
Transmembrane Cu(i) P-type ATPase pumps are electrogenic uniporters†
 a
Sameera
Abeyrathna,
a
M. Thomas
Morgan,
bc
Christoph J.
Fahrni
bc
and
Gabriele
Meloni
*a
a
Sameera
Abeyrathna,
a
M. Thomas
Morgan,
bc
Christoph J.
Fahrni
bc
and
Gabriele
Meloni
*a
Abstract
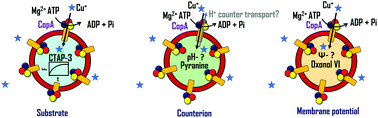
Cu(I) P-type ATPases are transmembrane primary active ion pumps that catalyze the extrusion of copper ions across cellular membranes. Their activity is critical in controlling copper levels in all kingdoms of life. Biochemical and structural characterization established the structural framework by which Cu-pumps perform their function. However, the details of the overall mechanism of transport (uniporter vs. cotransporter) and electrogenicity still remain elusive. In this work, we developed a platform to reconstitute the model Cu(I)-pump from E. coli (EcCopA) in artificial lipid bilayer small unilamellar vesicles (SUVs) to quantitatively characterize the metal substrate, putative counter-ions and charge translocation. By encapsulating in the liposome lumen fluorescence detector probes (CTAP-3, pyranine and oxonol VI) responsive to diverse stimuli (Cu(I), pH and membrane potential), we correlated substrate, secondary-ion translocation and charge movement events in EcCopA proteoliposomes. This platform centered on multiple fluorescence reporters allowed study of the mechanism and translocation kinetic parameters in real-time for wild-type EcCopA and inactive mutants. The maximal initial Cu(I) transport rate of 165 nmol Cu(I) mg−1 min−1 and KM, Cu(I) = 0.15 ± 0.07 μM was determined with this analysis. We reveal that Cu(I) pumps are primary-active uniporters and electrogenic. The Cu(I) translocation cycle does not require proton counter-transport resulting in electrogenic generation of transmembrane potential upon translocation of one Cu(I) per ATP hydrolysis cycle. Thus, mechanistic differences between Cu(I) pumps and other better characterized P-type ATPases are discussed. The platform opens the venue to study translocation events and mechanisms of transport in other transition metal P-type ATPase pumps.
- This article is part of the themed collection: New Talent: Americas
 Please wait while we load your content...
Please wait while we load your content...

