DOI:
10.1039/D0DT01271H
(Paper)
Dalton Trans., 2020,
49, 7701-7707
3d–4f heterometallic complexes by the reduction of transition metal carbonyls with bulky LnII amidinates†
Received
6th April 2020
, Accepted 1st May 2020
First published on 12th May 2020
Abstract
The redox chemistry between divalent lanthanide complexes bearing bulky amidinate ligands has been studied with 3d transition metal carbonyl complexes (iron and cobalt). The reaction of [(DippForm)2SmII(thf)2] (DippForm = N,N′-bis(2,6-diisopropylphenyl)formamidinate) with [Co2(CO)8] resulted in the formation of a tetranuclear Sm–Co complex, [{(DippForm)2SmIII(thf)}2{(μ-CO)2Co(CO)2}2]. The product of the reaction of [(DippForm)2YbII(thf)2] and [Co2(CO)8] gives the dinuclear Yb–Co complex [{(DippForm)2YbIII(thf)}{(μ-CO)Co(CO)3}] in toluene. The reaction of [(DippForm)2SmII(thf)2] was also carried with the neighbouring group 8 carbonyl complexes [Fe2(CO)9] and [Fe3(CO)12], resulting in a pentanuclear SmIII–Fe complex, [{(DippForm)2SmIII}2{(μ3-CO)2Fe3(CO)9}], featuring a triangular iron carbonyl cluster core.
Introduction
Heterometallic complexes have gained widespread attention due to the viable synergistic effect that may result from a judicious choice of metal centres.1,2 Ln–TM (TM = transition metal) heterometallic complexes have especially attracted interest due to their magnetic,3–5 and photophysical properties,6,7 as well as their catalytic activity.8,9 Among them, Ln–TM carbonyl complexes are a major class of compounds from both fundamental and application points of view.10–12 For example, Dy–TM carbonyl complexes have demonstrated single-molecule magnetic behaviour.13,14 Andersen and co-workers have synthesized several YbIII–TM isocarbonyl complexes, [YbIII-OC-TM], by reduction of TM carbonyl complexes with [Cp*2YbII(OEt2)].15–18 Edelmann and co-workers have isolated the formally SmIII–Fe0 complex [Cp*2Sm(μ-OC)2FeCp*]2 by reducing [Cp*Fe(CO)2]2 with [Cp*2Sm(thf)2].19 Some of us isolated the elusive [W2(CO)10]2− anion in a mixed-valent SmII/III calix[4]pyrrolide sandwich by the reduction of [W(CO)6] with a divalent samarium meso-octaethylcalix[4]pyrrolide.20 Also, LnII–TM carbonyl complexes have been accessed either by redox-transmetallation between Hg salts of TM carbonyl complexes and elemental Ln0 or by reduction of TM carbonyl complexes with Ln/Hg amalgam.10,21–26 Depending on the bonding situation between the two metal centres, Ln–TM carbonyl complexes can be divided into three major categories: (i) solvent-separated ion pairs,22,27 (ii) Ln–TM bonded compounds,11,12,14,23,28,29 and (iii) compounds with isocarbonyl-linkages10,16,30 between both metals. Isocarbonyl bridged Ln–TM complexes are the major representatives, which can be explained by the oxophilic nature of lanthanide cations and the additional stabilisation of the negative charge on the TM centre through π back-donation from TM(dπ) to C–O(π*).26 Usually, the reaction of divalent lanthanide complexes with TM-carbonyls leads to isocarbonyl-bridged systems, resulting from a single-electron transfer (SET) to the TM carbonyl moiety and leading to either cleavage or formation of TM–TM bonds.
The chemistry of divalent lanthanide complexes is dominated by the use of cyclopentadienyl-based ligands.31–33 Investigating the effects of the nature of the supporting ligands, we have examined the reactivity of the divalent lanthanide complexes [(DippForm)2LnII(thf)2] (Ln = Sm,35 Yb36) in the reduction of main group elements and compounds. By using different electronic and steric environments around divalent lanthanide elements, a different reactivity can be achieved with a same substrate.37–45 For example, Evans and co-workers have reported the formation of [{Cp*2SmIII}2(S3)] by reaction of [Cp*2SmII(thf)2] with elemental sulfur,46 whereas some of us have isolated lanthanide polysulfide coordination clusters, [(DippForm)3LnIII3S12] (Ln = Sm and Yb), by using [(DippForm)2LnII(thf)2] (Ln = Sm, Yb) as divalent lanthanide reagents.42 In addition, activation of white phosphorous and yellow arsenic by [(DippForm)2SmII(thf)2] led to the formation of [{(DippForm)2SmIII}2(μ-η4-Pn4)] (Pn = P, As).43 These products contrast with the cage-type molecules [{Cp*2SmIII}4(Pn)8] (Pn = P,37 As,44 and Sb47) that were obtained by reaction of [Cp*2SmII] with white phosphorous, nanoscale arsenic, and nanoscale antimony, respectively. We were then interested to extend the study of the influence of the ligands on the reductive behaviour of divalent lanthanide complexes from main group compounds to transition metal complexes. Very recently, some of us have reported the reduction of [Re2(CO)10] with [L2SmII(thf)2] (L = Cp* or DippForm) and, depending on the nature of the ligands around samarium, were able to isolate either a Fischer-type rhenacycle (A) or a novel [Re2(CO)8]2− dianion (B) in the coordination sphere of [L2SmIII]+ moieties (Scheme 1).34 Inspired by the above results, we further explored the reactivity of group 8 and 9 metal carbonyls towards divalent lanthanide complexes supported by the bulky amidinate DippForm ligand and report herein the synthesis and characterisation of the resulting 3d–4f metal complexes obtained.
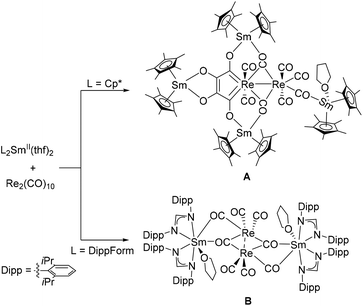 |
| | Scheme 1 Reactivity of [Re2(CO)10] towards Sm(II) complexes bearing different ligands.34 | |
Results and discussion
Ln–Co carbonyl complexes
The reaction of [(DippForm)2SmII(thf)2] with half an equivalent of [Co2(CO)8] in toluene at room temperature resulted in the formation of the heterometallic complex [{(DippForm)2SmIII(thf)}2{(μ-CO)2Co(CO)2}2] (1) (Scheme 2). After a short work-up, analytically pure yellow-coloured crystals of complex 1 were grown in a 56% yield by slow evaporation of toluene. The solid-state IR spectrum of complex 1 showed characteristic ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) CO bands at 2020 (m), 1951 (br), 1935 (br), 1922 (br), 1904 (br), 1842 (s), 1819 (s) and 1782 (s) cm−1. The low-frequency stretch at 1782 (s) cm−1 suggests the presence of bridging isocarbonyls between the Sm and Co atoms. The terminal CO stretches from 2020 to 1819 cm−1 and the low-frequency isocarbonyl stretch at 1782 cm−1 are apparent for complex 1. Similar low-frequency stretches at 1798 (m) and 1761 (s) cm−1 were observed for [{(Cp*)2YbIII(thf)}{(μ-CO)Co(CO)3}].15 Furthermore, the molecular structure of complex 1 was determined by single crystal X-ray crystallography. Complex 1 crystallizes in the monoclinic space group P21/n with half a molecule in the asymmetric unit. The solid-state structure showed that two [(DippForm)2SmIII(thf)]+ moieties are bridged by two [(μ-CO)2Co(CO)2]− units (Fig. 1). The formation of complex 1 can be rationalized by single-electron transfer steps from two [(DippForm)2SmII(thf)2] molecules to one molecule of [Co2(CO)8], resulting in the homolytic cleavage of the Co–Co bond and the formation of two [Co(CO)4]− anions along with two [(DippForm)2SmIII(thf)]+ cations.
CO bands at 2020 (m), 1951 (br), 1935 (br), 1922 (br), 1904 (br), 1842 (s), 1819 (s) and 1782 (s) cm−1. The low-frequency stretch at 1782 (s) cm−1 suggests the presence of bridging isocarbonyls between the Sm and Co atoms. The terminal CO stretches from 2020 to 1819 cm−1 and the low-frequency isocarbonyl stretch at 1782 cm−1 are apparent for complex 1. Similar low-frequency stretches at 1798 (m) and 1761 (s) cm−1 were observed for [{(Cp*)2YbIII(thf)}{(μ-CO)Co(CO)3}].15 Furthermore, the molecular structure of complex 1 was determined by single crystal X-ray crystallography. Complex 1 crystallizes in the monoclinic space group P21/n with half a molecule in the asymmetric unit. The solid-state structure showed that two [(DippForm)2SmIII(thf)]+ moieties are bridged by two [(μ-CO)2Co(CO)2]− units (Fig. 1). The formation of complex 1 can be rationalized by single-electron transfer steps from two [(DippForm)2SmII(thf)2] molecules to one molecule of [Co2(CO)8], resulting in the homolytic cleavage of the Co–Co bond and the formation of two [Co(CO)4]− anions along with two [(DippForm)2SmIII(thf)]+ cations.
 |
| | Scheme 2 Synthesis of complex 1. | |
 |
| | Fig. 1 Molecular structure of 1. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles [°]: Sm–O1 2.422(2), Sm′–O2 2.488(3), Sm–O3 2.552(3), Sm–N1 2.431(3), Sm–N2 2.449(3), Sm–N3 2.409(3), Sm–N4 2.449(3), Co–C1 1.733(4), Co–C2 1.787(4), Co–C3 1.747(4), Co–C4 1.782(4), O2–C1 1.175(4), O3–C3 1.170(4), O4–C2 1.138(5), O5–C4 1.145(5), N1–C5 1.322(4), N2–C5 1.318(4), N3–C6 1.330(5), N4–C6 1.326(5); O2′–Sm–O3 67.27(8), N1–Sm–N2 55.35(9), N1–Sm–N4 143.10(10), C1–Co–C2 108.8(2), C1–Co–C3 116.1(2), C1–Co–C4 110.1(2), C3–Co–C2 109.7(2), C3–Co–C4 105.8(2), C4–Co–C2 105.8(2), N2–C5–N1 118.4(3), N4–C6–N3 120.3(3). | |
Each samarium atom is heptacoordinated, surrounded by two bidentate amidinate ligands, two bridging isocarbonyls, and one coordinating thf. The average Sm–N bond length (2.434(3) Å) in complex 1 is significantly shorter than that in [(DippForm)2SmII(thf)2] (Sm–N(average) 2.573(3) Å). This shortening is consistent with a decrease in the ionic radius of the metal cation upon oxidation of SmII to SmIII.48 Besides, the shortening of the Sm–O1(thf) bond length, from 2.560(3) Å in [(DippForm)2SmII(thf)2] to 2.422(2) Å in 1, further supports the oxidation of the Sm centre to the +3 oxidation state. The cobalt centre in the bridging tetracarbonylcobaltate anion has a distorted tetrahedral coordination geometry with C–Co–C angles ranging from 105.8(2)° to 116.1(2)°. Owing to the coordination to the [(DippForm)2SmIII(thf)]+ moiety, the O2–C1 (1.175(4) Å) and O3–C3 (1.170(4) Å) bond distances are longer than the O4–C2 (1.138(5) Å) and O5–C4 (1.145(5) Å) analogues involving terminal CO ligands. A further effect of the formation of bridging isocarbonyls can be seen by analysis of the Co–C bond lengths: the Co–C(bridging) bonds are shortened and strengthened as compared to the Co–C(terminal) bonds (Co–C1 (1.733(4) Å) and Co–C3 (1.747(4) Å) vs. Co–C4 (1.782(4) Å) and Co–C2 (1.787(4) Å), respectively).15 To the best of our knowledge, complex 1 is a rare example of a SmIII–Co carbonyl complex featuring bridging isocarbonyls between the two metal centres. Mountford and co-workers have reported the SmIII–Co carbonyl complex [SmIII{(μ-CO)Co(CO)2(PCy3)}{Co(CO)3(PCy3)} (thf)3] (Cy = cyclohexyl) by a reaction between SmI2 with K[Co(CO)3(PCy3)(thf2)].25 Although the reduction of [Co2(CO)8] by [Cp*2SmII(thf)2] has been studied by Evans et al., no solid-state structure of the product has been reported.27 Besides, the reaction between [SmIII2(thf)2] and [Co2(CO)8] in thf led to the solvent-separated ion pair, [SmIIII2(thf)4][Co(CO)4], which was structurally characterized.27
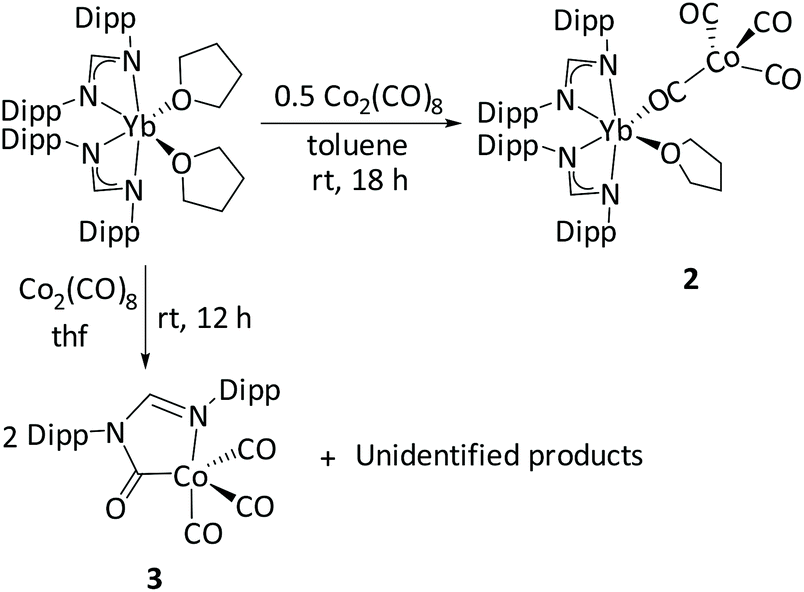
The ytterbium analogue of complex 1 was synthesized following a similar procedure (Scheme 3) and orange-coloured crystals were isolated in 63% yield. The IR spectrum of complex 2, [{(DippForm)2YbIII(thf)}{(μ-CO)Co(CO)3}], shows CO stretches in the carbonyl region from 2031 to 1748 cm−1, in a similar range to those of [{Cp*2YbIII}(thf)(μ-CO)Co(CO)3] (2023 to 1761 cm−1).15 The lowest stretch at 1748 cm−1 indicates bridging isocarbonyls between the tetracarbonylcobaltate anion and the [(DippForm)2YbIII(thf)]+ moiety. Similar CO stretches (2017 to 1780 cm−1) have been observed in the related complex [Cp2Lu(thf){Co(CO)4}].49 The solid-state structure was established by single-crystal X-ray crystallography (Fig. 2). Complex 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule in the asymmetric unit cell. In contrast to the dimeric form of complex 1, the solid-state structure of complex 2 reveals a monomeric arrangement. A possible reason for this difference could be a combined effect of the smaller ionic radius of Yb3+ as compared to Sm3+ and the sterically demanding nature of the DippForm ligands.48 The ytterbium centre is in a distorted octahedral environment, coordinated by two chelating amidinate ligands, one isocarbonyl O donor, and one thf ligand. In complex 2, the shortening of the Yb–N bond distances involving the amidinate ligands (2.315(2) Å, average) and of Yb–O5 (2.304(2) Å), in comparison to the corresponding separations in [(DippForm)2YbII(thf)2] (Yb–N (2.447 Å) and Yb–O (2.461 Å), average),36 is consistent with the oxidation of the Yb metal centre from the +2 to the +3 oxidation state.48 As observed in 1, the cobalt centre in the tetracarbonylcobaltate anion of 2 has a distorted tetrahedral coordination environment with C–Co–C angles varying from 104.6(2)° to 115.8(2)°. The Co–C1 bond length (1.705(3) Å) in 2 is almost the same as the Co–C(bridged) (1.699(3) Å) bond distance in [{Cp*2YbIII}(thf)(μ-CO)(Co(CO)3].15 The average Co–C(terminal) bond lengths in 2 and [{Cp*2YbIII}(thf)(μ-CO)Co(CO)3] are also similar (ca. 1.77 Å in both cases).
with one molecule in the asymmetric unit cell. In contrast to the dimeric form of complex 1, the solid-state structure of complex 2 reveals a monomeric arrangement. A possible reason for this difference could be a combined effect of the smaller ionic radius of Yb3+ as compared to Sm3+ and the sterically demanding nature of the DippForm ligands.48 The ytterbium centre is in a distorted octahedral environment, coordinated by two chelating amidinate ligands, one isocarbonyl O donor, and one thf ligand. In complex 2, the shortening of the Yb–N bond distances involving the amidinate ligands (2.315(2) Å, average) and of Yb–O5 (2.304(2) Å), in comparison to the corresponding separations in [(DippForm)2YbII(thf)2] (Yb–N (2.447 Å) and Yb–O (2.461 Å), average),36 is consistent with the oxidation of the Yb metal centre from the +2 to the +3 oxidation state.48 As observed in 1, the cobalt centre in the tetracarbonylcobaltate anion of 2 has a distorted tetrahedral coordination environment with C–Co–C angles varying from 104.6(2)° to 115.8(2)°. The Co–C1 bond length (1.705(3) Å) in 2 is almost the same as the Co–C(bridged) (1.699(3) Å) bond distance in [{Cp*2YbIII}(thf)(μ-CO)(Co(CO)3].15 The average Co–C(terminal) bond lengths in 2 and [{Cp*2YbIII}(thf)(μ-CO)Co(CO)3] are also similar (ca. 1.77 Å in both cases).
 |
| | Scheme 3 Synthesis of complex 2 and 3 (3 is not fully characterized). | |
 |
| | Fig. 2 Molecular structure of 2. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles [°]: Yb–O1 2.248(2), Yb–O5 2.304(2), Yb–N1 2.307(2), Yb–N2 2.337(2), Yb–N3 2.292(2), Yb–N4 2.324(2), Yb–C5 2.697(3), Yb–C6 2.686(3), Co–C1 1.705(3), Co–C2 1.783(5), Co–C3 1.787(4), Co–C4 1.763(3), O1–C1 1.200(3), O2–C2 1.143(5), O3–C3 1.152(5), O4–C4 1.140(4), N1–C5 1.317(3), N2–C5 1.315(3), N3–C6 1.319(3), N4–C6 1.315(3); O1–Yb–O5 81.70(8), N1–Yb–N2 58.23(7), N1–Yb N4 99.80(8), N3–Yb–N1 112.08(8), N3–Yb–N2 114.88(8), C1–Co–C2 111.8(2), C1–Co–C3 115.8(2), C1–Co–C4 109.6(2), C2–Co–C3 107.5(2), C4–Co–C2 104.6(2), C4–Co–C3 106.8(2). | |
In a different approach to synthesize complex 2, [(DippForm)2Yb(thf)2] was treated with cobalt carbonyl at room temperature in thf for 12 hours. In this case, we did not obtain the bimetallic species 2. Instead, crystals of the metallacyclic cobalt(I) complex [(DippFormCO)(CO)3Co] (3) were isolated (Scheme 3). Despite our attempts, complex 3 could not be obtained in analytically pure form. However, the isolation of 3 gave some insight into occurring side reactions. Complex 3 was unambiguously identified by single crystal X-ray diffraction studies. Complex 3 crystallizes in the monoclinic space group P21/n, with one molecule in the asymmetric unit (Fig. 3). This complex consists of a five-coordinate cobalt centre, connected to three carbonyl groups (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O), one –C(
O), one –C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)N donor and a nitrogen atom from the N–C–N backbone of the methanide ligand. Previously, Jones and co-workers have reported a very similar complex, [Co{C(O)N(Dipp)C(tBu)N(Dipp}(CO)3], by reacting CO gas with an amidinate stabilized cobalt(I) complex.50 In the IR spectrum of complex 3 taken from the reaction mixture bands at 2066 (s), 2009 (s), and 1983 (m) cm−1 can be attributed to C–O stretching frequencies of carbonyl groups. These values have close resemblance to the IR spectrum reported by Jones' group, where they identified 2064 (s), 2004 (s), and 1970 (br) cm−1 as C–O stretching frequencies. The bond lengths and angles of complex 3 and [Co{C(O)N(Dipp)C(tBu)N(Dipp}(CO)3] are very similar.50 The ligand in complex 3 showed a band at 1666 (s) cm−1 corresponding to the ketonic carbonyl –C(O)N stretching. Complex 3 presumably arises from [(DippForm)YbIII(μ-DippForm)(μ-OC)Co(CO)3], the latter formed by partial transmetallation from the bimetallic complex 2. The bridging DippForm is bound to the cobalt(I) centre through the imine N atom, providing an 18 electron Co species which undergoes CO insertion into the Co–N bond resulting in complex 3, where the new N,N′-(Dipp)formimidamidomethanide (C(O)N(Dipp)C(H)N(Dipp) now C,N′-chelates the cobalt.
O)N donor and a nitrogen atom from the N–C–N backbone of the methanide ligand. Previously, Jones and co-workers have reported a very similar complex, [Co{C(O)N(Dipp)C(tBu)N(Dipp}(CO)3], by reacting CO gas with an amidinate stabilized cobalt(I) complex.50 In the IR spectrum of complex 3 taken from the reaction mixture bands at 2066 (s), 2009 (s), and 1983 (m) cm−1 can be attributed to C–O stretching frequencies of carbonyl groups. These values have close resemblance to the IR spectrum reported by Jones' group, where they identified 2064 (s), 2004 (s), and 1970 (br) cm−1 as C–O stretching frequencies. The bond lengths and angles of complex 3 and [Co{C(O)N(Dipp)C(tBu)N(Dipp}(CO)3] are very similar.50 The ligand in complex 3 showed a band at 1666 (s) cm−1 corresponding to the ketonic carbonyl –C(O)N stretching. Complex 3 presumably arises from [(DippForm)YbIII(μ-DippForm)(μ-OC)Co(CO)3], the latter formed by partial transmetallation from the bimetallic complex 2. The bridging DippForm is bound to the cobalt(I) centre through the imine N atom, providing an 18 electron Co species which undergoes CO insertion into the Co–N bond resulting in complex 3, where the new N,N′-(Dipp)formimidamidomethanide (C(O)N(Dipp)C(H)N(Dipp) now C,N′-chelates the cobalt.
 |
| | Fig. 3 Molecular structure of 3. Hydrogen atoms and solvent of crystallisation have been omitted for clarity. Selected bond lengths (Å) and angles [°]: Co1–N2 1.968(3), Co1–C26 1.930(4), Co1–C27 1.755(6), Co1–C28 1.755(6), Co1–C29 1.799(5), C1–N1 1.346(5), C1–N2 1.285(3), N1–C26 1.430(5), C26–O1 1.201(5), C27–O2 1.135(6), C28–O3 1.134(7), C29–O4 1.138(6); C26–Co1–N2 82.7(2), C27–Co1–N2 121.6(2), C28–Co1–N2 117.7(2), C29–Co1–N2 93.6(2), C26–Co1–C27 86.2(2), C26–Co1–C28 86.0(2), C26–Co1–C29 176.1(2), C27–Co1–C28 118.5(3), C28–Co1–C29 96.7(2), N1–C1–N2 118.3(4). | |
Sm–Fe carbonyl complex
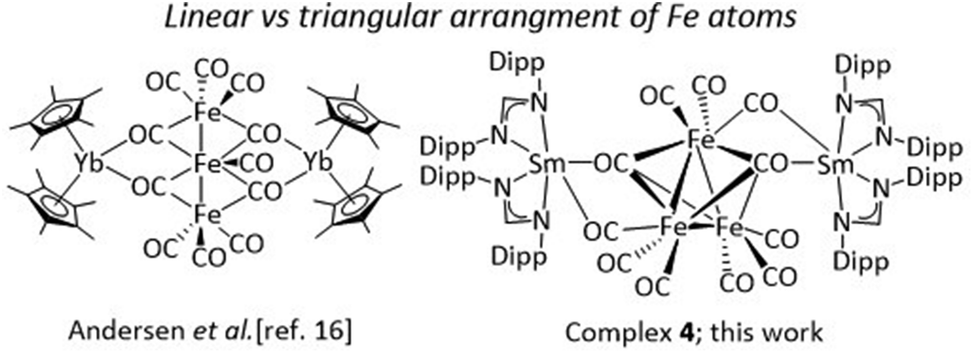
Further reactivity studies of [(DippForm)2SmII(thf)2] were carried out with iron-carbonyl complexes. The reaction between [(DippForm)2SmII(thf)2] and half an equivalent of [Fe3(CO)12] or one equivalent of [Fe2(CO)9] in thf at 60 °C resulted in the formation of [{(DippForm)2SmIII}2{(μ3-CO)2Fe3(CO)9}] (4) (Scheme 4). Red-coloured crystals were isolated in 33% yield after a short work-up. The solid-state IR spectrum showed characteristic CO absorption bands at 2011(vs), 1979 (vs), 1967 (s), 1878 (m), 1830 (w), and 1696 (w) cm−1. Similar CO absorption bands were observed for [(NEt4)2][Fe3(CO)11] at 1938, 1910, 1890, and 1670 cm−1.51 The low-frequency band at 1696 cm−1 is characteristic of the occurrence of bridging isocarbonyls. The solid-state structure of complex 4 was unambiguously determined by single crystal X-ray diffraction studies, revealing two [(DippForm)2SmIII]+ moieties associated together by a [(μ3-CO)2Fe3(CO)9]2− fragment via bridging isocarbonyls (Fig. 4). In complex 4, each Sm atom is hexacoordinated, surrounded by two bidentate amidinate ligands and two bridging isocarbonyls. Notably, the [(DippForm)2SmIII]+ moiety is free of coordinating thf molecules, which may result from the crystallisation of the complex from hot toluene and also the steric crowding around the Sm metal centre. The average Sm–N bond length (2.408(2) Å) in complex 4 is significantly shorter than in [(DippForm)2SmII(thf)2] (Sm–N(average) 2.573(3) Å).35 The [(μ3-CO)2Fe3(CO)9]2− moiety is formed by a two-electron reduction of [Fe3(CO)12] or [Fe2(CO)9] by two [(DippForm)2SmII(thf)2] molecules through SET steps. The [(μ3-CO)2Fe3(CO)9]2− moiety features three Fe atoms arranged in a triangular shape. Upon reaction, formally one CO group in [Fe3(CO)12] is replaced by a di-negative charge leading to the [(μ3-CO)2Fe3(CO)9]2− anion. In contrast, the related YbIII complex [(Cp*2YbIII)2((μ2-CO)4Fe3(CO)7)] isolated by Andersen and co-workers exhibits a linear arrangement for the three Fe atoms (Scheme 5).16 The possible reason for this different arrangement in the solid state may be the sterically demanding nature of the DippForm ligands. Due to disorder of the [(μ3-CO)2Fe3(CO)9]2− moiety the bond lengths and angles cannot be precisely discussed (Fig. S8, ESI†). Despite several attempts, the product formed upon the reaction between [Fe3(CO)12] and [(DippForm)2YbII(thf)2] could not be crystallized and its true identification remains unknown. The structural motive of [(μ3-CO)2Fe3(CO)9]2− observed in complex 4 is very rare as compared to the [(μ-CO)(μ3-CO)Fe3(CO)9]2− unit observed in [(NEt4)2][Fe3(CO)11].51 Previously, the [(μ3-CO)2Fe3(CO)9]2− moiety has been observed in coordination with main group elements such as Li,52 Ca,25 C,53 and B.54 For example, the Ca complex [Ca{(μ3-CO)2Fe3(CO)9}(MeCN)4]n, was obtained by reduction of [Fe3(CO)12] with Ca amalgam in liquid ammonia.25 In contrast, the reactions of [Fe3(CO)12] with Yb metal, resulted in [{(MeCN)3YbFe(CO)4}2·MeCN]n featuring a Yb–Fe bond,55 and that of [Fe3(CO)12] with [(Cp*2Yb(OEt2)], gave [(Cp*2YbIII)2((μ2-CO)4Fe3(CO)7)] (Scheme 5).16
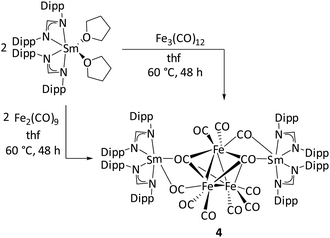 |
| | Scheme 4 Synthesis of complex 4via two routes. | |
 |
| | Fig. 4 Simplified view of the molecular structure of 4 in the solid state. H atoms are omitted for clarity. Selected bond distances (Å) and angles [°]: N1–Sm 2.409(2), N2–Sm 2.426(2), N3–Sm 2.381(2), N4–Sm 2.418(2), N1–C11 1.329(3), N2–C11 1.329(3), N3–C36 1.336(3), N4–C36 1.323(3); N1–C11–N2 117.87(2), N4–C36–N3 117.9(2), N1–Sm–N2 56.20(6), N1–Sm–N4 142.11(6), N3–Sm–N1 108.16(6), N3–Sm–N2 109.53(6), N3–Sm–N4 56.65(6). Discussion of the bond lengths and angles for the [Fe3(CO)11]2− moiety is precluded due to the disorder. | |
 |
| | Scheme 5 Different conformations of the [Fe3(CO)11]2− anion. | |
Conclusions
In conclusion, we have studied the reactivity of two divalent lanthanide (Sm and Yb) complexes coordinated by bulky amidinate ligands towards 3d transition metal carbonyl complexes of groups 8 and 9. The reaction of [(DippForm)2SmII(thf)2] with [Co2(CO)10] resulted in a tetranuclear SmIII–Co heterometallic complex (1) representing a rare example of a SmIII–Co carbonyl complex featuring a bridging isocarbonyl. An analogous reactivity was also observed when [(DippForm)2YbII(thf)2] was reacted with [Co2(CO)10] in toluene resulting in a dinuclear YbIII–Co heterometallic complex (2). Interestingly, the reaction between [(DippForm)2YbII(thf)2] and [Co2(CO)10] in thf resulted in a mononuclear cobalt(I) complex (3). The possible reason for the formation of complex 3 when using thf as solvent may be due to solvent induced displacement of DippForm from the Yb centre. The reaction of [(DippForm)2SmII(thf)2] was also carried out with two iron carbonyl complexes, [Fe2(CO)9] and [Fe3(CO)12]. In both cases, a pentanuclear SmIII–Fe heterometallic complex (4) was isolated. Complex 4 exhibits a [(μ3-CO)2Fe3(CO)9]2− anion with a triangular Fe core. The [(μ3-CO)2Fe3(CO)9]2− anion is sandwiched between two [(DippForm)2SmIII]+ moieties. Interestingly, the [(μ3-CO)2Fe3(CO)9]2− anion in complex 4 is in a triangular arrangement which is in sharp contrast to the linear arrangement observed in [(Cp*2YbIII)2((μ2-CO)4Fe3(CO)7)].16
Experimental
General procedures
All the manipulations of air- and water-sensitive reactions were performed with rigorous exclusion of oxygen and moisture in flame-dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−3 Torr) line or in an argon-filled MBraun glove box. Tetrahydrofuran was distilled under nitrogen from potassium benzophenoneketyl before storage in vacuo over LiAlH4. Hydrocarbon solvents were dried by using an MBraun solvent purification system (SPS 800) and degassed and stored in vacuo over LiAlH4. Elemental analyses were carried out with an Elementar vario Micro cube. IR spectra were obtained on a Bruker Tensor 37 spectrometer equipped with a room temperature DLaTGS detector and a diamond ATR (attenuated total reflection) unit. [(DippForm)2SmII(thf)2]35,41 and [(DippForm)2YbII(thf)2]36 were prepared according to literature procedure. [Fe3(CO)12], [Fe2(CO)9], and [Co2(CO8)] were purchased from commercial suppliers and used as received. No reasonable NMR could be obtained due to low solubility and paramagnetic character.
Synthesis of [{(DippForm)2SmIII(thf)}2{(μ-CO)2Co(CO)2}2] (1)56
To a mixture of [(DippForm)2SmII(thf)2] (205 mg, 0.20 mmol) and [Co2(CO)8] (34 mg, 0.10 mmol) was condensed toluene (15 mL) at −78 °C and then the reaction mixture was stirred for 18 hours at room temperature. The reaction mixture was filtered through P4 frit in a double ampule and flame sealed. Yellow-coloured crystals were grown by slow evaporation of toluene. Crystals were washed carefully with cold toluene and dried under vacuum. Yield: 127 mg, (based on crystals), 0.056 mmol, 56%. Anal. calcd for C116H156N8O10Co2Sm2 (2241.16): C, 62.17; H, 7.02; N, 5.00. Found: C, 62.34; H, 6.84; N, 4.81. IR (ATR) (cm−1): 2959 (s), 2927 (m), 2866 (m), 2020 (m), 1951 (br), 1935 (br), 1922 (br), 1904 (br), 1842 (s), 1819 (s), 1782 (s), 1665 (vs), 1636 (m), 1587 (m), 1527 (m), 1518 (m), 1464 (s), 1457 (s), 1439 (s), 1383 (s), 1361 (m), 1332 (m), 1314 (m), 1289 (m), 1272 (m), 1255 (m), 1236 (s), 1185 (m), 1107 (m), 1098 (m), 1057 (m), 1043 (m), 1016 (m), 934 (m), 912 (m), 865 (br), 800 (s), 753 (vs), 673 (s), 565 (w), 553 (s), 550 (s), 531 (s) 522 (s), 506 (s) 435 (w).
Synthesis of [{(DippForm)2YbIII(thf)}{(μ-CO)Co(CO)3}] (2)56
Following the procedure described above for 1, the reaction of [(DippForm)2YbII(thf)2] (209 mg, 0.20 mmol) and [Co2(CO)8] (34 mg, 0.10 mmol) afforded orange-coloured crystals of 2. Yield: 146 mg, (based on crystals), 0.127 mmol, 63%. Anal. calcd for C58H78N4O5CoYb (1143.27): C, 60.93; H, 6.88; N, 4.90. Found: C, 61.30; H, 6.89; N, 4.90. IR (ATR) (cm−1): 2960 (s), 2927 (m), 2869 (m), 2031 (m), 2016 (m), 1915 (br), 1792 (m), 1748 (w), 1665 (vs), 1636 (m), 1587 (s), 1521 (m), 1465 (m), 1458 (m), 1439 (s), 1383 (s), 1361 (m), 1331 (m), 1331 (m), 1319 (m), 1290 (m), 1268 (m), 1255 (w), 1236 (m), 1186 (m), 1107 (m), 1107 (w), 1097 (w), 1058 (w), 1043 (w), 1025 (w), 1007 (w), 934 (w), 883 (w), 871 (w), 822 (w), 799 (s), 767 (w), 753 (w), 712 (w), 673 (w), 564 (w), 551 (vs), 510 (m), 434 (m), 418 (m).
Synthesis of [Co(DippFormCO)(CO)3] (3)
THF (30 mL) was added to a Schlenk flask charged with [Yb(DippForm)2(thf)2] (418 mg, 0.40 mmol) under purified nitrogen Co2(CO)8 (160 mg, 0.47 mmol) was added to the orange solution with stirring and the mixture was stirred for 12 hours at ambient temperature. The colour of the solution turned from orange to dark brown. The solution was filtered to remove any solid materials and the volume reduced under vacuum. The solution was then stored at 3 °C for 3 days during which time needle like crystals formed. IR (Nujol) (cm−1): 2066 (s), 2009 (s), 1983 (m), 1897 (w), 1666 (s), 1587 (m), 1287 (m), 1234 (w), 1180 (m), 1098 (w), 1058 (w), 1000 (w), 935 (w), 821 (w), 799 (m), 753 (w), 767 (m), 722 (m). Complex 3 could not be obtained as analytically pure material.
Synthesis of [{(DippForm)2SmIII}2{(μ3-CO)2Fe3(CO)9}] (4)56
To a mixture of [(DippForm)2SmII(thf)2] (205 mg, 0.20 mmol) and [Fe3(CO)12] (51 mg, 0.10 mmol) or [Fe2(CO)9] (72 mg, 0.20 mmol) was condensed thf (15 mL) at −78 °C and the reaction mixture was stirred for 48 hours at 60 °C. All the volatiles were removed in vacuo. Toluene (15 mL) was added to the residue and refluxed for five minutes and the hot reaction mixture was filtered. Dark red-coloured crystals were obtained upon slowly cooling the filtrate to room temperature. The mother liquor was decanted off and the product was dried under vacuum. Yield: 74 mg (based on crystals), 0.033 mmol, 33% (average). Anal. calcd for C111H140N8O11Fe3Sm2 (2230.64): C, 59.77; H, 6.33; N, 5.02. Found: C, 59.77; H, 6.20; N, 4.81. IR (ATR) (cm−1): 2962 (vs), 2926 (m), 2869 (m), 2011 (vs), 1979 (vs), 1967 (s), 1878 (m), 1830 (w), 1696 (w), 1667 (s), 1640 (s), 1636 (m), 1586 (s), 1512 (s), 1464 (s) 1457 (m), 1437 (m), 1384 (s), 1362 (m), 1346 (m), 1332 (m), 1314 (s), 1278 (s), 1368 (m), 1254 (s), 1236 (m), 1186 (m), 1112 (w), 1098 (m), 1054 (m), 1042 (m), 1023 (w), 1004 (w), 947 (w), 935 (m), 823 (s), 800 (s) 768 (m), 753 (s), 681 (s), 645 (m), 612 (s), 585 (s), 508 (w), 474 (w), 458 (w), 444 (w), 438 (w), 433 (w), 420 (w).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
RY and PWR acknowledge funding for the current project from the SFB 1176 funded by the German Research Council (DFG). TS thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. PCJ and GBD thank the Australian research Council for grant DP190100798.
Notes and references
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- A. Bhunia, M. T. Gamer, L. Ungur, L. F. Chibotaru, A. K. Powell, Y. Lan, P. W. Roesky, F. Menges, C. Riehn and G. Niedner-Schatteburg, Inorg. Chem., 2012, 51, 9589–9597 CrossRef CAS PubMed.
- J.-L. Liu, J.-Y. Wu, Y.-C. Chen, V. Mereacre, A. K. Powell, L. Ungur, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2014, 53, 12966–12970 CrossRef CAS PubMed.
- J. Wu, L. Zhao, L. Zhang, X.-L. Li, M. Guo, A. K. Powell and J. Tang, Angew. Chem., Int. Ed., 2016, 55, 15574–15578 CrossRef CAS PubMed.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- T. D. Pasatoiu, C. Tiseanu, A. M. Madalan, B. Jurca, C. Duhayon, J. P. Sutter and M. Andruh, Inorg. Chem., 2011, 50, 5879–5889 CrossRef CAS PubMed.
- A. Bhunia, M. A. Gotthardt, M. Yadav, M. T. Gamer, A. Eichhöfer, W. Kleist and P. W. Roesky, Chem. – Eur. J., 2013, 19, 1986–1995 CrossRef CAS PubMed.
- M. Yadav, A. Bhunia, S. K. Jana and P. W. Roesky, Inorg. Chem., 2016, 55, 2701–2708 CrossRef CAS PubMed.
- C. E. Plečnik, S. Liu and S. G. Shore, Acc. Chem. Res., 2003, 36, 499–508 CrossRef PubMed.
- P. L. Arnold, J. McMaster and S. T. Liddle, Chem. Commun., 2009, 818–820 RSC.
- C. P. Burns, X. Yang, S. Sung, J. D. Wofford, N. S. Bhuvanesh, M. B. Hall and M. Nippe, Chem. Commun., 2018, 54, 10893–10896 RSC.
- T. Pugh, N. F. Chilton and R. A. Layfield, Angew. Chem., Int. Ed., 2016, 55, 11082–11085 CrossRef CAS PubMed.
- C. P. Burns, X. Yang, J. D. Wofford, N. S. Bhuvanesh, M. B. Hall and M. Nippe, Angew. Chem., Int. Ed., 2018, 57, 8144–8148 CrossRef CAS PubMed.
- T. D. Tilley and R. A. Andersen, J. Chem. Soc., Chem. Commun., 1981, 985–986 RSC.
- T. D. Tilley and R. Andersen, J. Am. Chem. Soc., 1982, 104, 1772–1774 CrossRef CAS.
- J. M. Boncella and R. A. Andersen, J. Chem. Soc., Chem. Commun., 1984, 809–810 RSC.
- J. M. Boncella and R. A. Andersen, Inorg. Chem., 1984, 23, 432–437 CrossRef CAS.
- A. Recknagel, A. Steiner, S. Brooker, D. Stalke and F. T. Edelmann, Chem. Ber., 1991, 124, 1373–1375 CrossRef CAS.
- G. B. Deacon, Z. Guo, P. C. Junk and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 8486–8489 CrossRef CAS PubMed.
- C. E. Plečnik, S. Liu, X. Chen, E. A. Meyers and S. G. Shore, J. Am. Chem. Soc., 2004, 126, 204–213 CrossRef PubMed.
- P. V. Poplaukhin, X. Chen, E. A. Meyers and S. G. Shore, Inorg. Chem., 2006, 45, 10115–10125 CrossRef CAS PubMed.
- M. P. Blake, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 15358–15361 CrossRef CAS PubMed.
- M. P. Blake, N. Kaltsoyannis and P. Mountford, Chem. Commun., 2013, 49, 3315–3317 RSC.
- M. P. Blake, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2015, 137, 12352–12368 CrossRef CAS PubMed.
- R. Green, A. C. Walker, M. P. Blake and P. Mountford, Polyhedron, 2016, 116, 64–75 CrossRef CAS.
- W. J. Evans, I. Bloom, J. W. Grate, L. A. Hughes, W. E. Hunter and J. L. Atwood, Inorg. Chem., 1985, 24, 4620–4623 CrossRef CAS.
- I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Chuklanova, N. I. Kirillova, A. K. Shestakova, I. N. Parshina, A. I. Gusev and G. K. I. Magomedov, J. Am. Chem. Soc., 1993, 115, 3156–3166 CrossRef CAS.
- C. Döring, A.-M. Dietel, M. V. Butovskii, V. Bezugly, F. R. Wagner and R. Kempe, Chem. – Eur. J., 2010, 16, 10679–10683 CrossRef PubMed.
- B. Oelkers, M. V. Butovskii and R. Kempe, Chem. – Eur. J., 2012, 18, 13566–13579 CrossRef CAS PubMed.
- W. J. Evans and B. L. Davis, Chem. Rev., 2002, 102, 2119–2136 CrossRef CAS PubMed.
- W. J. Evans, Organometallics, 2016, 35, 3088–3100 CrossRef CAS.
-
H. M. Nicholas and D. P. Mills, Lanthanides: Divalent Organometallic Chemistry, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, 2017 Search PubMed.
- R. Yadav, T. Simler, M. T. Gamer, R. Köppe and P. W. Roesky, Chem. Commun., 2019, 55, 5765–5768 RSC.
- M. L. Cole and P. C. Junk, Chem. Commun., 2005, 2695–2697 CAS.
- M. L. Cole, G. B. Deacon, C. M. Forsyth, P. C. Junk, K. Konstas, J. Wang, H. Bittig and D. Werner, Chem. – Eur. J., 2013, 19, 1410–1420 CrossRef CAS PubMed.
- S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, H. Schnöckel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740–5741 CrossRef CAS PubMed.
- T. Li, J. Wiecko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2011, 50, 9491–9495 CrossRef CAS PubMed.
- T. Li, M. T. Gamer, M. Scheer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2013, 49, 2183–2185 RSC.
- M. Kühling, R. McDonald, P. Liebing, L. Hilfert, M. J. Ferguson, J. Takats and F. T. Edelmann, Dalton Trans., 2016, 45, 10118–10121 RSC.
- C. Schoo, S. Bestgen, M. Schmidt, S. N. Konchenko, M. Scheer and P. W. Roesky, Chem. Commun., 2016, 52, 13217–13220 RSC.
- Y.-Z. Ma, S. Bestgen, M. T. Gamer, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 13249–13252 CrossRef CAS PubMed.
- C. Schoo, S. Bestgen, R. Köppe, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2018, 54, 4770–4773 RSC.
- C. Schoo, S. Bestgen, A. Egeberg, J. Seibert, S. N. Konchenko, C. Feldmann and P. W. Roesky, Angew. Chem., Int. Ed., 2019, 58, 4386–4389 CrossRef CAS PubMed.
- D. Werner, G. B. Deacon and P. C. Junk, Inorg. Chem., 2019, 58, 1912–1918 CrossRef CAS PubMed.
- W. J. Evans, G. W. Rabe, J. W. Ziller and R. J. Doedens, Inorg. Chem., 1994, 33, 2719–2726 CrossRef CAS.
- C. Schoo, S. Bestgen, A. Egeberg, S. Klementyeva, C. Feldmann, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 5912–5916 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Chuklanova, A. I. Gusev and G. K. Magomedov, Metalloorg. Khim., 1988, 1, 1383–1390 CAS.
- C. Jones, C. Schulten, R. P. Rose, A. Stasch, S. Aldridge, W. D. Woodul, K. S. Murray, B. Moubaraki, M. Brynda, G. La Macchia and L. Gagliardi, Angew. Chem., Int. Ed., 2009, 48, 7406–7410 CrossRef CAS PubMed.
- F. Y.-K. Lo, G. Longoni, P. Chini, L. D. Lower and L. F. Dahl, J. Am. Chem. Soc., 1980, 102, 7691–7701 CrossRef CAS.
- W. Petz and B. Neumüller, Z. Anorg. Allg. Chem., 2010, 636, 551–556 CrossRef CAS.
- A. A. Aradi, F. W. Grevels, C. Krueger and E. Raabe, Organometallics, 1988, 7, 812–818 CrossRef CAS.
- T. V. Laine, M. Klinga and M. Leskelä, Eur. J. Inorg. Chem., 1999, 1999, 959–964 CrossRef.
- H. Deng and S. G. Shore, J. Am. Chem. Soc., 1991, 113, 8538–8540 CrossRef CAS.
-
R. Yadav, Synthesis of Heterometallic Zinc-Gold and Lanthanide-Transition Metal carbonyl complexes and Reactivity Study of Pentaphosphaferrocene Towards Low-Valent Main Group Species, PhD Thesis, Karlsruhe Institute of Technology (KIT), Kalrsruhe, Germany, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra and X-ray crystallographic details of complexes 1–4, 1994647 (1), 1994648 (2), 1952438 (3), and 1994649 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt01271h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Md Elius
Hossain
a,
Md Elius
Hossain