
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Y4Be33Pt16 – a non-centrosymmetric cage superconductor with multi-centre bonding in the framework†
Alfred
Amon‡
 ,
Eteri
Svanidze
,
Yurii
Prots
,
Michael
Nicklas
,
Ulrich
Burkhardt
,
Alim
Ormeci
,
Andreas
Leithe-Jasper
* and
Yuri
Grin
,
Eteri
Svanidze
,
Yurii
Prots
,
Michael
Nicklas
,
Ulrich
Burkhardt
,
Alim
Ormeci
,
Andreas
Leithe-Jasper
* and
Yuri
Grin
Max-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Straße 40, Dresden 01187, Germany
First published on 25th June 2020
Abstract
The new ternary compound Y4Be33Pt16 was prepared from elements by arc melting, and its crystal structure was determined from single-crystal X-ray diffraction data (space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d, a = 13.4849(3) Å). The material is the first representative of a new structure type of complex intermetallic compounds and reveals a cage-like crystal structure. Analysis of chemical bonding by means of the electron localizabilty approach indicates ionic interaction of yttrium with the rest of the crystal structure, characteristic for cage compounds, in particular for clathrates. In contrast to the mostly two-centre bonding in the framework of clathrates, the new compound is characterized by a multi-centre interaction within the framework, caused by the demand of the valence electrons in the system. The non-centrosymmetric material enters the superconducting state at TC = 0.9 K.
3d, a = 13.4849(3) Å). The material is the first representative of a new structure type of complex intermetallic compounds and reveals a cage-like crystal structure. Analysis of chemical bonding by means of the electron localizabilty approach indicates ionic interaction of yttrium with the rest of the crystal structure, characteristic for cage compounds, in particular for clathrates. In contrast to the mostly two-centre bonding in the framework of clathrates, the new compound is characterized by a multi-centre interaction within the framework, caused by the demand of the valence electrons in the system. The non-centrosymmetric material enters the superconducting state at TC = 0.9 K.
Introduction
The diversity of intermetallic structures is fascinating. These compounds can have unit cells with one atom as in α-Po (Pearson symbol cP1) or with more than twenty thousand atoms as in Ta9063Al12827.56Cu1244.05 (Pearson symbol cF23134).1–3 The reasons for such complex behaviour are still under discussion. The compounds with crystal structures containing more than ca. 100 atoms in the unit cell are notated as complex metallic alloys (CMA) or complex intermetallic compounds.4 One of the characteristic features for CMA (but not for all) is the presence of different types of crystallographic disorder in crystal structures, which de facto means the violation of the translational symmetry in the strict sense. Recently, this was shown on examples of o-Al13Co4 (oP102-x)5 and ternary clathrate Ba7.81Ge40.67Au5.33 (cP54-x)6 by the combination of X-ray diffraction on single crystals with high-resolution transmission electron microscopy. One of the possible reasons for such complex behaviour may be the low valence electron concentration in these materials, which (i) does not allow to use traditional electron counting schemes for understanding the composition and the crystal structure, and (ii) leads to multi-centre atomic interactions in the structures, recently shown e.g. for Be21Pt5.7 Intermetallic compounds with complex crystal structures often show unusual physical behaviours. Physics and chemistry of CMA are discussed on the basis of the complexity of their crystal structures, whereby the latter is considered to be the origin of some specific behaviour of these substances. For instance, the intermetallic clathrates reveal unusual functionality in heat transport as a kind of phonon filters with the mean free path of phonons 10–100 nm.6,8,9From the position in the Periodic Table, beryllium contributes only two valence electrons for the formation of bonds, thus the compounds with high beryllium content should be a suitable field to search for new complex intermetallic materials. The recently described binary Be21Pt5 has a large unit cell with 416 atoms (cF416), but does not reveal any crystallographic disorder.7 The information about the ternary compounds of Be with other metals is rather scarce in the literature.10 Our initial phase diagram studies showed, that – despite the clear electronic demand – the R–Be–T systems are characterized by formation of several ternary compounds similar to the R–E–T systems with R being alkaline- or rare-earth metals, E – p-block elements, and T – late transition metals. In this work, we present crystal structure and bonding analysis of the new superconductor Y4Be33Pt16.
Results and discussion
The powder X-ray diffraction pattern of Y4Be33Pt16, shown in Fig. 1, was indexed using a cubic cell with a = 13.4849(3) Å. The structure was solved and refined from single-crystal diffraction data in the non-centrosymmetric space group I3d. The main crystallographic information is presented in Table 1, the atomic coordinates and isotropic atomic displacement parameters are shown in Table 2, the anisotropic displacement parameters for Y and Pt atoms as well as the interatomic distances are listed in ESI (Tables S1 and S2,† respectively).
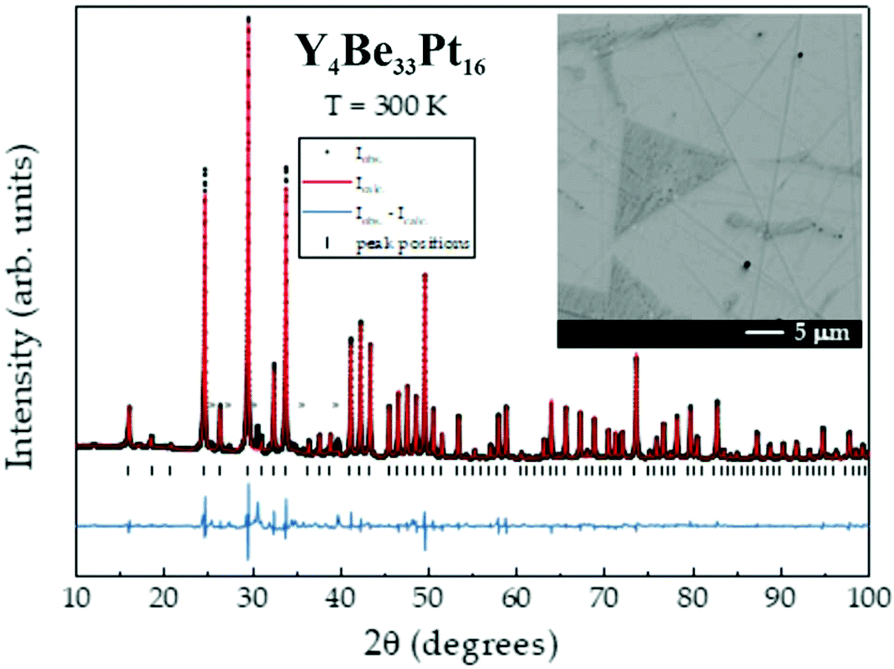 | ||
| Fig. 1 X-ray powder diffraction pattern for Y4Be33Pt16. Reflections of the minority phases (Be21Pt5, Y, and Pt) are marked by asterisks. Inset: Microstructure (material contrast image, SEM) shows the Y- and Pt-containing majority phase (light gray) and the Pt-poor minority phase (dark gray). | ||
| a Lattice parameter refined from the X-ray powder diffraction data (CuKα1 radiation). | |
|---|---|
| Crystal system | Cubic |
| Composition | Y4Be33Pt16 |
| Space group |
I3d |
| Pearson symbol | cI212 |
| Formula units per u. c. Z | 4 |
| Unit cell parameter a | 13.4849(3) Åa |
| Unit cell volume V | 2452.1(2) Å3 |
| Calculated density | 10.22 g cm−3 |
| Radiation, wavelength | MoKa, 0.71073 Å |
| Absorption coefficient | 105.2 mm−1 |
| Diffraction system | Rigaku AFC7 |
| 2θmax | 86.2° |
| N(hkl) measured | 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 218 218 |
| N(hkl) unique | 1477 |
| N(hkl) observed | 1446 |
| R int; Rs | 0.045; 0.032 |
| Refined parameters | 28 |
| R F; wRF2 | 0.044; 0.088 |
| Weighting scheme | w i = [ln(Fobs,i4)]−1 |
| Extinction coefficient17 | 0.000062(9) |
| Flack parameter | −0.005(10) |
| Residual electron density max; min | +2.3; −1.9 e Å−3 |
| Atom | Site | x/a | y/b | z/c |
B
eq/isoa (Å−2) |
|---|---|---|---|---|---|
| a B eq = ⅓[B11 × a*2 × a2 + ⋯ + 2B23 × b* × c* × b × c × cos(α)]. | |||||
| Y | 16c | 0.8097(1) | x | x | 0.66(2) |
| Pt1 | 16c | 0.93652(4) | x | x | 0.594(6) |
| Pt2 | 48e | 0.48917(4) | 0.89865(4) | 0.77608(4) | 0.607(8) |
| Be1 | 12a | ⅝ | 0 | ¾ | 1.2(6) |
| Be2 | 24d | 0.914(3) | 0 | ¾ | 1.1(4) |
| Be3 | 48e | 0.056(2) | 0.989(2) | 0.664(2) | 1.1(3) |
| Be4 | 48e | 0.620(2) | 0.919(2) | 0.893(2) | 1.0(3) |
The atoms in the crystal structure of Y4Be33Pt16 reveal coordination polyhedrons characteristic for intermetallic compounds in the ternary systems of rare-earth and related metals as R component with transition metals and p-block elements. The yttrium atoms are located within a spacious polyhedron with 20 vertices, being very similar to that of Ca in the CaCu5 structure type and its several derivatives11,12 (Fig. 2, top panel). Interestingly, there are three additional Be1 ligands at the distance of 3.666 Å, which is much longer than other yttrium contacts (Table S2†) and – from distance analysis – these three do not necessarily belong to the first coordination sphere (cf. analysis of chemical bonding below). While the platinum atoms have environments derived from the Frank–Kasper polyhedrons with coordination numbers of CN(Pt1) = 14 and CN(Pt2) = 13, Be1 has a bisdisphenoid-like environment with eight closer neighbours (and additional four yttrium species at a much longer distance), all remaining beryllium species have 12 or 13 ligands with their coordination environments derived from an icosahedron (Fig. 2, top panel).
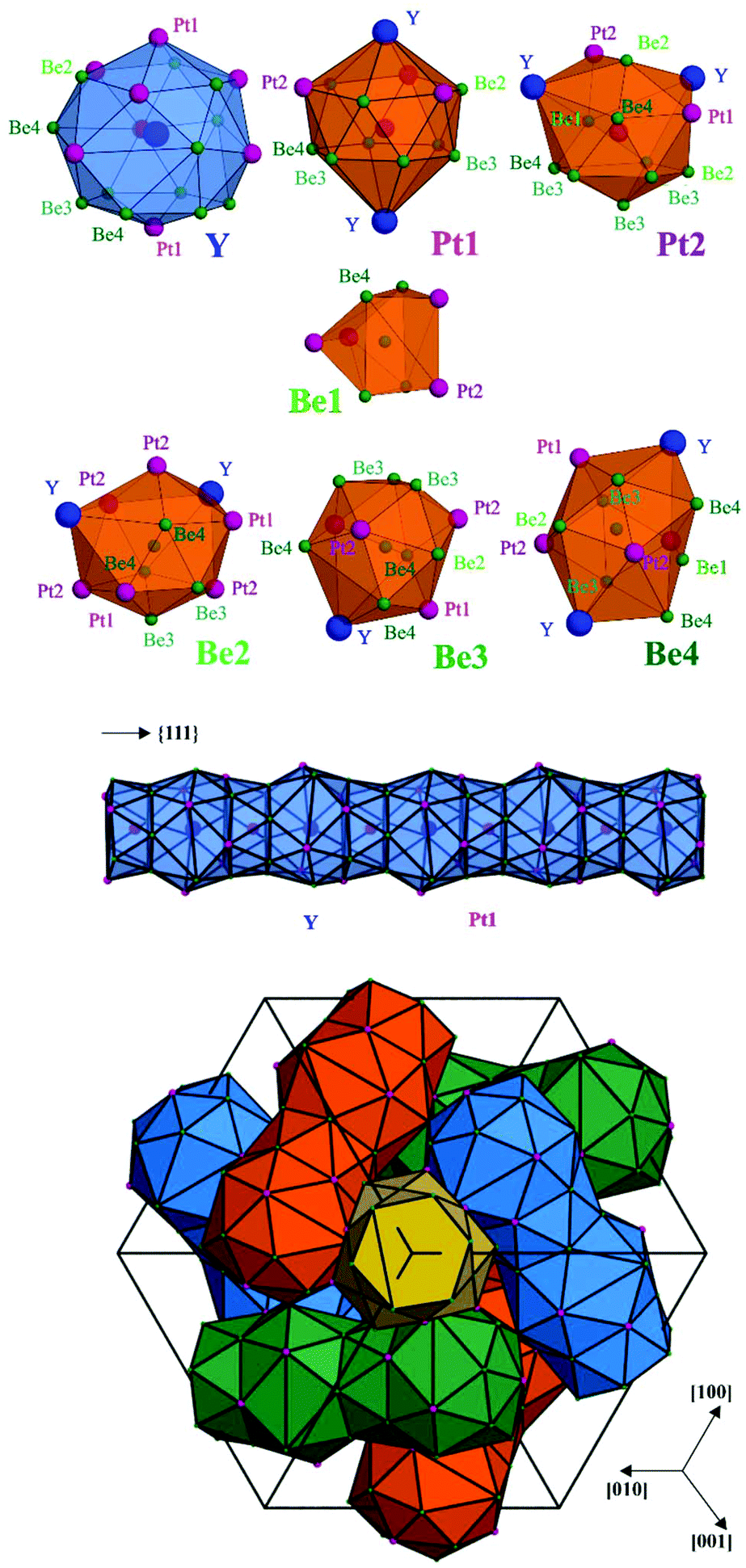 | ||
| Fig. 2 Crystal structure of Y4Be33Pt16 with the Y, Pt, and Be atoms represented by blue, pink, and green spheres, respectively: (top) coordination polyhedrons of atoms as obtained from analysis of interatomic distances; (middle) interpenetrating polyhedrons of Y and Pt1 along the three-fold axes; (bottom) rod packing of the interpenetrating polyhedron arrangements in the unit cell, the different colours visualize rods on different three-fold axes. | ||
The crystal structure of Y4Be33Pt16 can be visualized by considering the coordination environment of yttrium. Adjacent coordination polyhedrons of Y are vertex-sharing via the Pt1 position. This results in the formation of a linear rod-like arrangement of interpenetrating Y and Pt1 polyhedrons, as shown in Fig. 2 (middle panel), along the three-fold axes of the cubic unit cell ({111} directions). The resultant rod packing is presented in Fig. 2 (bottom panel) and was first described as crystallographic object in ref. 13. The existence of voids between the rods is due to an incomplete space filling. These voids are located at (⅞ 0 ¼) (Wyckoff site 12b) and (⅜ 0 ¼) (Wyckoff site 12a). The former are empty in Y4Be33Pt16, as they are too small to contain an atom. The voids located at (⅜ 0 ¼) having the shape of a bisdisphenoid are occupied by Be1.
Among the CaCu5 derivatives, the crystal structure of Y4Be33Pt16 is closely related to the ferromagnet Nd2Fe23B3 (space group I3d, a = 14.19 Å).14–16 For a comparison, the compositions can be written as Nd4Fe30(B2)3Fe16 and Y4Be30Be3Pt16. In the Nd2Fe23B3 structure, the Y site is occupied by Nd, all other positions beside Be1 – by iron. While the bisdisphenoid in Y4Be33Pt16 is occupied by a single Be1 atom (Wyckoff site 12a), the same coordination environment is occupied in Nd2Fe23B3 by a B2 pair with the centre at the 12a site (Fig. S1†). In this way, the B atoms belong to the coordination sphere of Nd with d(B1–Nd1) = 3.32 Å in Nd2Fe23B3, in contrast to Be1 in Y4Be33Pt16 which shows much longer distance of d(Be1–Y) = 3.666 Å.
The complex intermetallic structures with large cubic unit cell, in particular derivatives of the gamma-brass structural pattern, can often be interpreted from the crystallographic point of view as packings of nested polyhedrons.18 Recently, this was explained on example of Be21Pt5 by the formation of multi-atomic bonding interactions centred at the middle points of the nested polyhedrons.7 Interestingly, this approach does not work reasonably well for Y4Be33Pt16. This observation was the starting point for the analysis of chemical bonding in Y4Be33Pt16 employing the electron localizability approach.
As for most intermetallic beryllium compounds, a characteristic feature of Y4Be33Pt16 is the very low valence electron concentration (VEC) of ca. 1.4 e per atom, which is even lower than in the gamma-brass derivative Be21Pt5 (1.62 e per atom). An important factor stabilizing the latter structure was found to be the charge transfer from Be to Pt.7 In order to study the role of the ionic contributions to the bonding, the electron density in Y4Be33Pt16 was investigated applying the QTAIM (Quantum Theory of Atoms In Molecules19) approach. The zero-flux surfaces in the distribution of electron density define the shape of the atoms in the QTAIM representation. Integration of electron density within the atomic shapes yields their electronic populations and effective charges (Fig. 3). The yttrium atoms in Y4Be33Pt16 in the QTAIM representation have a shape close to a spherical one, which is characteristic for cage compounds, e.g. clathrates of Ba or Sr.9,20 Their effective charge of +1.70 is also characteristic for the earth-alkaline and rare-earth atoms in intermetallic clathrates.20,21 The shapes of the QTAIM atoms of platinum and beryllium are far from a sphere. They have closer to plane or even concave faces. While beryllium atoms – as expected from the electronegativity differences – reveal positive effective charges between +1.30 and +1.40, the platinum species have large negative charges of −2.72 and −3.31 for Pt1 and Pt2, respectively. This confirms strong charge transfer in the compound according to the scheme [Y+1.70]4[Be1+1.36]3[Be2+1.30]6[Be3+1.36]12[Be4+1.31]12[Pt1–2.72]4[Pt2−3.31]12.
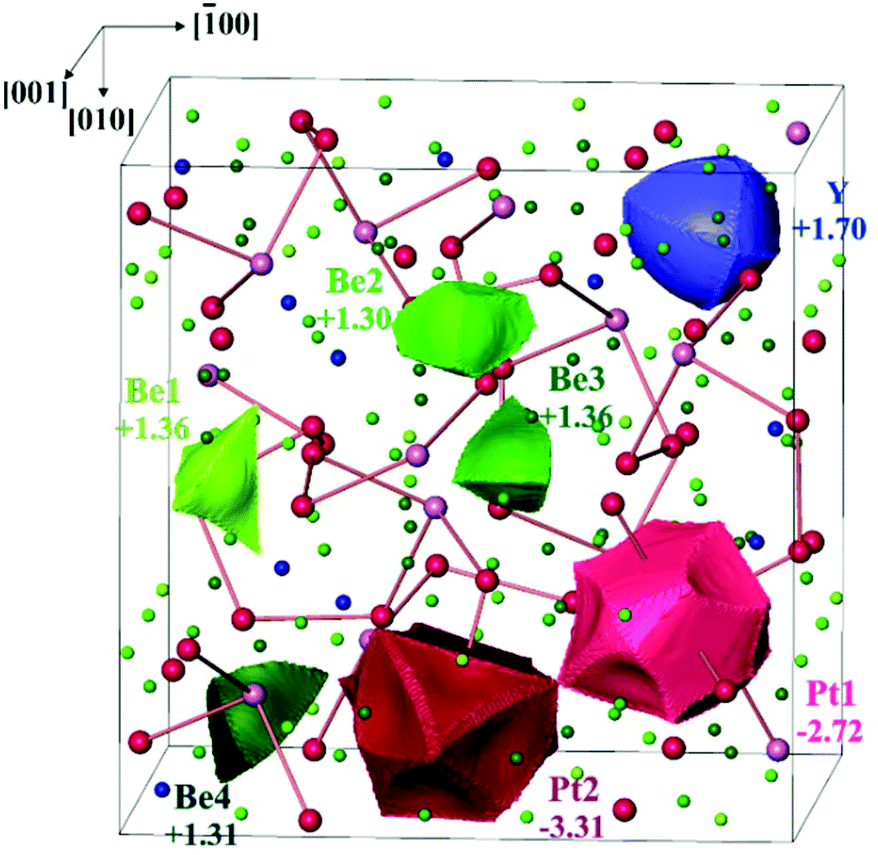 | ||
| Fig. 3 QTAIM atoms and their effective charges in Y4Be33Pt16. The Pt–Pt contacts with 2.82 Å < d(Pt–Pt) < 2.91 Å are shown to emphasize the volume representation. | ||
Further information about atomic interactions was obtained by applying the electron localizability approach. The distribution of the electron localizability indicator (ELI-D) in the vicinity of the yttrium nuclei is close to a spherical one, whereby the valence shell (4th) is absent. The light structuring of the penultimate shell indicates the participation of the electrons from this shell in the bonding. The intersection with the QTAIM yttrium atom reveals that the latter bears only the inner shells, confirming the ionic interaction of yttrium with the Be–Pt framework (Fig. 4, top panel). In the cage wall, there are two bonds involving Be2, Be4 and Pt2. Populations are 1.63 and 1.36 electrons, respectively. The interesting aspect is that Pt1 also contributes to them. Its contribution is comparable to those of Be2 and Be4. Hence, each attractor describes a four-atomic bond, two Pt and two Be. This reflects the characteristic multi-atomic interaction in the whole Be–Pt framework. The distribution of ELI-D in the (Be1–Pt2–Be4) triangle reveals one maximum located within the triangle. The intersection of the ELI-D basin of this maximum with the QTAIM shapes of the neighbouring atoms (Fig. 4, middle panel) shows the Be1, Be4, and Pt2 atoms contributing 0.11, 0.15, and 1.45 of the total population of 1.84 electrons for this basin. This confirms the mainly three-atomic (3a) character of the interaction in this part of the structure. The remaining 0.13 electrons originate from two other Pt2, one Be3, and even one yttrium neighbours, suggesting that even more atoms are participating in this multi-atomic bonding. The Pt atoms contribute the majority of the population for the bonding basins, which makes the multi-centre interactions strongly polar. The bonding analysis reveals that the Be1 atoms should be included into the coordination sphere of yttrium despite a large interatomic distance (cf. distance-based considerations above and comparison of the Y-polyhedrons in Fig. S1(e)†). The Y-centred cages – as obtained from the bonding analysis – within the three-dimensional Be–Pt framework fill-up the large part of the unit cell of Y4Be33Pt16 (Fig. 4, bottom panel). In summary, analysis of the chemical bonding classifies Y4Be33Pt16 as a cage compound, e.g. a material with its crystal structure in the form of a 3D framework with large cages. Ionic interaction of cationic yttrium species in the cages of the anionic Be–Pt framework is the bonding characteristic of this cage structure. In contrast to other representatives of cage compounds, e.g. intermetallic clathrates with mainly two-centre (non-polar or polar) bonds in the framework, the framework in Y8Be66Pt32 is formed by multi-atomic (polar) interaction. In agreement with the chemical bonding picture, the calculated electronic density of states of Y4Be33Pt16 is characterized by the intermixing of Pt and Be states in the whole energy range and shows three main regions (Fig. 5). The low-energy range (E < −6.7 eV) is formed mainly by the s states of Be and Pt with small contributions of the s states of yttrium, reflecting the bonding in the Be–Pt framework. The middle-energy region (−6.7 eV < E < −2.3 eV) is dominated by d states of platinum atoms. The region below the Fermi level (EF) contains contributions of all essential atomic states. Interestingly, a pseudo gap is found above the Fermi level (due to the band dispersion along Γ–H in the Brillouin zone, Fig. S2†), in contrast to the typical DOS distribution in cage compounds with two-centre bonds in the framework, where the pseudo gap appears typically close to EF.9 The presence of non-filled states agrees with the electron-deficient situation in this material.
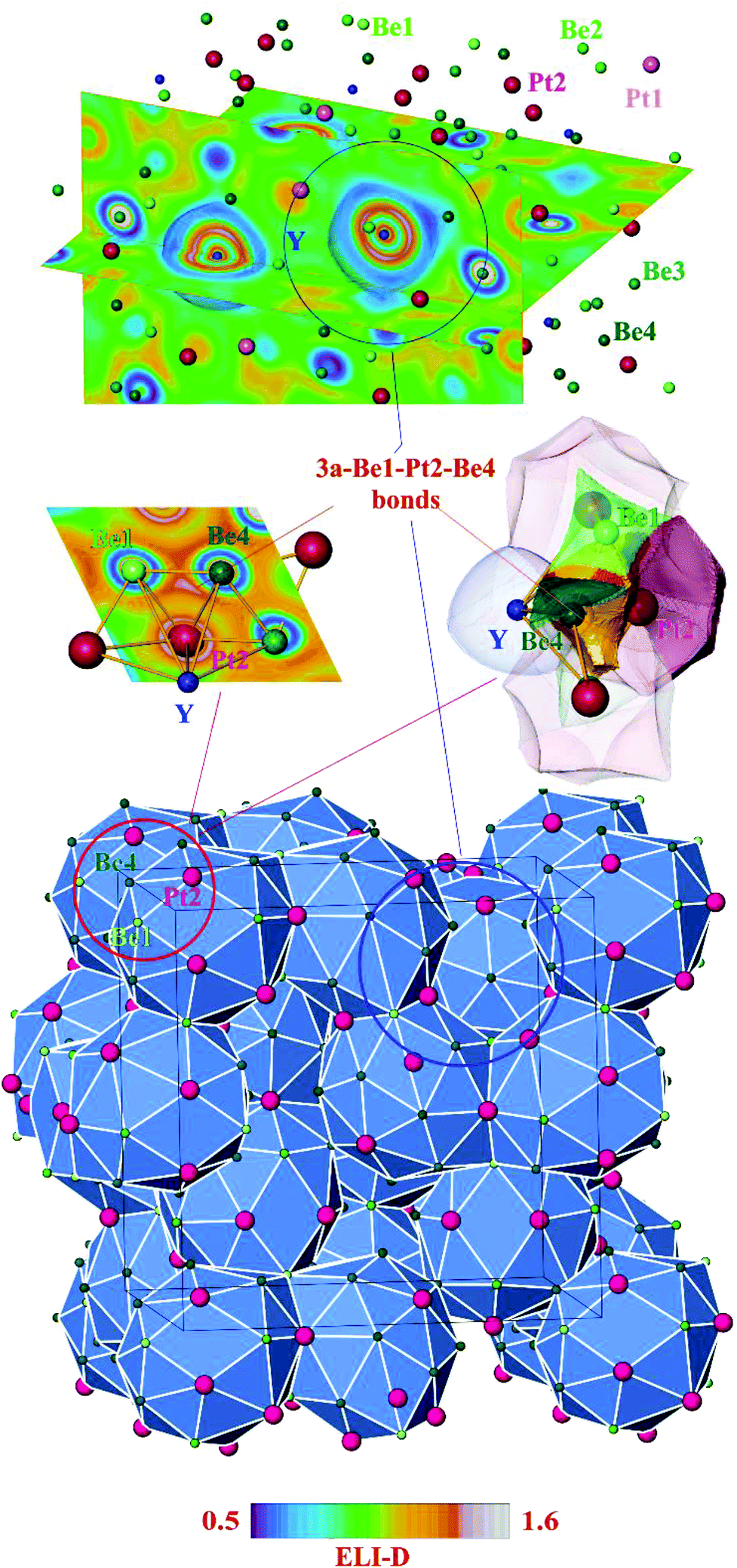 | ||
| Fig. 4 Atomic interactions in Y4Be33Pt16 by electron localizability approach: (top panel) distribution of the ELI-D in the plane crossing two neighbouring yttrium atoms; (middle left panel) distribution of ELI-D in one of the face planes of the coordination polyhedron of yttrium with the Pt2, Be1 and Be4 atoms; (middle right panel) bonding basin of the three-atomic interaction Pt2–Be1–Be4 (yellow bulk) intersected by the QTAIM atomic basins of the participating atoms (transparent), three main contributing atoms are labelled; (bottom panel) crystal structure of Y4Be33Pt16 as a packing of the coordination polyhedrons of Y (blue) as obtained from the bonding analysis. | ||
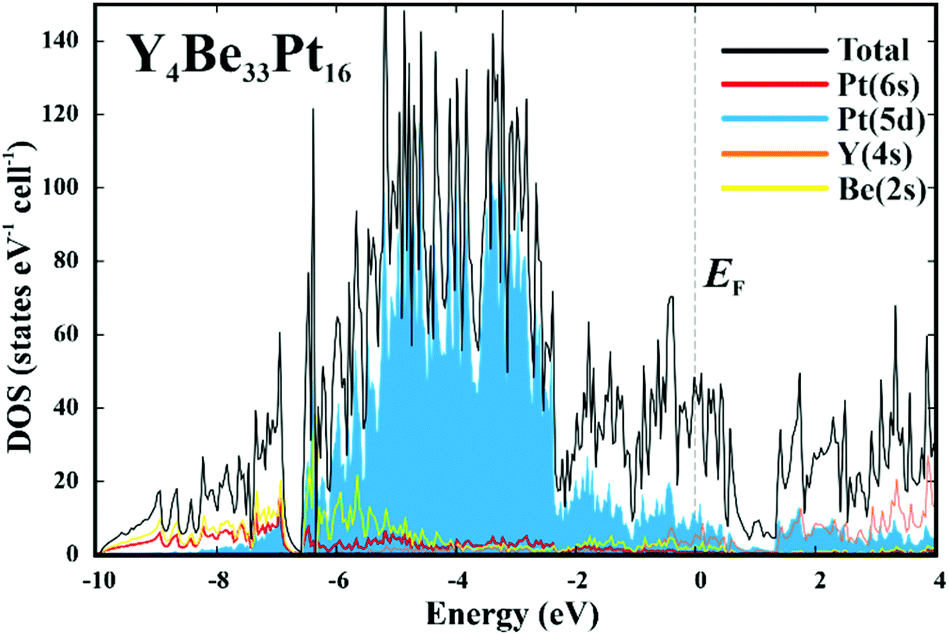 | ||
| Fig. 5 Calculated electronic density of states (DOS) for Y4Be33Pt16. The total DOS (black line) is shown together with the contributions of essential atomic states. | ||
The non-centrosymmetric compound Y4Be33Pt16 enters a superconducting state below TC = 0.9 K, as evidenced by the BCS-like anomaly observed in specific heat data, shown in Fig. 6. A fit to the specific heat data above the superconducting transition (dashed line), can be used to estimate the value of the normal state electronic specific heat coefficient γn = 18 mJ molY−1 K−2, as well as cubic term coefficient β = 4.5 mJ molY−1 K−3. Using the value of γn, it is then possible to estimate the effective mass m* from the following relation:22m* = (γnħ2kF2)/(π2nkB2) ≈ me, indicating lack of electron mass enhancement (equivalently, the theoretically computed value of DOS at the Fermi energy yields γcalc = 16.3 mJ molY−1 K−2). From the value of β, the Debye temperature θD can be calculated using θD = [(14π4NArkB)/(5β)]1/3 = 358 K.22 Consequently, the value of θD can then be used to estimate the strength of the electron–phonon coupling, employing the McMillan's formula.23 Typically, materials with λe–p → 1 are classified as strongly-coupled superconductors, while λe–p → 0.5 indicates weak coupling.23 For Y4Be33Pt16, 0.38 ≤ λe–p ≤ 0.47, indicating weakly-coupled superconductivity. Once the phonon contribution to the specific heat (βT3) has been subtracted from the specific heat data, an entropy conserving construction (not shown) yields the ratio ΔCe/γnTC ≈ 1.80, which is comparable to the BCS value of ΔCe/γnTC = 1.44, consistent with Y4Be33Pt16 being a weakly-coupled superconductor. As expected, the critical temperature TC is gradually suppressed by the application of a magnetic field, consistent with type-II superconductivity, allowing to construct the H−T phase diagram (inset of Fig. 6). A relatively small value of the upper critical field Hc2 = 0.44 T, extracted from a Ginzburg–Landau fit22 (dashed line), further confirms weak coupling in Y4Be33Pt16. The upper critical fields of non-centrosymmetric superconductors are often rather high (on the order of several or even dozens of Tesla), but Y4Be33Pt16 belongs to the smaller family of non-centrosymmetric superconductors with the upper critical fields on the order of few hundred Oersted. The other examples are AuBe (Hc2 = 335 Oe),24 Mo2Al3C (Hc2 = 50 Oe),25 or LaRhSi3 (Hc2 = 180 Oe).26
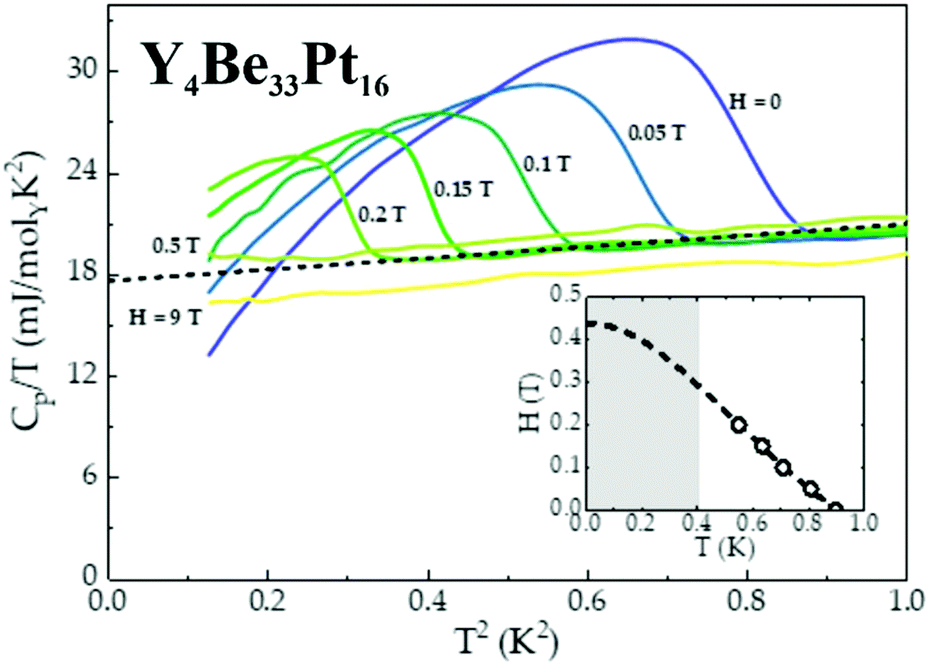 | ||
| Fig. 6 Superconducting properties of Y4Be33Pt16: specific heat data, scaled by temperature T, CP/T as a function of T2 in applied magnetic fields 0 ≤ H ≤ 9 T. The dashed line represents a fit from which the value of γn was extracted. Inset: H–T phase diagram for Y4Be33Pt16 with the values of Hc2(T) taken from specific heat data. | ||
The fact, that – despite of absence of inversion symmetry – no distinct deviations from BCS theory are observed, points towards the lack of strong correlation effects.27,28 However, a more comprehensive analysis of the superconducting state in Y4Be33Pt16 is currently underway. In particular, because several in-depth muon spin-rotation, relaxation, and resonance experiments performed on another Be-containing non-centrosymmetric superconductor BeAu24 revealed that not only type-I character,29 but also a multi-gap nature of superconductivity30 may exist in such unconventional systems.
Experimental
Synthetic procedures
All sample preparation was performed in a laboratory specialized for work with Be, inside a argon-atmosphere glove box system (MBraun, p(H2O/O2) < 0.1 ppm).Caution! Beryllium is considered (according to UN GHS criteria) a health risk and experiments require appropriate infra structure.
Polycrystalline samples around the composition Y4Be33Pt16 were synthesized by arc melting Y (pieces, Ames, >99.9%), Pt (foil, Chempur, >99.9%), and Be (sheet, Heraeus, >99.9%) in the 10:30:60 ratio. The samples did not exhibit any marked air or moisture sensitivity. Small amounts of impurities were found to be present in all samples. The minority phases are Be21Pt5 (superconductor with TC = 2.06 K (ref. 7)), Be5Pt (semiconductor31), as well as elemental Pt in the amount of <3%. Temperature treatment, as well as slight variation of the initial composition did not result in an improved sample quality.
Crystallography
Powder X-ray diffraction was performed on a Huber G670 Image plate Guinier camera with a Ge-monochromator (CuKα1 radiation, λ = 1.54056 Å). Phase identification was done using the WinXPow software.32 All crystallographic calculations were done with the WinCSD software package33 Single-crystal X-ray diffraction data have been collected on a Rigaku AFC7 diffractometer equipped with a Saturn 724 + CCD detector using MoKα radiation (λ = 0.71073 Å).Chemical analysis
Chemical composition of polished samples was studied using energy-dispersive X-ray spectroscopy (EDS) with an UltraDry EDS detector (ThermoFisher NSS7) attached to a Jeol JSM 6610 scanning electron microscope (SEM). The semi-quantitative analysis was performed with 25 keV acceleration voltage and ≈3 nA beam current.Physical properties measurements
Heat capacity was measured in the temperature range from 0.4 K to 10 K, in magnetic fields up to H = 9 T using a QD Physical Property Measurement System.Calculation procedures
Electronic structure calculations were performed by using the all-electron, full-potential local orbital (FPLO) method.34 All main results were obtained within the local density approximation (LDA) to the density functional theory through the Perdew–Wang parametrization for the exchange–correlation effects.35 Application of the generalized gradient approximation did not reveal essential differences in the electronic structure below and in vicinity of the Fermi level. Chemical bonding analysis in position space was performed within the approach of combined topological analysis of electron density (ED) and electron localizability indicator (ELI). The former type of analysis forms the basis of the quantum theory of atoms in molecules (QTAIM).19 ELI was calculated in the ELI-D representation36–38 by a module implemented in the FPLO package.39 Topological analysis of the ED and the ELI-D were carried out by the program DGrid.40Conclusions
The ternary compound Y4Be33Pt16 is the first representative of a new prototype of intermetallic cage compounds with an unusual combination of structural and bonding features. The cubic crystal structure with 212 atoms in the unit cell is – in contrast to many other complex intermetallic structures – completely ordered and does not show violation of the translational symmetry, characteristic for CMA materials. Analysis of chemical bonding in position space reveals mainly ionic interaction of Y with the remaining parts of the structure, indicating an analogy to intermetallic clathrates and classifying the new atomic arrangement as a cage-like one. In contrast to other cage materials – clathrates – with mainly two-centre bonding in the framework, the framework atoms in Y4Be33Pt16 participate only in multi-centre interactions. On top of the interesting structure-bonding relationship, the new compound is a non-centrosymmetric BCS-type superconductor with TC = 0.9 K.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors acknowledge W. Schnelle for valuable discussions, H. Borrmann and S. Hückmann for assistance in powder diffraction experiments, K. Vanatko and R. Gellrich for support of preparative work. Open Access funding provided by the Max Planck Society.References
- R. J. de Sando and R. C. Lange, J. Inorg. Nucl. Chem., 1966, 28, 1837 CrossRef CAS.
- T. Weber, J. Dshemuchadse, M. Kobas, M. Conrad, B. Harbrecht and W. Steurer, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 308 CrossRef CAS PubMed.
- Yu. Grin, in Comprehensive Inorganic Chemistry II, Elsevier, Oxford, 2013, vol. 2, p. 359 Search PubMed.
- Complex Metallic Alloys Fundamentals and Applications, ed. J.-M. Dubois and E. Belin-Ferré, Wiley-VCH, Weinheim, 2011 Search PubMed.
- P. Simon, I. Zelenina, R. Ramlau, W. Carrillo-Cabrera, U. Burkhardt, H. Borrmann, R. Cardoso Gil, M. Feuerbacher, P. Gille and Yu. Grin, J. Alloys Compd., 2020, 820, 153363 CrossRef CAS.
- P. F. Lory, S. Pailhès, V. M. Giordano, H. Euchner, H. D. Nguyen, R. Ramlau, H. Borrmann, M. Schmidt, M. Baitinger, M. Ikeda, P. Tomeš, M. Mihalkovič, C. Allio, M. R. Johnson, H. Schober, Y. Sidis, F. Boudarot, L. P. Regnault, J. Ollivier, S. Paschen, Yu. Grin and M. de Boissieu, Nat. Commun., 2017, 8, 1 CrossRef CAS PubMed.
- A. Amon, A. Ormeci, M. Bobnar, L. G. Akselrud, M. Avdeev, R. Gumeniuk, U. Burkhardt, Yu. Prots, C. Hennig, A. Leithe-Jasper and Yu. Grin, Acc. Chem. Res., 2018, 51, 214 CrossRef CAS PubMed.
- H. Euchner, S. Pailhès, L. T. Nguyen, W. Assmus, F. Ritter, A. Haghighirad, Yu. Grin, S. Paschen and M. de Boissieu, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 224303 CrossRef.
- H. Zhang, H. Borrmann, N. Oeschler, C. Candolfi, W. Schnelle, M. Schmidt, U. Burkhardt, M. Baitinger, J.-T. Zhao and Yu. Grin, Inorg. Chem., 2011, 50, 1250 CrossRef CAS PubMed.
- O. Janka and R. Pöttgen, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 143 Search PubMed.
- W. B. Pearson, The crystal Chemistry and Physics of metals and Alloys, Wiley-Interscience, N.Y., 1972, p. 643 Search PubMed.
- P. I. Krypiakevich, Structure Types of Intermetallic Compounds, Nauka, Moscow, 1977 (in Russian) Search PubMed.
- M. O'Keeffe and S. Andersson, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1977, 33, 914 CrossRef.
- K. H. Buschow, D. B. de Mooij and H. M. van Noort, J. Less-Common Met., 1986, 125, 135 CrossRef CAS.
- D. de Mooij and K. H. J. Buschow, Philips J. Res., 1986, 41, 400 CAS.
- C. Gou, Z. X. Cheng, D. F. Chen, S. W. Niu, Q. W. Yan, P. L. Zhang, B. G. Shen and L. Y. Yang, J. Magn. Magn. Mater., 1993, 128, 26 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- B. Chabot, K. Cenzual and E. Parthé, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1981, 37, 6 CrossRef.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Clarendon Press, 1994 Search PubMed.
- J. Hübner, Yu. Prots, W. Schnelle, M. Bobnar, M. König, M. Baitinger, P. Simon, W. Carrillo-Cabrera, A. Ormeci, E. Svanidze, Yu. Grin and U. Schwarz, Chem. – Eur. J., 2020, 26, 756 CrossRef.
- A. Ormeci and Yu. Grin, J. Thermoelectr., 2015, 6, 16 Search PubMed.
- M. Tinkham, Introduction to Superconductivity, McGraw-Hill, New York, 1996 Search PubMed.
- W. L. McMillan, Phys. Rev., 1968, 167, 331 CrossRef CAS.
- A. Amon, E. Svanidze, R. Cardoso-Gil, M. N. Wilson, H. Rosner, M. Bobnar, W. Schnelle, J. W. Lynn, R. Gumeniuk, C. Hennig, G. M. Luke, H. Borrmann, A. Leithe-Jasper and Yu. Grin, Phys. Rev. B, 2018, 97, 014501 CrossRef CAS.
- A. B. Karki, Y. M. Xiong, I. Vekhter, D. Browne, P. W. Adams, D. P. Young, K. R. Thomas, J. Y. Chan, H. Kim and R. Prozorow, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 064512 CrossRef.
- N. Kimura, H. Ogi, K. Satoh, G. Ohsaki, K. Saitoh, H. Iida and H. Aoki, JPS Conf. Proc., 2014, 3, 015011 Search PubMed.
- Non-centrosymmetric superconductors, Lecture Notes in Physics, ed. E. Bauer and M. Sigrist, 2012, vol. 847, p. 3 Search PubMed.
- F. Kneidinger, E. Bauer, I. Zeiringer, P. Rogl, C. Blaas-Schenner, D. Reith and R. Podloucky, Phys. C, 2015, 514, 388 CrossRef CAS.
- J. Beare, M. Nugent, M. N. Wilson, Y. Cai, T. J. S. Munsie, A. Amon, A. Leithe-Jasper, Z. Gong, S. L. Guo, Z. Guguchia, Yu. Grin, Y. J. Uemura, E. Svanidze and G. M. Luke, Phys. Rev. B, 2019, 99, 134510 CrossRef CAS.
- R. Khasanov, R. Gupta, D. Das, A. Amon, A. Leithe-Jasper and E. Svanidze, Phys. Rev. Res., 2020, 2, 023142 CrossRef.
- A. Amon, E. Svanidze, A. Ormeci, M. König, D. Kasinathan, D. Takegami, Yu. Prots, Y. F. Liao, K. D. Tsuei, L. H. Tjeng, A. Leithe-Jasper and Yu. Grin, Angew. Chem., Int. Ed., 2019, 58, 15928 CrossRef CAS PubMed.
- STOE Powder Diffraction Software WinXPow (version 2), STOE and Cie GmbH, Darmstadt, 2001 Search PubMed.
- L. Akselrud and Yu. Grin, J. Appl. Crystallogr., 2014, 47, 803 CrossRef CAS.
- K. Koepernik and H. Eschrig, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1743 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef PubMed.
- M. Kohout, Int. J. Quantum Chem., 2004, 97, 651 CrossRef CAS.
- M. Kohout, F. R. Wagner and Yu. Grin, Int. J. Quantum Chem., 2006, 106, 1499 CrossRef CAS.
- M. Kohout, Faraday Discuss., 2007, 135, 43 RSC.
- A. Ormeci, H. Rosner, F. R. Wagner, M. Kohout and Yu. Grin, J. Phys. Chem. A, 2006, 110, 1100 CrossRef CAS PubMed.
- M. Kohout, DGrid, version 4.6, Radebeul, 2011 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1996617. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt01374a |
| ‡ Current address: Department of Chemistry, University College London. |
| This journal is © The Royal Society of Chemistry 2020 |