
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA ferrocene-templated Pd-bearing molecular reactor†
Artur
Kasprzak
 * and
Piotr A.
Guńka
* and
Piotr A.
Guńka
Faculty of Chemistry, Warsaw University of Technology, Noakowskiego Str. 3, 00-664 Warsaw, Poland. E-mail: akasprzak@ch.pw.edu.pl
First published on 12th May 2020
Abstract
High-yield, chromatography-free syntheses of a ferrocene-templated molecular cage and its Pd-bearing derivative are presented. The formation of a symmetric cage-type structure was confirmed by single-crystal X-ray diffraction analysis. The Pd-bearing cage was used as an innovative catalyst for the efficient synthesis of 1,1′-biphenyls under mild conditions. The presented catalyst is reusable and 1,1′-biphenyls can be obtained efficiently in a gram scale process.
The design of supramolecular systems comprising π-conjugated building blocks attracts unflagging interest in supramolecular chemistry.1–9 An important example of application of supramolecular frameworks is their use as catalysts.10–15 Such machines, termed molecular reactors, may enable the acceleration of the catalytic reaction or may induce stereo- or regioselectivity of the process. Commonly, the driving force responsible for this feature is the ability to recognize aromatic molecules.
1,3,5-Triphenylbenzene frameworks or their nitrogen-containing derivatives are the widely used motifs for the construction of supramolecular frameworks possessing various prospective applications.16–19 In recent years, an emerging field of application of 1,3,5-triphenylbenzene-type motifs has been the synthesis of symmetric molecular cages.20–28 Such structures are characterized by the organized cavity. In fact, the synthesis of such symmetric molecular cages may be challenging; reported reaction yields vary between 15 and 80%. However, the efforts are worthwhile, because an appropriate selection of the building blocks results in a tunable cavity size and properties. For example, in 2019 Stoddart et al. reported the synthesis of a triazine-templated cage.20 This cage was obtained with a combined yield of 14%. The specific inter-platform distance of 11.0 Å enabled accommodation of several molecules within confined space.
We envisioned that the 1,1′-ferrocene motif is the ideal candidate for the construction of a novel symmetric molecular cage bearing the 1,3,5-triphenylbenzene moiety. Herein, we report high-yield and chromatography-free methods for the construction of such a cage compound and its Pd-bearing derivative, exhibiting the ability to bind aromatic molecules. The Pd-decorated cage was employed as the innovative catalyst for the fast and efficient synthesis of 1,1′-biphenyls. We envision that our findings will stimulate the progress in the design, chemistry and applications of supramolecular frameworks.
The synthesis of the ferrocene-templated cage (3) is presented in Fig. 1a, step I. Acid-catalysed imine-bond formation between 1,1′-diformylferrocene (1) and 1,3,5-tris(4-aminophenyl)benzene (2) afforded 3 in 95% yield.29 Such high synthesis yield is an exceptional value in molecular cage chemistry. Additionally, pure 3 was obtained by filtration, without the need to perform further chromatographic purification. The selective formation of 3 was confirmed with a series of characterization techniques; a combination of NMR spectroscopy, Fourier-transform infrared spectroscopy (FT-IR), UV-Vis spectroscopy, high-resolution mass spectrometry (HRMS) and elemental analysis confirmed the formation of pure cage 3.30 The 1H NMR spectrum of 3 (Fig. 2c) comprises six groups of signals, only. It suggests that the obtained molecular cage is highly symmetric (average D3h symmetry in solution).
 | ||
| Fig. 1 (a) Synthesis of 3 (step I) and 5 (step II); molecular structure of 3: (b) top view and (c) side view. Thermal ellipsoids are drawn at 50% probability level. Hydrogen atoms were omitted for clarity in (c). | ||
 | ||
| Fig. 2 (a) Structures of the aromatic molecules tested; (b) atomic labels for NMR investigations with 3; (c) 1H NMR spectrum of 3 (top) and 3 in the presence of G-2 (800 mol%; bottom); (d) inset of the 1H–1H ROESY NMR spectrum of 3 in the presence of G-2 (800 mol%; crucial-cross correlations are marked); (e) 1H DOSY NMR spectrum of 3 in the presence of G-2 (800 mol%). | ||
Cage 3 crystallizes in the rhombohedral R![[3 with combining macron]](https://www.rsc.org/images/entities/i_char_0033_0304.gif) space group with a third of the molecule in the asymmetric unit (Fig. 1b and c).31 The molecule exhibits the symmetry of the C3 point group and the imine moieties are in the E configuration. The central benzene ring is practically coplanar with peripheral cyclopentadienyl rings on one side of the molecule with a root mean square displacement of carbon atoms from the average plane of only 0.081 Å. A root mean square displacement (RMSD) of carbon atoms from an average plane of the rings on the other side of the molecule is significantly larger (0.270 Å). Similarly as in 1,3,5-triphenylbenzene,32 the middle phenyl rings are twisted out of the planes of the central rings. This is due to steric repulsion between hydrogen atoms of the middle rings and that of the central ring as well as imine-type hydrogen atoms, as evidenced by the dihedral angles between average ring planes on the top (27.89(12)°) and bottom (23.06(12)°) sides of 3. The middle phenyl rings from the top and bottom parts are close to being parallel with a dihedral angle of 4.88(12)° which maximizes π stacking interactions as well as minimizes steric repulsion between hydrogen atoms. The central benzene rings are parallel with a dihedral angle of 0.00(11)°. The separation between their centres equals 3.7342(17) Å. This value is larger than the interlayer separation of graphene layers in graphite (3.354 Å). The distance between imine nitrogen atoms is shorter (3.680(3) Å).33 Molecules pack in layers via dispersion interactions and C–H⋯N hydrogen bonds, forming two symmetry-independent types of smaller and larger voids in 1
space group with a third of the molecule in the asymmetric unit (Fig. 1b and c).31 The molecule exhibits the symmetry of the C3 point group and the imine moieties are in the E configuration. The central benzene ring is practically coplanar with peripheral cyclopentadienyl rings on one side of the molecule with a root mean square displacement of carbon atoms from the average plane of only 0.081 Å. A root mean square displacement (RMSD) of carbon atoms from an average plane of the rings on the other side of the molecule is significantly larger (0.270 Å). Similarly as in 1,3,5-triphenylbenzene,32 the middle phenyl rings are twisted out of the planes of the central rings. This is due to steric repulsion between hydrogen atoms of the middle rings and that of the central ring as well as imine-type hydrogen atoms, as evidenced by the dihedral angles between average ring planes on the top (27.89(12)°) and bottom (23.06(12)°) sides of 3. The middle phenyl rings from the top and bottom parts are close to being parallel with a dihedral angle of 4.88(12)° which maximizes π stacking interactions as well as minimizes steric repulsion between hydrogen atoms. The central benzene rings are parallel with a dihedral angle of 0.00(11)°. The separation between their centres equals 3.7342(17) Å. This value is larger than the interlayer separation of graphene layers in graphite (3.354 Å). The distance between imine nitrogen atoms is shorter (3.680(3) Å).33 Molecules pack in layers via dispersion interactions and C–H⋯N hydrogen bonds, forming two symmetry-independent types of smaller and larger voids in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio.34 The layers in turn form 3D crystals via dispersion and π stacking interactions.35
:3 ratio.34 The layers in turn form 3D crystals via dispersion and π stacking interactions.35
We envisaged that 3 might be capable of binding aromatic molecules by means of non-covalent forces. This hypothesis was based on the presence of two, π-conjugated 1,3,5-triphenylbenzene motifs that may induce non-covalent interactions with other π-electron systems. We did not anticipate the formation of host–guest complexes with the inclusion of guest molecules within the cavity of 3, since its X-ray structure revealed that there is no space to entrap the aromatic molecules there. Thus, we considered arrangements of the aromatic molecules on the cage or along the rim of the triphenylbenzene moiety, e.g., parallel displaced π–π stacking and/or aromatic-aromatic interactions at peripheral parts of the cage.37 To uncover binding modes, interactions between 3 and various aromatics (G-1–G-6, Fig. 2a) were tracked by NMR spectroscopy (the spectra for the representative interactions between 3 and G-1 are presented in Fig. 236a). Upon the addition of the aromatic molecule (800 mol%), the upfield shifts for the Ha, Hb and Hc were observed in the 1H NMR spectrum of 3 (Fig. 2c). This feature was ascribed to the π–π interactions between 3 and aromatic molecules.20 Simple benzene derivatives (G-1 and G-2) underwent higher upfield shifts in comparison with the highly conjugated, unsubstituted aromatics (G-3–G-5).38 The presence of the electron withdrawing substituent in the pyrene skeleton (G-6) provided stronger binding with 3 than for the native pyrene (G–5). The interactions with G-2 and G-6 were also tracked by 1H-1H ROESY NMR and 1H DOSY NMR (Fig. 2d and e for representative G-139). The cross-correlations between Ha, Hb and Hc and the protons of G-1–G-6 were observed in the 1H–1H ROESY NMR spectrum (Fig. 2d). It means that the distance between the interacting protons (H{G-1–G-6} ↔ Ha, Hb and Hc) is shorter than 4–4.5 Å. Thus, we anticipate that the aromatic molecules were accommodated near {Ha, Hb and Hc}-oriented sites of 3 and were located further (>4.5 Å) from the Fc moiety. We anticipate that this binding mode follows a parallel displaced π-π stacking geometry.37 The lowering of the diffusion coefficient for 3 after the addition of the aromatic molecule was observed in the 1H DOSY NMR spectra (Fig. 2e). These features were previously reported to stand for the non-covalent interactions.20,40 DOSY NMR experiments suggested the formation of single non-covalent systems (all peaks showed the same diffusion coefficient).
To gain an insight into the binding parameters of these non-covalent systems, association constants (Kapp) and free energies (ΔG) were estimated based on PL experiments (UV-Vis and PL titration experiments were performed; PL was found to be a more sensitive technique).36b The studied non-covalent systems featured the lowering of the emission intensity for 3 after the addition of further portions of an aromatic molecule. The change in the emission intensity differed between the systems. It was a result of different Kapp and ΔG. The association constant values were estimated.36b,c The non-covalent systems exhibited good binding parameters with ΔG equal to −16.1 to −14.2 kJ mol−1 at 298.15 K (Table 1). G-6 exhibited the highest binding parameters (Table 1, entry 6). For the non-covalent system of G-6 exhibiting the highest Kapp and the lowest ΔG, these parameters evaluated from PL experiments were also compared with the respective values obtained from the 1H DOSY NMR experiment. These results were highly consistent (PL analysis: 650 M−1, 1H DOSY NMR analysis: 658 M−1).36bG-1 and G-2 were characterized by lower ΔG values than G-3–G-5 (Table 1, entries 1–5). It suggested that simple benzene derivatives were bound stronger by 3 in comparison with unsubstituted, conjugated aromatics. This trend is consistent with the outcomes from 1H NMR assays.
| Entry | Aromatic molecule | Non-covalent system stoichiometry (3:aromatic molecule) |
K
appa,b |
ΔG/kJ mol−1 |
|---|---|---|---|---|
| a Because of some complexity of the studied non-covalent systems, Kapp values should be treated as the approximate ones and they elucidate the trend of these interactions. b Standard errors were estimated based on the outcomes from the least squares regression method for the I0/I = f(Car) linear fits (standard error of the slope of curve). | ||||
| 1 | G-1 | 1:3 |
6.1(1) × 102 M−3 | −15.9 |
| 2 | G-2 | 1:3 |
5.9(2) × 102 M−3 | −15.8 |
| 3 | G-3 | 1:1 |
3.1(2) × 102 M−1 | −14.2 |
| 4 | G-4 | 1:1 |
3.5(3) × 102 M−1 | −14.5 |
| 5 | G-5 | 1:1 |
4.0(3) × 102 M−1 | −14.9 |
| 6 | G-6 | 1:1 |
6.5(2) × 102 M−1 | −16.1 |
Job's plot analyses suggested 1:3 binding stoichiometry for the interactions between 3 and G-1 or G-2, whilst 1:1 stoichiometry was found for G-3–G-6.36b Following these estimated stoichiometries, the formation of the non-covalent systems was further confirmed with ESI-MS analysis.41a,b The mixtures of 3 and G-1–G-6 were analysed. Besides the peaks coming from the native cage 3 and unbound G-1–G-6, the peaks originating from the presence of the non-covalent systems were clearly found.41c It is noteworthy that ESI-MS studies supported the systems’ stoichiometries estimated from PL (1:1 stoichiometry for G-3–G-6 and 1:3 stoichiometry for G-1 and G-2).
In order to exclude any possible non-covalent interaction between an aromatic molecule and a 1,3,5-triphenylbenzene-templated compound, the 1H NMR, 1H–1H ROESY NMR and 1H DOSY NMR spectra of 1,3,5-tris(4-aminophenyl)benzene (2) were measured in the presence of G-6 (800 mol%). No significant changes between the spectra of native 2 and after addition of G-6 were found.41d These findings revealed that the cage-type architecture of 3 is essential for providing the binding property. To further support this claim, the ESI-MS spectrum of the mixture of 2 and G-6 (300 mol%) was measured.41c If a non-covalent system was formed, the significant peak of m/z > 580 shall be observed. The peaks that were ascribed to native 2 (m/z = ca. 230) and native G-6 (m/z = ca. 352) were found, only. No peak coming from the non-covalent system (m/z > 580) was found.
Cage 3 comprises the imine-type nitrogen atoms located in a vertical orientation. We envisioned that this part of 3 might play a role of a metal coordination site.43–48 Due to the importance of Pd in many fields of applied science, e.g., catalysis, our goal was to incorporate three Pd residues to 3. The chromatography-free protocol for the synthesis of the Pd-decorated cage (5) was successfully developed (Fig. 1a, step II). Treatment of 3 with bis(acetonitrile)dichloropalladium(II) (4) afforded 5 in high yield (98%).29 Pure 5 was obtained by means of filtration and was fully characterized.30 NMR spectroscopy revealed that the symmetric cage-type structure of the cage was retained after the incorporation of the PdCl2 residues; similarly to the 1H NMR spectrum of 3, the 1H NMR spectrum of 5 comprises six groups of signals, indicating the average D3h symmetry in solution.42
With the Pd-bearing cage 5 at hand, we began to engineer its application. A breakthrough at this point of our project occurred when we had noted the structural similarity between 5 and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), a commercially used catalyst for the Suzuki–Miyaura reaction.43–45 Inspired by this resemblance, we decided to study the pioneering application of 5 as the catalyst for 1,1′-biphenyl synthesis. The optimization experiments gave remarkable results.49 These trials revealed that 1,1′-biphenyls can be obtained in 99% yield under mild conditions (room temperature) in 20 minutes, only (Table 2, entry 1).50 Importantly, 0.5 mol% of 5 was used, only. Having achieved for the first time such exceptional results of the Suzuki–Miyaura reaction employing an easy-to-perform procedure without the use of a sophisticated instrument (e.g., microwave reactor), we were curious whether our method can be applied for other starting materials. To our delight, the substituted 1,1′-biphenyls were efficiently obtained (96–98%) in very short reaction times (Table 2, entries 2–5). We believe that the designed cage architecture is a new feature of good ligands of Pd catalysts. Secondly, we showed above that the designed cage non-covalently interacts with aromatic molecules. In the light of calculated binding parameters, one might also consider the influence of non-covalent interactions with aromatic molecules on providing encouraging reaction parameters. We hypothesised that the designed cage architecture giving rise to the non-covalent interaction feature facilitated the contact between the reagents towards the catalytic process. To support these hypotheses, the reaction rates under 1H NMR conditions were estimated for the reaction between phenylboronic acid and chlorobenzene (Fig. 3; the Michaelis–Menten method was employed).50b,c No reaction was found without the addition of a catalyst. In contrast, the rate constant for the reaction in the presence of 5 (kcage) was found to be ca. 8.5 × 10−3 M−1 s−1. This feature elucidated the usefulness of the designed catalyst and was the first premise of the accelerating effect of non-covalent interactions during the catalytic process. In order to further support this claim, the respective reaction was monitored in the presence of binding competing aromatic molecule G-6 (Fig. 3). We envisioned that G-6 might prevent the non-covalent interactions between our cage-type catalyst and phenylboronic acid or chlorobenzene, because of the competitive ΔG values between these systems (compare the ΔG values for G-1, G-2 and G-6; Table 1). In other words, G-6 might “block” the appropriate arrangement of phenylboronic acid or chlorobenzene in their system with 5, and thus, highly limits the acceleration of the catalytic process. Any inhibiting effect of G-6 shall result in lowering of the reaction rate of the studied catalytic process. With the presence of this competing molecule, the reaction rate was indeed ca. 100-fold lower (kcage+G-6 = ca. 8.2 × 10−5 M−1 s−1). This feature was ascribed to the inhibiting effect of G-6; in other words, non-covalent interactions between 5 and G-6 highly prevented binding of the reactants (chlorobenzene, phenylboronic acid) by 5. These experiments clearly suggested an influence of aromatic molecule binding on the designed catalytic process. We also anticipate that the presence of three units of Pd per one unit of cage might play a role in this process.
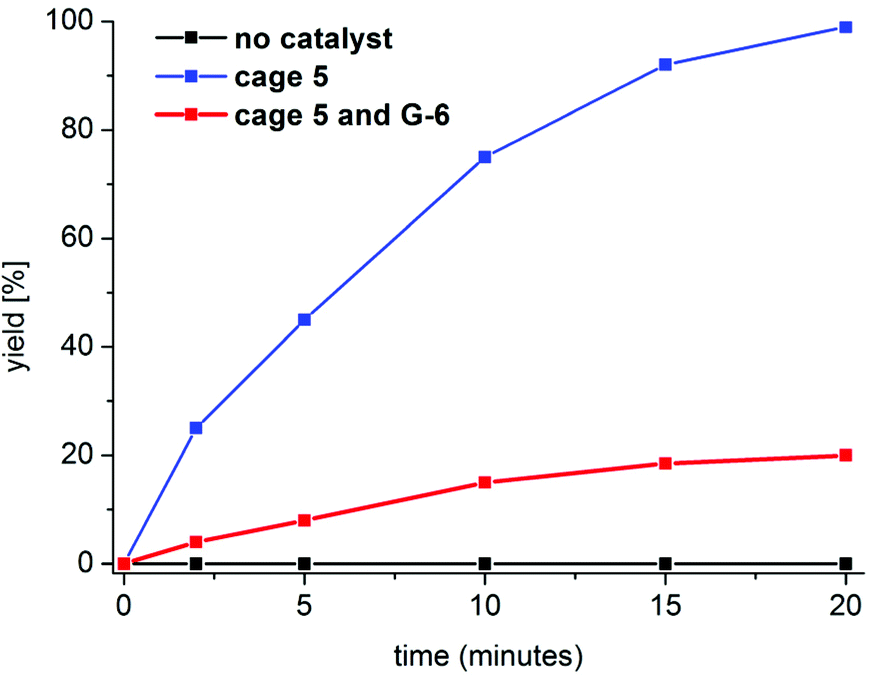 | ||
| Fig. 3 Kinetic curves for the designed catalytic process. | ||

Encouraged by the above-presented results, we further studied the designed methodology towards its prospective offering to industrial users. We found that 1,1′-biphenyls can be obtained in a gram scale reaction (Table 2, entry 6). Moreover, the catalyst can be easily recovered from the reaction mixture (after the catalytic reaction 5 was precipitated and filtered51) and reused in the next ten reaction cycles without any loss of its high catalytic activity.52 In general, the process parameters (that is, the reaction yield and time and the amount of the catalyst) remain unchanged between the experiments (compare entries 1 and 6 in Table 2). These prominent results showed that scaling-up the reaction and the use of the recovered catalyst did not affect its excellent catalytic performance. To the best of our knowledge, such parameters have never been studied before in molecular cage chemistry.
Conclusions
In conclusion, the efficient, easy-to-perform and chromatography-free synthesis of the new type of molecular cage comprising the ferrocene and 1,3,5-triphenylbenzene motifs was obtained. The constructed molecular cage exhibits the property to bind aromatic molecules. This cage can be used as the effective ligand of Pd catalysts. The Pd-decorated cage (‘molecular reactor’) was applied for the very efficient and fast synthesis of various 1,1′-biphenyls under mild conditions. The developed catalyst is also interesting towards its prospective use in industrial practice. We believe that this work significantly improves the state-of-the-art of the chemistry of supramolecular systems and sheds light on applications of such new types of cage compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from Warsaw University of Technology (WUT) is acknowledged. We thank Dr Magdalena Poplawska (WUT) for helpful discussions, Dr Krzysztof M. Borys (WUT) for providing 1,1′-diformylferrocene and Dr Maciej Dranka (WUT) for his assistance in single-crystal X-ray analyses. The authors would like to thank the reviewers for their important and constructive comments.Notes and references
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed.
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635 RSC.
- Z.-T. Li and J. Beilstein, Org. Chem., 2015, 11, 2057 CAS.
- T. Ogoshi, T.-a. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937 CrossRef CAS PubMed.
- M. M. Cetin, Y. Beldjoudi, I. Roy, O. Anamimoghadam, Y. J. Bae, R. M. Young, M. D. Krzyaniak, C. L. Stern, D. Philp, F. M. Alsubaie, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 18727 CrossRef CAS PubMed.
- S. R. Scottwell and J. D. Crowley, Chem. Commun., 2016, 52, 2451 RSC.
- Y. Yakiyama, T. Hasegawa and H. Sakurai, J. Am. Chem. Soc., 2019, 141, 18099 CrossRef CAS PubMed.
- P. Li, P. Li, M. R. Ryder, Z. Liu, C. L. Stern, O. K. Farha and J. F. Stoddart, Angew. Chem., Int. Ed., 2019, 58, 1664 CrossRef CAS PubMed.
- M. Kołodziejski, A. R. Stefankiewicz and J.-M. Lehn, Chem. Sci., 2019, 10, 1836 RSC.
- C. J. Easton, S. F. Lincoln, L. Barr and H. Onagi, Chem. – Eur. J., 2004, 10, 3120 CrossRef CAS PubMed.
- A. S. Lubbe, T. van Leeuwen, S. J. Wezenberg and B. L. Feringa, Tetrahedron, 2017, 73, 4837 CrossRef CAS.
- J. Teyssandier, S. De Feyter and K. S. Mali, Chem. Commun., 2016, 52, 11465 RSC.
- M. S. Shad, P. V. Santhini and W. Dehaen, Beilstein J. Org. Chem., 2019, 15, 2142 CrossRef CAS PubMed.
- L. Barr, S. F. Lincoln and C. J. A. Easton, Supramol. Chem., 2005, 17, 547 CrossRef CAS.
- J. Kang and J. Rebek, Nature, 1997, 385, 50 CrossRef CAS PubMed.
- K. Gontarczyk, W. Bury, J. Serwatowski, P. Wieciński, K. Woźniak, K. Durka and S. Luliński, ACS Appl. Mater. Interfaces, 2017, 9, 31129 CrossRef CAS PubMed.
- M. Taddei, F. Costantino, R. Vivani, S. Sabatini, S.-H. Lim and S. M. Cohen, Chem. Commun., 2014, 50, 5737 RSC.
- D. Kaleeswaran, P. Vishnoi and R. Murugavel, J. Mater. Chem. C, 2015, 3, 7159 RSC.
- V. K. Maka, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Nanoscale, 2018, 10, 22389 RSC.
- T. Jiao, K. Cai, Z. Liu, G. Wu, L. Shen, C. Cheng, Y. Feng, C. L. Stern, J. F. Stoddart and H. Li, Chem. Sci., 2019, 10, 5114 RSC.
- Z. Wang, Y. Luo, Y. T.-L. Zhai, H. Ma, J.-J. Chen, Y. Shu and C. Zhang, Org. Lett., 2016, 18, 4574 CrossRef CAS PubMed.
- C. Mejuto, G. Guisado-Barrios, D. Gusev and E. Peris, Chem. Commun., 2015, 51, 13914 RSC.
- Q.-Q. Wang, N. Luo, X.-D. Wang, Y.-F. Ao, Y.-F. Chen, J.-M. Liu, C.-Y. Su, D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2017, 139, 635 CrossRef CAS PubMed.
- Z. Wang, H. Ma, T.-L. Zhai, G. Cheng, Q. Xu, J.-M. Liu, J. Yang, Q.-M. Zhang, Q.-P. Zhang, Y.-S. Zheng, B. Tan and C. Zhang, Adv. Sci., 2018, 5, 1800141 CrossRef PubMed.
- B. M. Schmidt, T. Osuga, T. Sawada, M. Hoshino and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 1561 CrossRef CAS PubMed.
- W. Cullen, H. Takezawa and M. Fujita, Angew. Chem., Int. Ed., 2019, 58, 9171 CrossRef CAS PubMed.
- Y. Zhou, H. Li, T. Zhu, T. Gao and P. Yan, J. Am. Chem. Soc., 2019, 141, 19634 CrossRef CAS PubMed.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850 CrossRef CAS PubMed.
- For the experimental details, see Section S1, ESI.†.
- For the spectra and data, see Sections S1–S4, ESI.†.
- Crystal Data for cage 3: C84H60Fe3N6 (M = 1320.94 g mol−1), trigonal, space group R
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (no. 148), color dark pink, a = 24.9627(11) Å, c = 20.5795(11) Å, V = 11105.8(11) Å3, Z = 6, T = 293 K, μ(Mo Kα) = 0.626 mm−1, Dcalc = 1.185 g cm−3, 11433 reflections measured (6.778° ≤ 2Θ ≤ 53.998°), 5389 unique (Rint = 0.0362, Rsigma = 0.0644) which were used in all calculations. The final R1 was 0.0467 (I > 2σ(I)) and wR2 was 0.1154 (all data); GoF = 0.964. For additional structure determination and crystal packing data, see Section S5, ESI.† CCDC 1970365 contains supplementary crystallographic data for this paper.†.
(no. 148), color dark pink, a = 24.9627(11) Å, c = 20.5795(11) Å, V = 11105.8(11) Å3, Z = 6, T = 293 K, μ(Mo Kα) = 0.626 mm−1, Dcalc = 1.185 g cm−3, 11433 reflections measured (6.778° ≤ 2Θ ≤ 53.998°), 5389 unique (Rint = 0.0362, Rsigma = 0.0644) which were used in all calculations. The final R1 was 0.0467 (I > 2σ(I)) and wR2 was 0.1154 (all data); GoF = 0.964. For additional structure determination and crystal packing data, see Section S5, ESI.† CCDC 1970365 contains supplementary crystallographic data for this paper.†. - For the crystal structure of 1,3,5-triphenylbenzene, see: M. S. Farag, Acta Crystallogr., 1954, 7, 117 CrossRef CAS.
- The separation between centers of cyclopentadienyl anions interacting with iron cations is even shorter (3.306(5) Å).
- For molecular packing in layers, see Fig. S9, ESI.†.
- See Fig. S10, ESI.†.
- (a) For the NMR data for the interactions of 3 with G-2–G-6, see Section S5, ESI;†; (b) For the additional discussion, spectra and calculation of binding parameters, see Section S9, ESI;†; (c) Stern–Volmer method was used, for details of this methodology, see for example: A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. C. Barton, J. Am. Chem. Soc., 1989, 111, 3051 CrossRef CAS; (d) A. Kasprzak and H. Sakurai, Dalton Trans., 2019, 48, 17147 RSC.
- (a) R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523 CrossRef CAS; (b) R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2014, 16, 3238 RSC; (c) S. E. Wheeler, T. J. Seguin, Y. Gua and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061 CrossRef CAS PubMed; (d) D. B. Ninković, J. P. Blagojević Filipović, M. B. Hall, E. N. Brothers and S. D. Zarić, ACS Cent. Sci., 2020, 6, 420 CrossRef PubMed.
- See the data in Table S2, ESI.†.
- For the spectra on binding studies with G-6, see Fig. S17 and S18, ESI.†.
- A. Kasprzak, K. Borys, S. Molchanov and A. Adamczyk-Wozniak, Carbohydr. Polym., 2018, 198, 294 CrossRef CAS PubMed.
- (a) Q. He, Y.-F. Ao, Z.-T. Huang and D.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 11785 CrossRef CAS PubMed; (b) E. Darii, S. Alves, Y. Gimbert, A. Perret and J.-C. Tabet, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1047, 45 CrossRef CAS PubMed; (c) For the ESI-MS spectra, see Section S11, ESI;†; (d) For the spectra, see Fig. S19–S23, ESI.†.
- See Fig. S13, ESI.†.
- T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158 CrossRef CAS.
- T. J. Colacot, H. Qian, R. Cea-Olivares and S. Hernandez-Ortega, J. Organomet. Chem., 2001, 637–639, 691 CrossRef CAS.
- R. S. Chauhan, D. B. Cordes, A. M. Z. Slawin, S. Yadav and C. Dash, Inorg. Chim. Acta, 2018, 478, 125 CrossRef CAS.
- J. Wiedermann, K. Mereiter and K. Kirchner, J. Mol. Catal. A: Chem., 2006, 257, 67 CrossRef CAS.
- G. R. Owen, A. J. P. White and R. Vilar, Organometallics, 2009, 28, 5783 CrossRef CAS.
- H. Hu, D. Chen, H. Gao, L. Zhong and Q. Wu, Polym. Chem., 2016, 7, 529 RSC.
- Optimization experiments: Table S1, ESI.†.
- (a) For the experimental conditions, see Subsection S1.6, ESI;†; (b) For details of this methodology, see for example: J. Jiao, Z. Li, Z. Qiao, X. Li, Y. Liu, J. Dong, J. Jiang and Y. Cui, Nat. Commun., 2018, 9, 4423 CrossRef PubMed; (c) For the additional discussions and experimental details, see Section S10, ESI.†.
- For details, see Subsection S1.6, ESI.†.
- For the data on the reusability studies, see Fig. S31, ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and compounds characterization data, crystal data, molecular recognition experiments, calculation of binding parameters, reaction kinetics data. CCDC 1970365. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/D0DT01366H |
| This journal is © The Royal Society of Chemistry 2020 |