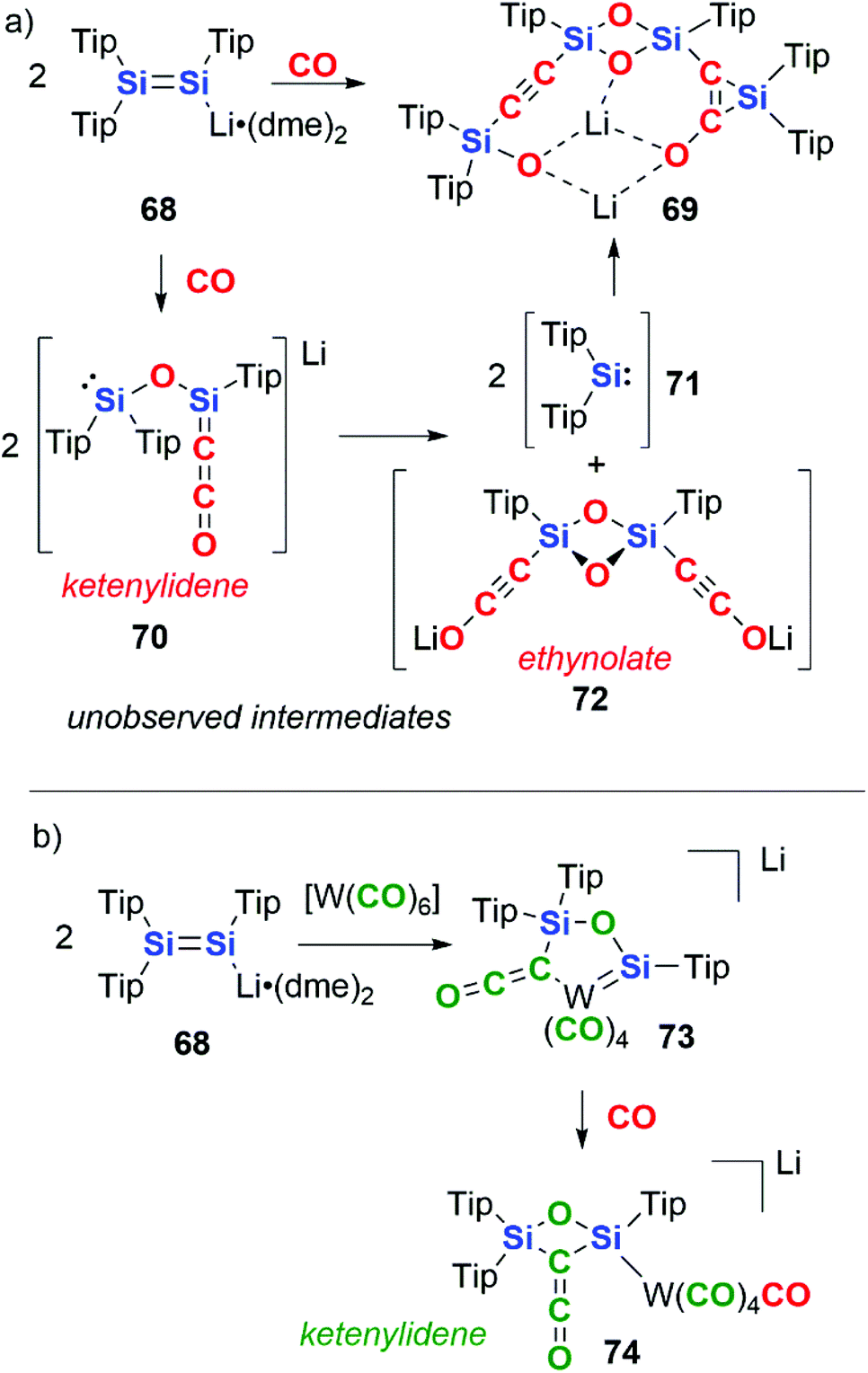 | ||
| Scheme 18 Disilenide reactivity with (a) CO and (b) tungsten hexacarbonyl. Tip = 2,4,6-tri-iso-propylphenyl, dme = 1,2-dimethoxyethane. | ||
Altering the source of CO to a transition metal carbonyl complex changes the observed reactivity (Scheme 18b).45 Hence, reaction of 68 with [W(CO)6], allows for the isolation of a ketenylidene-ligated group 6 metal–silylene complex, 73. Reaction of 73 with a further equivalent of CO gas liberates the ketenylidene ligand from the transition metal, resulting in the formation of a 2,4-disilaoxetane 74. In these reactions, the carbon units that make up the chain can be derived from both atmospheric CO as well as transition-metal bound CO units.
In 2013, Braunschweig and co-workers documented the reductive coupling of four CO units at BB bond through reaction of CO with diboryne 75 (Scheme 19). The reaction is unusual due to the head-to-tail coupling of two CO units to form an ethenediolate species, 76. Most examples of CO homologation involve head-to-head dimerization and C–C bond formation rather than C–O bond formation. The stoichiometric addition of 1 equiv. of CO to 75 affords a coordination compound 77 in which CO act as a ligand. The CO ligand is coordinated to the two boron centres in a semi-bridging fashion. The long CO bond distance (1.249(2) Å) provides structural evidence for back-donation from the B2 unit to the π* orbital of CO.46 A number of related examples of this type of interaction of CO with boron sites are known.47 The N-heterocyclic carbene (NHC) ligands on the diboryne unit play an important role in determining the reactivity toward CO.48 The stabilisation of the singlet ground state of the B2 permits back-donation from the triple bond in B2 into the π* antibonding of CO, allowing both binding and activation of CO akin to transition-metal reactivity.49 Addition of excess CO to 77 affords the homologation product 76.
 | ||
| Scheme 19 CO reduction and homologation at a diboryne centre. Dip = 2,6-di-iso-propyl-phenyl. Compound 77 has been represented as reported in ref. 46. | ||
Summary and perspective
Cooperative strategies have emerged as a key feature in CO homologation. Through the use of two or more metal centres, the discrete coordination, reduction, and finally C–C bond formation steps can be achieved. In many cases either proposed transition states or intermediates involve the cooperative action of two or more reactive sites with CO. This approach also provides a coordination framework made of multiple sites which can support the growing carbon chain.The mechanism of any chain growth sequence using CO (or CO2) as a C1 building block to form Cn chains can be conceptualised in terms of initiation, propagation, and termination events. In the long term, the detailed understanding of each of these events may allow for the development of methods to convert CO and CO2 to higher-value products with greater selectivity and ultimately pave the way for catalytic methods.
The initial activation of CO often involves the formation of a metal–carbon bond. Formation of the metal–carbon bonds occurs most commonly through coordination of CO to a metal-centre. In a number of cases described herein, metal carbonyls have been computationally shown to be key intermediates. Isolable transition metal–carbonyl complexes have also been used as the CO source in homologation strategies. In the case of the latter approach, geometric and electronic restrictions on CO reactivity provide insight into the C–C bond formation process on heterogeneous F–T catalysts. Geometrically, a cis carbonyl relationship has been observed in numerous systems to be critical in transition-metal mediated chain-growth steps. Electronically, transition-metal bound CO units are shown to react with low-oxidation state compounds where atmospheric CO is inert.
Following the generation of a reactive organometallic intermediate from CO, the formation of C–C bonds occurs in the propagation stage, resulting in growth of a carbon chain. In almost all studies, this is studied computationally as these reactive intermediates are transient species and only the final chain-growth products are experimentally isolated and characterised.
For the model complexes described herein, the termination of chain-growth usually involves the formation of stable M–O bonds. Comparatively few studies have demonstrated the dissociation of the carbon chain from the metal sites. The onward reaction of the carbon chain and liberation from the metal fragments is essential for the development of catalytic processes. Trialkylsilyl halides have been used as hydrogen surrogates to liberate carbon chains from metal complexes. The hydrogenative coupling of CO-derived bis(alkylidyne) ligands reported by Suess and Peters is the only example to-date where dihydrogen is used to displace a transition-metal bound carbon chain. A detailed mechanistic understanding of the reactivity of dihydrogen with metal-bound carbon chains is currently unknown and also critical to both developing F–T models, as well as catalytic homogeneous chemistry.
As the field develops, we anticipate that more strategies will arise to couple CO units together at metal centres. The study of these numerous disparate systems will build a clearer understanding of CO reactivity. Ultimately this understanding has the potential to culminate in catalytic methodologies where the controlled coupling of CO to value-added products is achieved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Imperial College London for the award of a President's Scholarship, the Royal Society of Chemistry for the award of a Sir Geoff Wilkinson Dalton Poster Prize that led to this article, and the EPSRC (EP/S036628/1).References
- S. S. Ail and S. Dasappa, Renewable Sustainable Energy Rev., 2016, 58, 267–286 CrossRef CAS.
- R. B. Anderson, R. A. Friedel and H. H. Storch, J. Chem. Phys., 1951, 19, 313–319 CrossRef CAS.
- N. M. West, A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Coord. Chem. Rev., 2011, 255, 881–898 CrossRef CAS.
- J. Liebig, Liebigs Ann., 1834, 11, 182 Search PubMed.
- W. Büchner and E. Weiss, Helv. Chim. Acta, 1964, 47, 1415–1423 CrossRef.
- W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728–3730 CrossRef CAS.
- W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176–14184 CrossRef CAS PubMed.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829–831 CrossRef CAS PubMed.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602–9603 CrossRef CAS PubMed.
- A. S. Frey, F. G. N. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816–13817 CrossRef CAS PubMed.
- N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353–1362 CrossRef CAS.
- D. McKay, A. S. P. Frey, J. C. Green, F. G. N. Cloke and L. Maron, Chem. Commun., 2012, 48, 4118–4120 RSC.
- J. Parry, E. Carmona, S. Coles and M. Hursthouse, J. Am. Chem. Soc., 1995, 117, 2649–2650 CrossRef CAS.
- W. J. Evans, S. A. Kozimor, G. W. Nyce and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 13831–13835 CrossRef CAS PubMed.
- P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2010, 2, 77–79 RSC.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed.
- H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757 CrossRef PubMed.
- M. A. Pearsall and R. West, J. Am. Chem. Soc., 1988, 110, 7228–7229 CrossRef CAS.
- R. Becerra and R. Walsh, J. Am. Chem. Soc., 2000, 122, 3246–3247 CrossRef CAS.
- H. Bornemann and W. Sander, J. Organomet. Chem., 2002, 641, 156–164 CrossRef CAS.
- C. Ganesamoorthy, J. Schoening, C. Wölper, L. Song, P. R. Schreiner and S. Schulz, Nat. Chem., 2020 DOI:10.1038/s41557-020-0456-x.
- Y. Wang, A. Kostenko, T. J. Hadlington, M.-P. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2019, 141, 626–634 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, T. Szilvási, A. Ruzicka and M. Driess, Chem. Commun., 2020, 56, 747–750 RSC.
- A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- D. H. Berry, J. E. Bercaw, A. J. Jircitano and K. B. Mertes, J. Am. Chem. Soc., 1982, 104, 4712–4715 CrossRef CAS.
- P. A. Bianconi, I. D. Williams, M. P. Engeler and S. J. Lippard, J. Am. Chem. Soc., 1986, 108, 311–313 CrossRef CAS.
- P. A. Bianconi, R. N. Vrtis, C. Pulla Rao, I. D. Williams, M. P. Engeler and S. J. Lippard, Organometallics, 1987, 6, 1968–1977 CrossRef CAS.
- R. N. Vrtis and S. J. Lippard, Isr. J. Chem., 1990, 30, 331–341 CrossRef CAS.
- R. N. Vrtis, C. P. Rao, S. Warner and S. J. Lippard, J. Am. Chem. Soc., 1988, 110, 2669–2670 CrossRef CAS.
- F. Calderazzo, U. Englert, A. Guarini, F. Marchetti, G. Pampaloni and A. Segre, Angew. Chem., Int. Ed. Engl., 1994, 33, 1188–1189 CrossRef.
- F. Calderazzo, U. Englert, A. Guarini, F. Marchetti, G. Pampaloni, A. Segre and G. Tripepi, Chem. – Eur. J., 1996, 2, 412–419 CrossRef CAS.
- D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12580–12583 CrossRef CAS PubMed.
- J. A. Buss and T. Agapie, Nature, 2016, 529, 72–75 CrossRef CAS PubMed.
- J. A. Buss and T. Agapie, J. Am. Chem. Soc., 2016, 138, 16466–16477 CrossRef CAS PubMed.
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614–13617 CrossRef CAS PubMed.
- R. Yadav, T. Simler, M. T. Gamer, R. Köppe and P. W. Roesky, Chem. Commun., 2019, 55, 5765–5768 RSC.
- B. B. Wayland, A. E. Sherry and V. L. Coffin, J. Chem. Soc., Chem. Commun., 1989, 662–663 RSC.
- W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS PubMed.
- B. Wang, G. Luo, M. Nishiura, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16967–16973 CrossRef CAS PubMed.
- B. Wang, X. Kang, M. Nishiura, Y. Luo and Z. Hou, Chem. Sci., 2015, 7, 803–809 RSC.
- K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS PubMed.
- K. Yuvaraj, I. Douair, D. D. L. Jones, L. Maron and C. Jones, Chem. Sci., 2020, 11, 3516–3522 RSC.
- M. Majumdar, I. Omlor, C. B. Yildiz, A. Azizoglu, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2015, 54, 8746–8750 CrossRef CAS PubMed.
- H. Braunschweig, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer, I. Krummenacher, J. Mies, A. K. Phukan and A. Vargas, Nat. Chem., 2013, 5, 1025–1028 CrossRef CAS.
- H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS.
- N. Holzmann, M. Hermann and G. Frenking, Chem. Sci., 2015, 6, 4089–4094 RSC.
- H. Zhang, Z. Cao, W. Wu and Y. Mo, Angew. Chem., Int. Ed., 2018, 57, 13076–13081 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |