
Dual switchable molecular tweezers incorporating anisotropic MnIII–salphen complexes†
 ‡a
Olivier
Cador,
c
Fabrice
Pointillart,
c
Wolfgang
Wernsdorfer,
d
Valérie
Marvaud,
a
Bernold
Hasenknopf
a
and
Guillaume
Vives
*a
‡a
Olivier
Cador,
c
Fabrice
Pointillart,
c
Wolfgang
Wernsdorfer,
d
Valérie
Marvaud,
a
Bernold
Hasenknopf
a
and
Guillaume
Vives
*a
Abstract
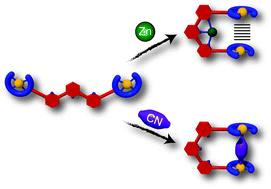
An alternative strategy for the synthesis of terpyridine based switchable molecular tweezers has been developed to incorporate anisotropic Mn(III)–salphen complexes. The free ligand was synthesized using a building block strategy based on Sonogashira coupling reactions and was then selectively metalated with manganese in a last step. The conformation of the tweezers was switched from an open ‘W’ shaped form to a closed ‘U’ form by Zn(II) coordination to the terpyridine unit bringing the two Mn–salphen moieties in close spatial proximity as confirmed by X-ray crystallography. An alternate switching mechanism was observed by the intercalation of a bridging cyanide ligand between the two Mn–salphen moieties that resulted in the closing of the tweezers. These dual stimuli are attractive for achieving multiple controls of the mechanical motion of the tweezers. A crystallographic structure of unexpected partially oxidized closed tweezers was also obtained. One of the two Mn–salphen moieties underwent a ligand-centered oxidation of an imino to an amido group allowing an intramolecular Mn–Oamide–Mn linkage. The magnetic properties of the manganese(III) dimers were investigated to evaluate the magnetic exchange interaction and analyze the single molecule magnet behavior.
 Please wait while we load your content...
Please wait while we load your content...

