
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn 8-hydroxyquinoline–proline hybrid with multidrug resistance reversal activity and the solution chemistry of its half-sandwich organometallic Ru and Rh complexes†
János P.
Mészáros
 ab,
Jelena M.
Poljarević
ac,
István
Szatmári
d,
Oszkár
Csuvik
d,
Ferenc
Fülöp
d,
Norbert
Szoboszlai
e,
Gabriella
Spengler
bf and
Éva A.
Enyedy
*ab
ab,
Jelena M.
Poljarević
ac,
István
Szatmári
d,
Oszkár
Csuvik
d,
Ferenc
Fülöp
d,
Norbert
Szoboszlai
e,
Gabriella
Spengler
bf and
Éva A.
Enyedy
*ab
aDepartment of Inorganic and Analytical Chemistry, Interdisciplinary Excellence Centre, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: enyedy@chem.u-szeged.hu
bMTA-SZTE Lendület Functional Metal Complexes Research Group, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
cFaculty of Chemistry, University of Belgrade, Studentski trg. 12-16, 11000 Belgrade, Serbia
dInstitute of Pharmaceutical Chemistry and Stereochemistry Research Group of Hungarian Academy of Sciences, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary
eLaboratory for Environmental Chemistry and Bioanalytics, Institute of Chemistry, Eötvös Lóránd University, Pázmány Péter stny. 1/A, H-1117 Budapest, Hungary
fDepartment of Medical Microbiology and Immunobiology, University of Szeged, Dóm tér 10, H-6720 Szeged, Hungary
First published on 14th May 2020
Abstract
Herein the design and synthesis of a new 8-hydroxyquinoline derivative, (S)-5-chloro-7-((proline-1-yl)methyl)8-hydroxyquinoline (HQCl-Pro), with good water solubility and multidrug resistance reversal activity are reported. In this work the proton dissociation processes of HQCl-Pro and its complex formation with [Rh(η5-C5Me5)(H2O)3]2+, [Ru(η6-p-cymene)(H2O)3]2+ and [Ru(η6-toluene)(H2O)3]2+ were investigated by the combined use of pH-potentiometry, UV-visible spectrometry and 1H NMR spectroscopy. Our results revealed the prominent solution stability of the complexes in all cases. The lipophilicity of the complexes increased with the chloride ion concentration, and the complexes showed moderate log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D values (−0.8 to +0.4) at pH 7.4 at all tested Cl− concentrations. The formation of mixed hydroxido complexes from the aqua complexes was characterized by relatively high pKa values (8.45–9.62 in chloride-free medium). Complexation processes are much slower with the Ru(η6-arene) triaqua cations than with [Rh(η5-C5Me5)(H2O)3]2+. Both the pKa values and H2O/Cl− exchange constants of the Ru-complexes are lower by 0.5–1.0 orders of magnitude than those of the Rh analogue. Arene loss (p-cymene and toluene) and oxidation were found in the case of Ru-complexes when an excess of HQCl-Pro and aromatic (N,N) bidentate ligands was added. The cytotoxicity and antiproliferative effect of HQCl-Pro and its complexes were assayed in vitro. In contrast to the structurally familiar 8-hydroxyquinoline, HQCl-Pro and its Rh(η5-C5Me5) complex were somewhat more effective against drug resistant Colo 320 adenocarcinoma human cells compared to the drug sensitive Colo 205 cells. The Ru- and Rh-complexes showed a similar metal uptake level after 4 h, while a longer incubation time resulted in higher cellular Rh concentration.
D values (−0.8 to +0.4) at pH 7.4 at all tested Cl− concentrations. The formation of mixed hydroxido complexes from the aqua complexes was characterized by relatively high pKa values (8.45–9.62 in chloride-free medium). Complexation processes are much slower with the Ru(η6-arene) triaqua cations than with [Rh(η5-C5Me5)(H2O)3]2+. Both the pKa values and H2O/Cl− exchange constants of the Ru-complexes are lower by 0.5–1.0 orders of magnitude than those of the Rh analogue. Arene loss (p-cymene and toluene) and oxidation were found in the case of Ru-complexes when an excess of HQCl-Pro and aromatic (N,N) bidentate ligands was added. The cytotoxicity and antiproliferative effect of HQCl-Pro and its complexes were assayed in vitro. In contrast to the structurally familiar 8-hydroxyquinoline, HQCl-Pro and its Rh(η5-C5Me5) complex were somewhat more effective against drug resistant Colo 320 adenocarcinoma human cells compared to the drug sensitive Colo 205 cells. The Ru- and Rh-complexes showed a similar metal uptake level after 4 h, while a longer incubation time resulted in higher cellular Rh concentration.
Introduction
During the administration of conventional chemotherapeutic agents, multidrug resistant (MDR) cancer phenotypes are often developed manifesting resistance to related and unrelated classes of compounds, which is one of the major impediments to successful treatment.1 The ATP-binding cassette transporter P-glycoprotein (P-gp, ABCB1) is the most known transmembrane transporter that is associated with this resistance phenomenon; however, multidrug resistance protein 1 and 2 (MRP1 and MRP2) and breast cancer resistance protein (BCRP) also cause an elevated efflux of toxic compounds.1 Potent P-gp inhibitors were explored for overcoming the MDR (e.g. verapamil and cyclosporine A), although their use is accompanied by undesirable side effects.1 Szakács et al. reported a group of anticancer compounds which are more cytotoxic against MDR cancer cells than drug sensitive ones.2 These molecules are not the inhibitors of P-gp, and their mechanism of action is still not revealed in detail. 8-Hydroxyquinolines were found to be an important family of MDR-selective compounds. It was shown that the CH2–N subunit at position 7 plays an important role in the collateral selectivity, which is present in compounds e.g. NSC1014, NSC297366, NSC693871 and NSC57969.2 The 8-hydroxyquinoline (HQ) scaffold itself has the potential for various structural modifications including changes due to pharmaceutical purposes.3–5 HQ and its numerous derivatives are well-known compounds and have been used in analytical chemistry for many decades due to their simple chemical structure and strong coordination ability to metal ions. E.g. clioquinol (5-chloro-7-iodo-8-hydroxyquinoline) was used as an antiprotozoal and antifungal drug; moreover, it was explored as a treatment for Alzheimer's and Parkinson's diseases.3,5Compounds with the 8-hydroxyquinoline moiety were also reported to exhibit anti-inflammatory, antiviral and antiparasitic effects and numerous HQ derivatives with various substituents have been developed and tested as anticancer agents in the last few decades.3–5 Among them halogenated compounds were often found to be more efficient.5,6
Not only 8-hydroxyquinolines but their particular metal complexes show anticancer activity as well. The most prominent example is the orally active tris(8-hydroxy-quinolato)gallium(III) (KP46), which is now under clinical trials and was successfully tested in phase I on renal cancer.7,8
Other examples are the complexes of platinum group organometallic cations bearing an aromatic ligand in a half-sandwich configuration (usually p-cymene (p-cym) or 1,2,3,4,5-pentamethyl-cyclopentadienyl (C5Me5) ligand). The complexes of these cations usually have various physico-chemical properties: the low oxidation state of the metal center (e.g. Ru2+) was stabilized and the lipophilicity was increased by the arene ligand; their kinetic lability can be associated with the strong π-donor/acceptor ability of the arene ligand as well.9,10 Moreover, the fine-tuning of the 3D structure and electronic properties can be easily achieved by functionalization of the arene or the remaining facial ligands. The interaction with the chloride content of the medium can change charge and lipophilicity. Nevertheless, new reaction pathways with biomolecules can be reached, which differ from the ligand–biomolecule interactions: e.g. catalysis of GSH oxidation.9 There are two main types of active complexes: in the first class, a monodentate ligand binds strongly to the metal centre and the leaving group(s) is/are present on two other coordination sites. Widely known examples are the RAPTA complexes where a strong P-donor, 1,3,5-triaza-7-phosphaadamantane (PTA), is the monodentate ligand.11 In the other class, the strongly coordinated bidentate ligand occupies two sites, and a leaving group is bound in a monodentate way. The dissociation of the latter can be fine-tuned well with the change of donor atoms. For example, the target biomolecule is switched from DNA to protein, depending on the nature of the bidentate ligand: (N,N) or (O,O).12 This field contains a plethora of studied complexes, and an early example is the group of RAED complexes which are in vivo active compounds showing activity on cisplatin-resistant cell lines.13 Though structure–activity relationships have been reported for these complexes, there is still no organoruthenium containing drug in clinical use.14,15
Half-sandwich organometallic complexes of the (N,O) donor 8-hydroxyquinolines are also often investigated. The organo-ruthenium complexes formed with clioquinol and other halogenated 8-hydroxyquinoline derivatives were studied thoroughly by Turel et al., and the inhibition of cathepsin B16 and the anticancer,17 antileukaemic18 and antibacterial effects19 were reported for these compounds. Nitroxoline (8-hydroxy-5-nitroquinoline) derivatives showed the anti-metastatic effect, which was improved by coordination to a half-sandwich Ru(p-cym) cation.20 An earlier study demonstrated the anticancer and antibacterial activities of the half-sandwich Rh and Ir complexes of HQ as well.21 These complexes are characterized by fairly low IC50 values (∼10 μM or less), which are promising results; however, they suffer from poor water solubility.6 The limited water solubility is a major problem encountered with drug formulation since it makes the administration difficult and might restrain the attainment of the desired concentration in the blood circulatory system. Movassaghi et al. introduced a more polar aromatic ligand (N-acetyl-L-phenylalanine ethyl ester) instead of p-cymene, and with this modification the solubility could be improved, while the cytotoxicity remained at the low-micromolar level.22
The hydrophilic nature of the complexes can be increased via the improved water solubility of the coordinating HQ derivative by the introduction of polar functional groups. However, the use of 8-hydroxyquinoline-5-sulfonate with excellent water-solubility caused the loss of the anticancer activity reported in our previous study.23 Therefore, the lipophilic nature of the HQ ligand should be optimized very carefully, and finding the optimal balance between lipophilicity and water-solubility is a very important endeavour in the development of novel anticancer compounds. Therefore, in this work we aimed to design an HQ–proline hybrid with elevated water solubility containing the CH2–N subunit at position 7 for the expected MDR-selectivity. It is also interesting how the coordination to metal ions has an effect on the cytotoxicity and MDR selectivity. When the MDR selective ligand 7-(1-piperidinylmethyl)-8-hydroxyquinoline (NSC57969) was combined with organometallic half-sandwich rhodium and ruthenium cations, the MDR selectivity remained only in the case of the Rh(η5-C5Me5) complex, while the coordination to Ru(η6-p-cym) resulted in the loss of this property.23 Herein the synthesis and solution chemical characterization of a new, water-soluble derivative of HQ, (S)-5-chloro-7-((proline-1-yl)methyl)8-hydroxyquinoline (HQCl-Pro, Scheme 1), is reported. Its complex formation with half-sandwich cations [Rh(η5-C5Me5)(H2O)3]2+, [Ru(η6-p-cym)(H2O)3]2+ and [Ru(η6-toluene)(H2O)3]2+ (abbreviated as [Ru(η6-tol)(H2O)3]2+) is also characterized in solution. In addition, the measurement of the lipophilicity of the compounds depending on the actual chloride ion concentration was performed: in the different biofluids the concentration of this coordinating ion may change charge and may have serious effects. The cytotoxicity of HQCl-Pro was monitored in sensitive and in multidrug resistant cancer cells and the effect of complexation with the selected organometallic triaqua cations on the biological activity was assayed as well.
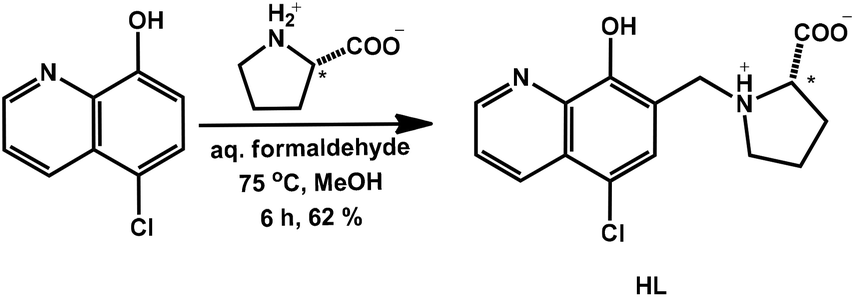 | ||
| Scheme 1 Synthetic route for the HQCl-Pro ligand. | ||
Results and discussion
Synthesis of HQCl-Pro and its complexes
A novel derivative of 5-chloro-8-hydroxyquinoline (HQCl) was designed with increased water solubility via the incorporation of the zwitterionic proline amino acid moiety. L-Proline was attached to the HQCl scaffold at position 7 via a methylene linker providing the CH2–N subunit that is reported to be crucial for the MDR reversal activity.2,23 The coupling of HQCl with L-proline in the presence of formaldehyde via a modified Mannich reaction was achieved in methanol under reflux conditions (Scheme 1). The formed (S)-5-chloro-7-((proline-1-yl)methyl)8-hydroxyquinoline (HQCl-Pro) was isolated by crystallization from the cooled reaction mixture.The half-sandwich organometallic complexes with Rh(η5-C5Me5), Ru(η6-p-cym) and Ru(η6-tol) were prepared by mixing the ligand with a half-equivalent amount of the dimeric precursors [M(arene)Cl2]2 in methanol solution for 1 h at room temperature, then the solution was concentrated and precipitation was completed with the addition of diethyl ether. The formed complex was filtered out, washed with n-hexane and dried. Complexes were collected in good yields (84–90%). HQCl-Pro and its complexes were characterized by 1H and 13C NMR spectroscopy (attached proton test (APT)) and electrospray ionization mass spectrometry (ESI-MS). Mass spectra and NMR spectra are shown in the ESI (Fig. S1–S12†). The NMR spectra showed a double set of peaks in CD3OD, which can be explained by diastereomer formation (as was found in ref. 22 for complexes with chiral arene) or the appearance of a rigid structure (as it appears also in water for deprotonation vide infra). The differences of the two sets are shown in Fig. S13† peak-by-peak, projected on the proposed structures of complexes. The biggest differences are found in positions 14, 6 and 9, which can be connected with the diastereomer formation: in the complex, next to the chiral carbon atom, the Ru centre and the N of the protonated amino group become chiral, and these positions (6,9,14) are close to these atoms.
Additionally, the increased solubility and stability of the ligand and complexes were checked in phosphate buffer at pH = 7.4. All compounds were water-soluble; 10 mM concentration exceeds the limits of solubility of HQ, HQCl and their complexes. The compounds were stable for 1 week (not shown), except for the ligand itself, which showed a new set of peaks in the aliphatic region in a ∼20% ratio (see Fig. S14†) after 6 days.
Hydrolysis of the organometallic cations and proton dissociation processes of HQCl-Pro
Knowing the stability constants of a metal complex and proton dissociation processes of a molecule is necessary to understand the role of different forms of biologically active compounds in the biological environment. It can give information about the response to pH and explain the active and inactive forms of a prodrug. In order to describe the solution speciation of the metal complexes, characterization of the hydrolysis of the organometallic cations and the (de)protonation equilibrium processes of the ligand is needed under the conditions used. The hydrolytic behaviour of the selected organometallic cations ([Rh(η5-C5Me5)(H2O)3]2+, [Ru(η6-p-cym)(H2O)3]2+ and [Ru(η6-tol)(H2O)3]2+) has been studied in detail previously.24,25 Overall stability constants were reported for the μ-hydroxido bridged dinuclear species formed24,25 and these constants are used in this work. According to Buglyó et al., the hydrolysis of the [Ru(η6-arene)(H2O)3]2+ organometallic cations can be adequately described by the formation of the hydrolysis product [(Ru(η6-arene))2(μ-OH)3]+ in chloride-free medium (I = 0.20 M (KNO3)).25 However, in the case of [Rh(η5-C5Me5)(H2O)3]2+ the formation of [(Rh(η5-C5Me5))2(μ-OH)2]2+ and [(Rh(η5-C5Me5))2(μ-OH)3]+ was reported.24 The interaction between the metal ions and the hapto ligands is strong, and as a result the release of the aromatic ligands was not detected in the studied pH range (pH = 0.7–11.5) (only in the case of some competition reactions vide infra). These organometallic cations are considered as units and denoted as ‘M(arene)’ hereinafter.The knowledge of the pKa value of a bioactive compound is needed not only for speciation studies, but it is also a key parameter affecting the pharmacokinetic properties, since with the pKa values the actual protonation state and charge of the molecule at a given pH can be calculated. In HQCl-Pro the incorporation of the L-prolinylmethyl substituent results in two additional dissociable protons besides the quinolinium-NH+ (NqH+) and the hydroxyl group (Scheme 2) of the HQ scaffold. N-methyl-L-proline (N-Me-Pro) is structurally similar to this substituent and it has two pKa values for the carboxylic acid and amino functions (Table 1).26
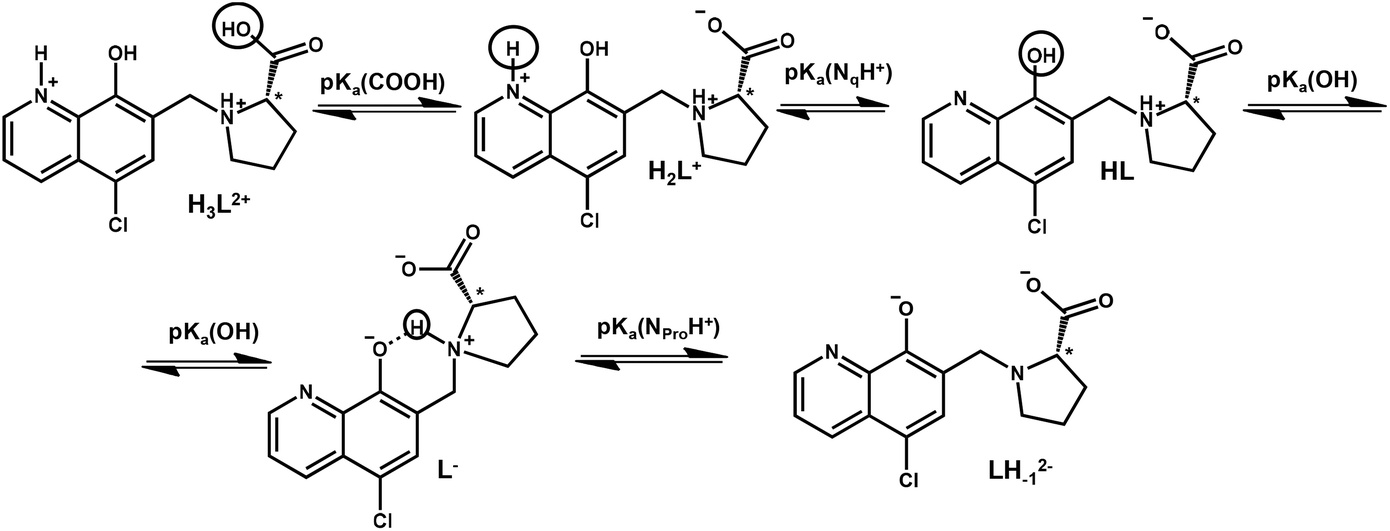 | ||
| Scheme 2 Proton dissociation processes of the ligand HQCl-Pro. | ||
| Method | pKa (COOH) | pKa (NqH+) | pKa (OH) | pKa (NProH+) | |
|---|---|---|---|---|---|
| a I = 0.20 M (KCl), taken from ref. 27. b Calculated with Marvin (ref. 28). c Estimated from the data obtained by UV-Vis titrations, pH = 2.0–11.5 (I = 0.20 M (KNO3)). d I = 0.10 M (KCl), taken from ref. 26. | |||||
| HQ | pH-potentiometry | — | 4.99a | 9.51a | — |
| HQCl | Predicted | 4.01b | 8.37b | — | |
| UV-Vis | 3.8c | 7.6c | |||
| N-Me-Pro | pH-potentiometry | 1.75d | — | — | 10.36d |
| HQCl-Pro | pH-potentiometry | — | 2.36 ± 0.02 | 7.76 ± 0.01 | >11.5 |
| UV-Vis | — | — | 7.63 ± 0.01 | — | |
| 1H NMR | ≪2 | 2.22 ± 0.02 | 7.62 ± 0.01 | — | |
In order to characterize the proton dissociation processes of HQCl-Pro pH-potentiometric, UV-visible (UV-Vis) and 1H NMR titrations were performed. Although this compound has four dissociable protons, only two deprotonation processes could be determined adequately (with acceptable standard deviation) in the studied pH range by pH-potentiometry. The assignment of the deprotonation processes to the functional groups was done by the interpretation of the 1H NMR and UV-Vis spectral changes (Fig. 1, S15† and Table 1). Notably, HL denotes the neutral (zwitterionic) form of the ligand.
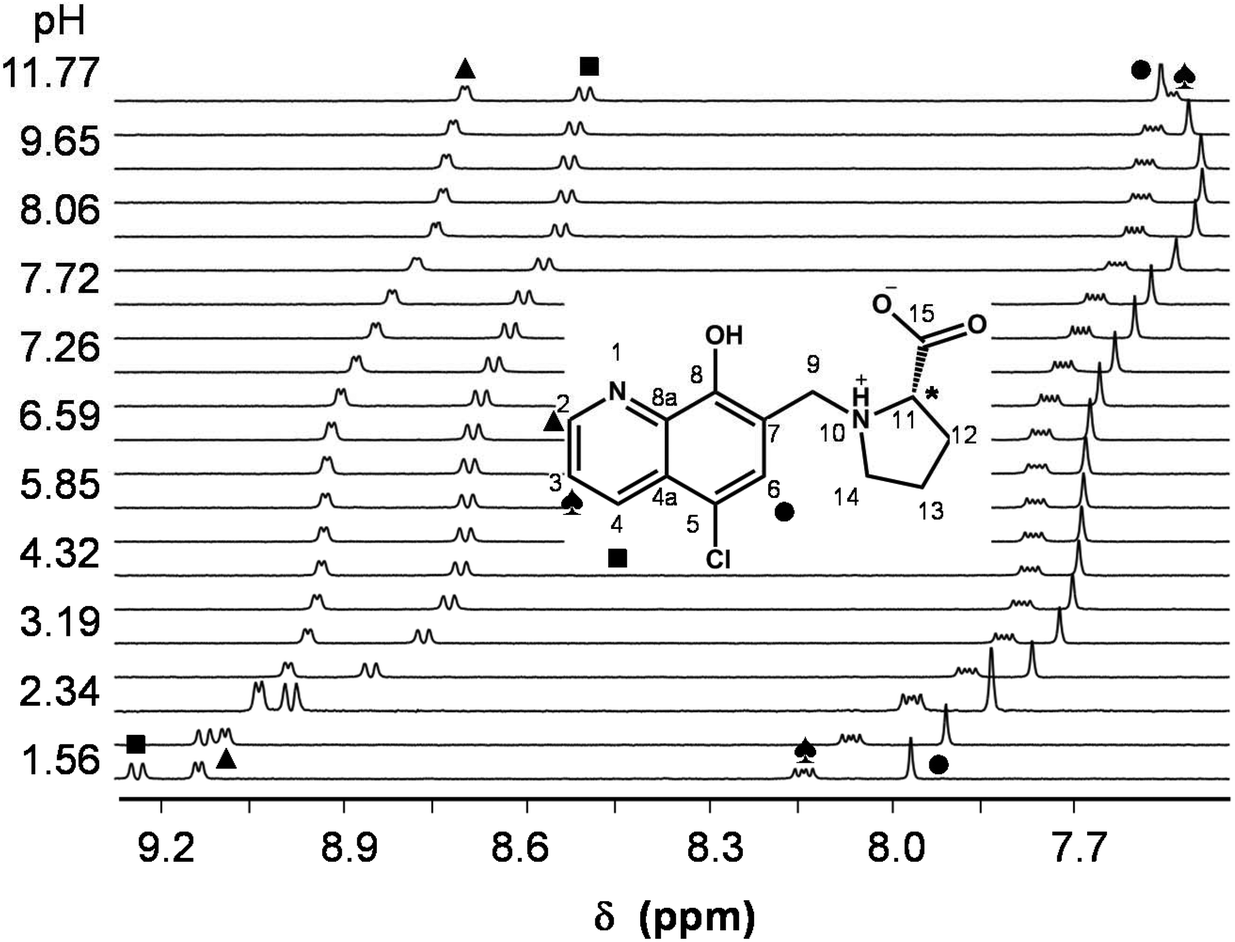 | ||
| Fig. 1 1H NMR spectra of HQCl-Pro recorded at pH = 1.6–11.8 (low-field region). Numbering of the positions and peak assignment is shown on the central structure. {c(HQCl-Pro) = 480 μM; solvent: 90% H2O/10% D2O; T = 25.0 °C; I = 0.20 M (KNO3)}. | ||
The most acidic pKa (≪2) of HQCl-Pro belongs to the carboxylic group of the prolinyl substituent, which caused shifts of the peaks only in the aliphatic region of the 1H NMR spectra (not shown). The deprotonation of the quinolinium NqH+ (up to pH 4) and OH (pH 5.5–9.5) groups was ascertained as high-field shifts of all the CH proton peaks (Fig. 1). The pKa values of these groups are much lower than those of HQ (see in Table 1) as a consequence of the presence of the two electron-withdrawing substituents (chlorine and the protonated CH2–NProH+ moiety). The structurally more related HQCl suffers from very poor solubility in aqueous medium and we could not determine its pKa values accurately enough in pure water by UV-Vis titrations even at rather low concentrations (5–10 μM) (see the estimated values in Table 1). Both the experimentally obtained and the predicted values for HQCl represent significantly lower values compared to those of HQ due to the electron withdrawing effect of the chlorine substituent as it is expected. Based on the pKa values it can be observed that the prolinyl amino group deprotonates at a much higher pH than the amino group in N-Me-Pro.
This can be the result of an intramolecular H-bond between the deprotonated hydroxylate and the NProH+ moiety (Scheme 2). A similar hydrogen bond was found in the crystal structures between the hydroxylate group and a protonated morpholine or piperidinyl nitrogen in HQ derived Mannich bases, and these substituents also had a similar effect on the pKa values.29
According to the 1H NMR spectra, only the singlet C6H peak seems to be sensitive to this deprotonation and a high-field shift was observed at pH > 10. The UV-Vis spectra, recorded at various pH values (Fig. 2), revealed three deprotonation processes. However, only one pKa value could be computed accurately by the deconvolution of the spectra (Table 1) as in the other two cases the whole deprotonation processes could not be seen.
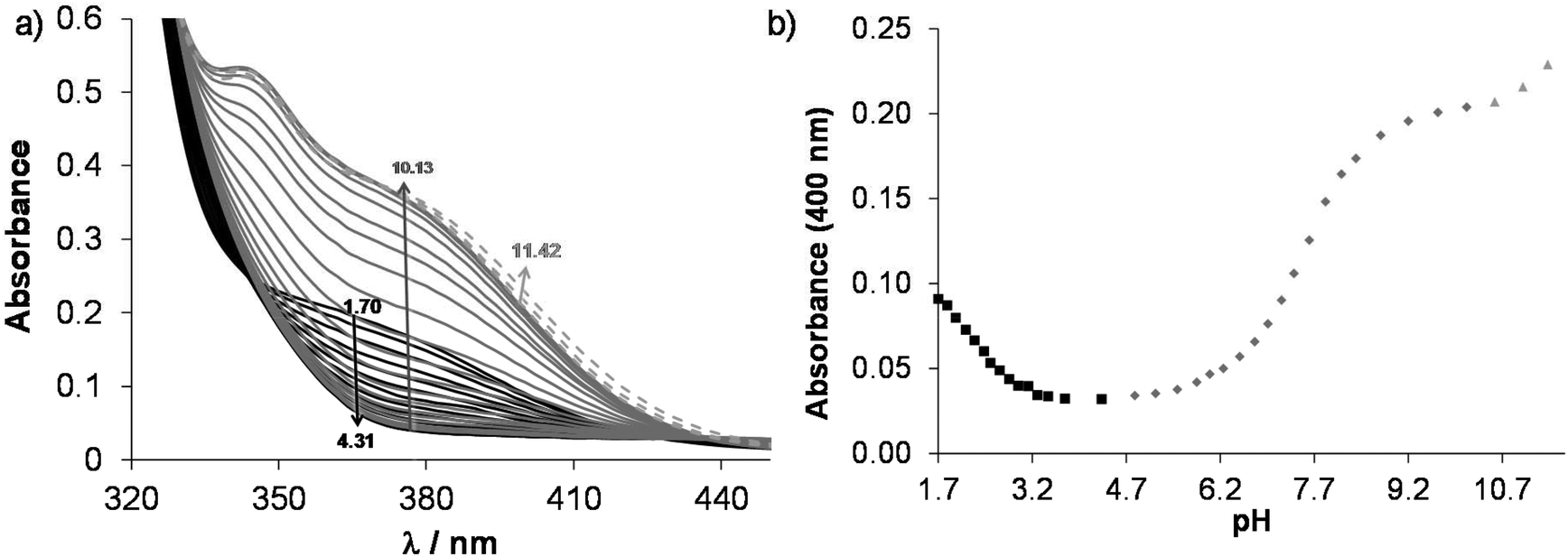 | ||
Fig. 2 (a) UV-Vis absorption spectra of HQCl-Pro at pH = 1.7–11.4. (b) Absorbance values at 400 nm as a function of pH. The separated deprotonation steps are shown with different symbols (■, ◆, ▲). {c(HQCl-Pro) = 130 μM; I = 0.20 M (KNO3); T = 25.0 °C; ![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) = 1 cm}. = 1 cm}. | ||
On increasing the pH, the first two deprotonation processes detected in the UV-Vis spectra belong to the NqH+ and the hydroxyl groups, since changes in their protonation state have a considerable effect on the electron density of the aromatic rings. Thus, their deprotonation results in significant spectral changes, especially in the case of the hydroxyl moiety, namely the emerging strong bands in the range 330–430 nm originate from the more extended conjugated π-electron system in the deprotonated form. Surprisingly, the deprotonation of the prolinyl amino group also affects the spectra; it is accompanied by a minor bathochromic shift (see changes at pH > 10 in Fig. 2). Proton dissociation constants determined on the basis of the UV-Vis spectrophotometric and pH-potentiometric data are in good agreement with those obtained by the 1H NMR spectroscopic studies (Fig. S16† and Table 1).
On the basis of the obtained pKa values, species distribution was calculated at pH 7.4 and 63.5% of the ligand is present in its neutral form (HL+/−) that has notably a zwitterionic structure. In 36.5% the hydroxyl group is deprotonated (L−) resulting in the excellent water solubility of the compound at physiological pH (vide infra lipophilicity characterization).
Complex formation equilibria of HQCl-Pro with [Rh(η5-C5Me5)(H2O)3]2+ and [Ru(η6-arene)(H2O)3]2+
Complex formation of the studied half-sandwich organometallic triaqua cations with the HQ-type ligands is simple (Scheme S1†) as generally only mono complexes are formed. The HQ-type ligands coordinate in a (N,O−) bidentate mode based on the crystallographic and solution speciation studies.6,16–20,23 Firstly, the kinetics of the complex formation was investigated spectrophotometrically. Based on the spectral changes in the [Rh(η5-C5Me5)(H2O)3]2+–HQCl-Pro system the complexation reaction was found to be fairly fast at pH 3.6 (Fig. S17†), while in the case of Ru(η6-arene) complexes it was much slower, and at least 1 h was needed to reach the equilibrium as the spectral changes show in Fig. 3.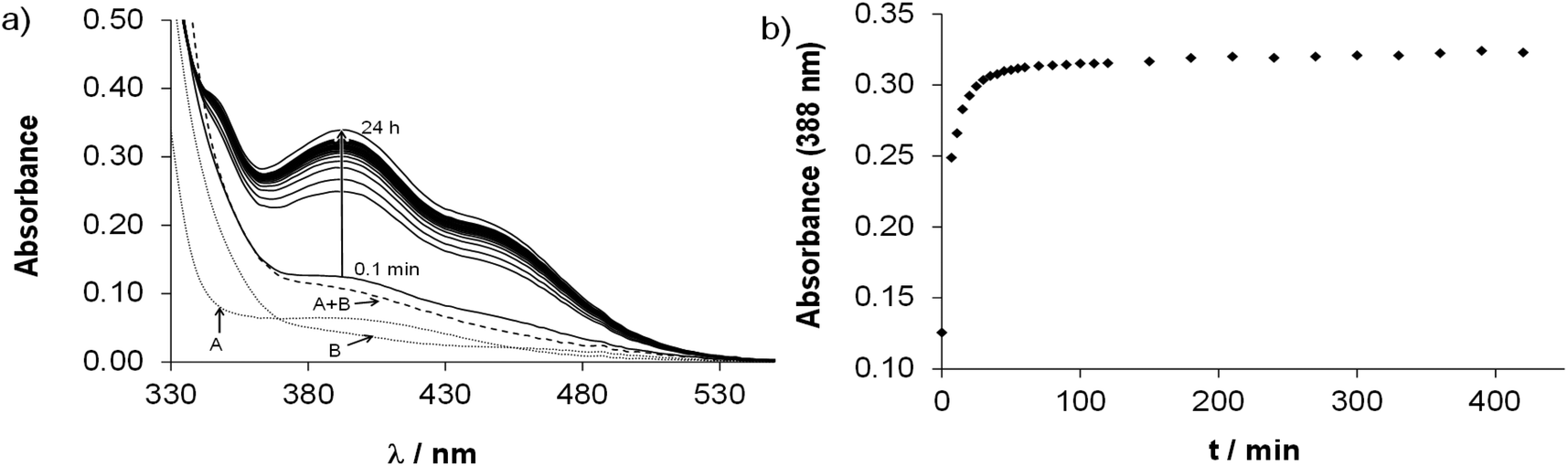 | ||
| Fig. 3 (a) Time-dependent UV-Vis absorption spectra of the Ru(η6-tol)–HQCl-Pro (1:1) system at pH = 3.64 (solid lines). Dotted curves show the absorption spectra of the triaqua organometallic metal ion (A) and HQCl-Pro ligand (B); the dashed curve shows their additive spectrum (A + B). (b) Absorbance values at 388 nm as a function of time. {c([Ru(η6-tol)(H2O)3]2+) = c(HQCl-Pro) = 101 μM; pH = 3.65; I = 0.20 M (KNO3); T = 25.0 °C; = 1 cm}. | ||
The 1H NMR spectrum recorded for the [Rh(η5-C5Me5)(H2O)3]2+–HQCl-Pro system at pH 2 revealed peaks belonging only to a metal complex, and neither a free organometallic triaqua ion nor an unbound ligand was detected (not shown). It suggests the formation of significantly highly stable complexes. Even though the ligand has carboxylate and amino functions that might coordinate to another metal center, there is no sign of the formation of any dinuclear species based on the recorded 1H NMR spectra, and only the mono complex [Rh(η5-C5Me5)(L)(H2O)]+ is formed (where L is the coordinated form of HQCl-Pro). The Ru(η6-arene) complexes of HQCl-Pro behaved similarly. Fig. 4 shows the suggested structures for complexes [M(arene)(L)(H2O)]+ in water.
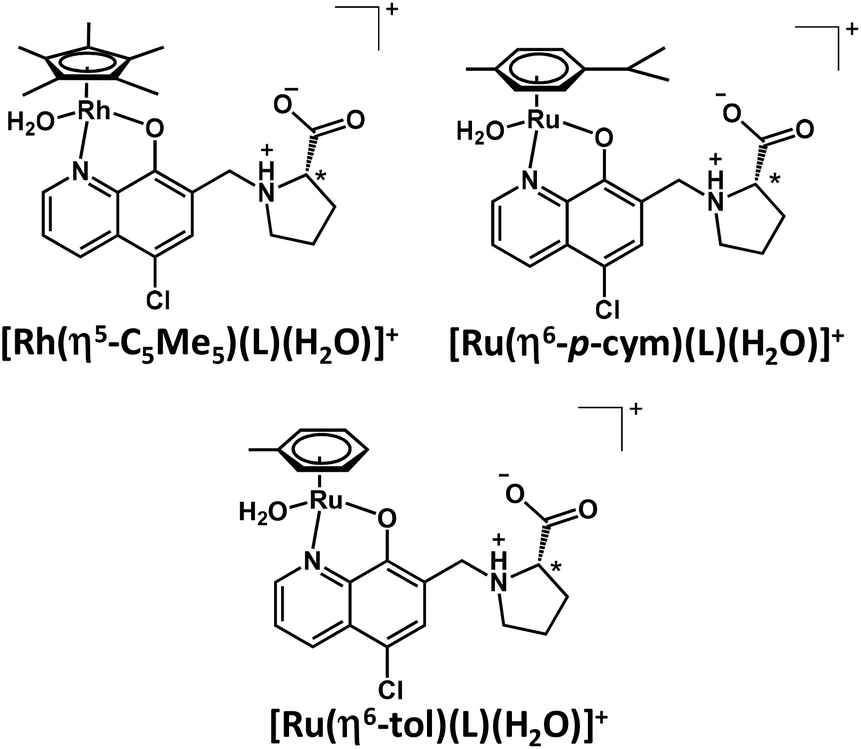 | ||
| Fig. 4 The schematic representation of the mono complexes [M(arene)(L)(H2O)]+ formed with HQCl Pro and the various organometallic cations. | ||
However, the formation of the hydrogen bond is not indicated in Fig. 4, which may occur between the coordinated hydroxylate group and the protonated prolinyl nitrogen (O−⋯H+N) as it was determined for the Zn(II) complexes of various 8-hydroxyquinolines.30 A sign of this hydrogen bond may appear in methanol: the doubling found in the 13C NMR spectra may belong to the isomers formed after this hydrogen bond (Fig. S13†); the biggest differences are seen in the positions, which are close to this suggested hydrogen bond.
In order to determine the stability constants of the [M(arene)(L)] complexes, the UV-Vis spectra of acidic samples were recorded (pH = 0.8–2.8) to force the complex dissociation. The spectra obtained for the [Rh(η5-C5Me5)(H2O)3]2+–HQCl-Pro system were practically identical in the entire monitored pH range, however they are significantly different from the spectra of the unbound organometallic ion and ligand (Fig. 5a). It indicates that the complex formation is already complete at pH = 0.8, which hindered the calculation of the stability constant, and similarly no constants could be determined for the Ru(η6-arene) species (Fig. 5b). However, this approach was successfully used previously for the Rh(η5-C5Me5) and Ru(η6-p-cym) 8-hydroxyquinolinato complexes.23 In this work a logK [M(arene)(L)] = 16.45 ± 0.02 was obtained for the complex [Ru(η6-tol)(8-hydroxyquinolinato)(H2O)]+ based on the pH-dependent UV-Vis spectra (pH = 1.2–2.8, Fig. S18†) for comparison. Notably, this constant is slightly smaller than that of [Ru(η6-p-cym)(8-hydroxyquinolinato)(H2O)]+.23
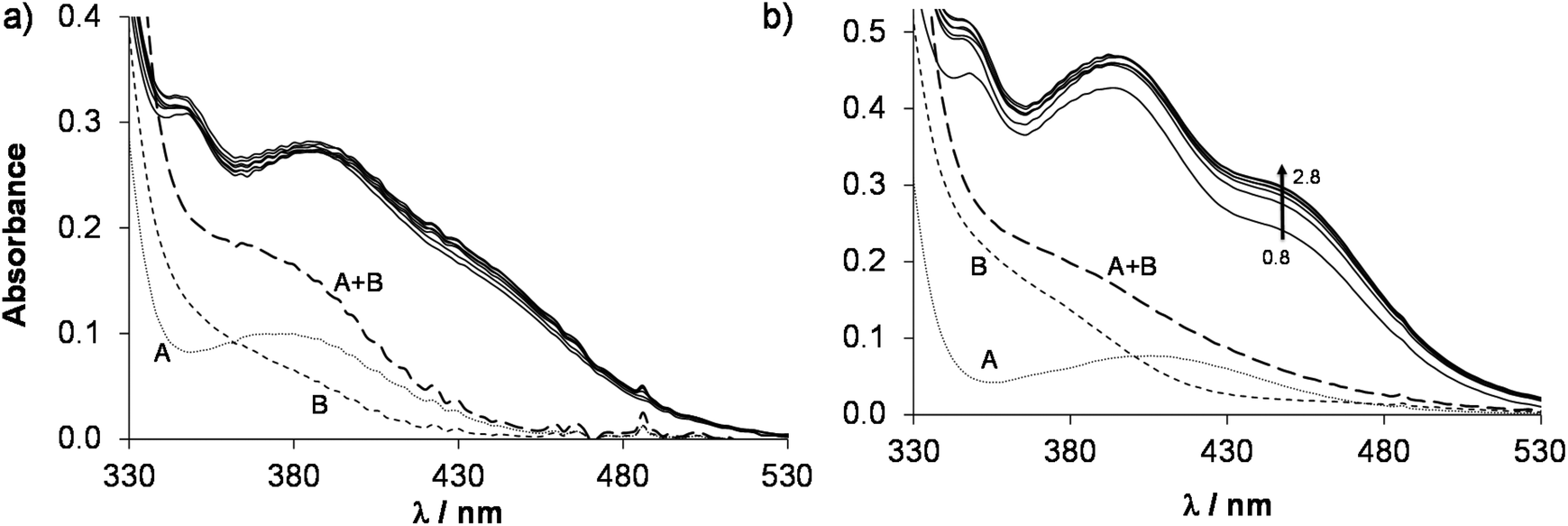 | ||
| Fig. 5 UV-Vis absorption spectra (solid lines) of the (a) Rh(η5-C5Me5)–HQCl-Pro system at pH = 0.8–2.0 and of the (b) Ru(η6-p-cym)–HQCl–Pro system at pH = 0.8–2.8. The dotted curve shows the absorption spectrum of the free organometallic cation (A), the dashed curve (B) shows the spectrum of the free HQCl-Pro ligand and the long dashed curve (A + B) shows their additive spectrum. {c([Rh(η5-C5Me5)(H2O)3]2+) = c(HQCl-Pro, in Fig. 4a) = 60 μM; c([Ru(η6-p-cym)(H2O)3]2+) = c(HQCl-Pro, in Fig. 4b) = 115 μM; I = 0.20 M (KNO3); T = 25.0 °C; = 1 cm}. | ||
Therefore, displacement reactions were performed to determine the stability constants of these highly stable complexes. First, ethylenediamine (en) was chosen as a competitor ligand. It has no absorbance in the 200–800 nm wavelength range, which makes it an attractive choice for these studies. However, mixed-ligand complex formation was detected according to the 1H NMR spectra (Fig. S19†) hindering the constant determination. The peaks belonging to the methyl hydrogens in the C5Me5 moiety reflect the various chemical environments of the organometallic fragment. E.g. at an excess of 23 equivalents of ethylenediamine not only the peaks of binary complexes [Rh(η5-C5Me5)(en)(H2O)]2+ and [Rh(η5-C5Me5)(L)(H2O)]+ appear (L: coordinated form of HQCl-Pro), but two unexpected peaks were also observed, signed with ♠ and ♣ in Fig. S19.† Addition of chloride ions decreased the ratio of these peaks (38% → 6%), which indicates that the Cl− and ethylenediamine compete for the third coordination site. Only ternary complex formation occurred in the case of the [Ru(η6-p-cym)(L)(H2O)]+ complex even at a huge excess (70 equivalents) of ethylenediamine, and at this high c(en)/c(HQCl-Pro) ratio ca. 50% of Ru(η6-p-cym) is found in the mixed-ligand complex.
In the next step, 2-picolylamine (pin) was selected and found to be an appropriate competitor in the case of the rhodium complex. Since pin has an intense ligand band in the UV region, only the use of 1H NMR spectroscopy was helpful to determine the speciation. Therefore, 1H NMR spectra were recorded for the [Rh(η5-C5Me5)(H2O)3]2+–HQCl-Pro–pin ternary system at various complex-to-pin ratios (Fig. 6a). The spectra revealed that while the amount of free HQCl-Pro and [Rh(η5-C5Me5)(pin)(H2O)]2+ is increasing with the 2-pin excess, the amount of the [Rh(η5-C5Me5)(L)(H2O)]+ complex is decreasing. Based on the integrals of the peaks, fractions of the different compounds were calculated (Fig. 6b) and the stability constant was determined (Table 2) using the stability constant of [Rh(η5-C5Me5)(pin)(H2O)]2+ taken from our previous work.31
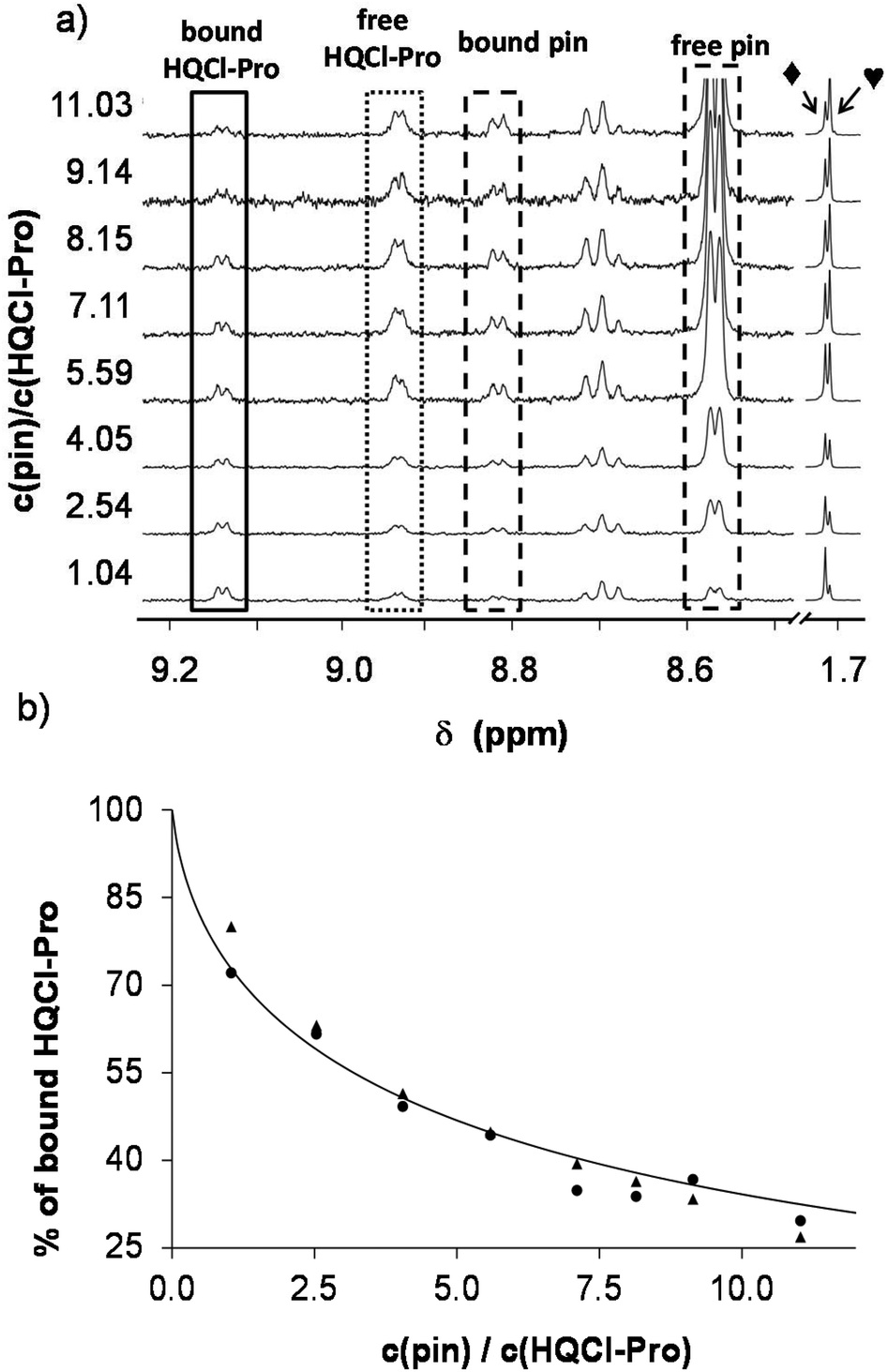 | ||
| Fig. 6 (a) 1H NMR spectra of the [Rh(η5-C5Me5)(L)(H2O)]+–2-picolylamine system recorded at various c(pin)/c(HQCl-Pro) ratios (L is the coordinated form of HQCl-Pro). The aromatic region (with increased intensity) and C5Me5 methyl protons are shown. Assignment: ◆ = [Rh(η5-C5Me5)(L)(H2O)]+; ♥ = [Rh(η5-C5Me5)(pin)(H2O)]+. (b) Fitted (solid line) and measured bound HQCl-Pro ratio values calculated from the integrals of HQCl-Pro (●) and C5Me5 (▲). {c([Rh(η5-C5Me5)(L)(H2O)]+) = 100 μM; c(pin) = 0.104–1.520 mM; solvent: 90% H2O/10% D2O; pH = 5.91 (20 mM phosphate buffer); T = 25.0 °C; I = 0.20 M (KNO3)}. | ||
| Ru(η6-p-cym) | Ru(η6-tol) | Rh(η5-C5Me5) | |
|---|---|---|---|
| a For the [Rh(η5-C5Me5)(L)(H2O)]2+ + pin ⇌ [Rh(η5-C5Me5)(pin)(H2O)]2+ + L equilibrium determined at various c(pin) concentrations by 1H NMR spectroscopy. b Determined by UV-Vis spectroscopy at pH 2.0–11.5. c pH = 5.50 (phosphate buffer). For the [M(arene)(L)(H2O)]+ + Cl− ⇌ [M(arene)(L)Cl] + H2O equilibrium determined at various total chloride ion concentrations by UV-Vis spectrophotometry. | |||
| logK [M(arene)(L)] |
— | — | 13.41 ± 0.02a |
| pKa [M(arene)(L)]b | 8.62 ± 0.04 | 8.45 ± 0.03 | 9.62 ± 0.04 |
| logK′ (H2O/Cl−)c |
1.21 ± 0.01 | 1.09 ± 0.01 | 1.57 ± 0.01 |
However, in the case of [Ru(η6-arene)(H2O)3]2+ complexes the original yellow colour of the samples changed to pink and the peaks of the coordinated HQCl-Pro, p-cymene and toluene disappeared in the 1H NMR spectra at an excess of the competitor 2-picolylamine ligand, which can be explained by arene loss and the probable oxidation of the Ru centre (Fig. S20†). Thus, determination of the stability constants failed in these cases.
This side reaction aroused our interest as other competitor ligands (including e.g. ligands from biofluids) may also cause arene loss and may affect bioactivity. Therefore, ligand displacement reactions were studied by UV-Vis spectroscopy. First the effect of HQCl-Pro itself was studied using different conditions. The addition of two equivalents of ligand to the complex [Ru(η6-p-cym)(L)(H2O)]+ resulted in a too slow reaction at pH 2 to detect any changes during 1 h (while O2 passed through the solution). On the other hand, considerable changes of the spectra can be seen at physiological pH in Fig. 7a. The shape of the spectrum changes markedly, and a strong band developed at 420 nm, which is most probably related to the loss of the arene ligand and binding of the second and third HQCl-Pro. The rising broad band at higher wavelengths (λ > 500 nm) directly indicates the presence of the Ru(III) compound as it was found for [Ru(III)(8-hydroxyquinolate)3].32 Thus the change at 640 nm provides information about the rate of the Ru(III) complex formation (see Fig. 7b). In the literature the loss of arene (benzene, p-cymene) and the formation of [Ru(III)(L)3] were also found in the case of 8-hydroxyquinoline.23,32 As competitor ligands, deferiprone (1,2-dimethyl-3-hydroxy-pyridin-4(1H)-one) as an (O,O) model and 1,10-phenanthroline (phen) as a representative of (N,N) bidentate ligands were chosen. Addition of deferiprone to the complex [Ru(η6-p-cym)(L)(H2O)]+ did not result in spectral changes, while the reaction with phen was undoubtedly fast, and there was a significant difference between the initial solution (see the additive spectrum) and the spectrum recorded after 7 s for the mixed reactants (Fig. S21†). The yellow solution turned red and a strong band developed with λmax = 502 nm. After the first reaction step seemed to be completed in ∼90 s, another process started at ∼150 s and the main band shifted to 440 nm. The sign of a tiny amount of Ru(III) was also observed at higher wavelengths (A(690 nm) ∼ 0.05). When this experiment was repeated under argon, similar spectral changes were observed; however, the formation of Ru(III) could be successfully avoided. To explain the two main processes, the experiment was repeated with only 1 equiv. of phen under aerobic conditions (Fig. S22a†). The first step accompanied by the development of a band with 502 nm maximum was similar and was completed within 7 min (Fig. S22b†). Although the second process was different, most probably oxidation took place as the development of the strong band at 694 nm indicates (Fig. S22c†). Based on these findings the first step is most probably the loss of p-cymene and the formation of the mixed-ligand complex [Ru(II)(phen)(L)(H2O)2]+ (λmax = 502 nm, shown in gray rhombuses in Fig. S21b, S22b and c†). This is followed by the slow coordination of the second phen forming [Ru(II)(phen)2(L)]+ when 2 equiv. of phen were provided, while the slow oxidation occurs without the second phen ligand forming [Ru(III)(phen)(L)(H2O)2]2+ (shown in orange squares in Fig. S21b, S22b and c†).
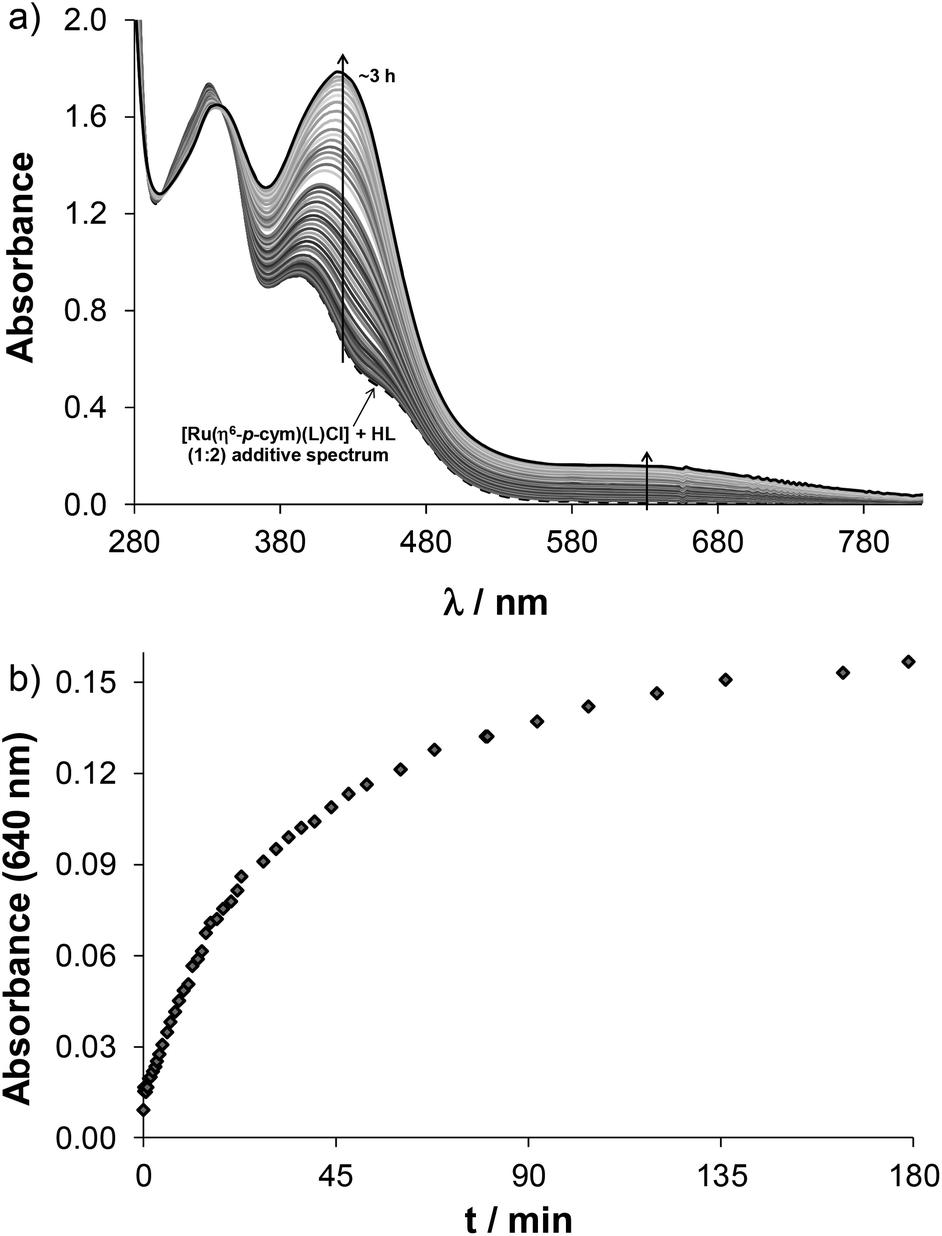 | ||
| Fig. 7 Time-dependent UV-Vis absorption spectra of the [Ru(η6-p-cym)(L)(H2O)]+–HQCl-Pro (1:2) system at pH = 7.40 in the presence of O2 (L: the coordinated form of HQCl-Pro). The dashed curve shows the additive spectrum of the metal complex and 2 equivalents of the HQCl-Pro ligand. The inserted figure shows absorbance values at 640 nm as a function of time. {c([Ru(η6-p-cym)(L)(H2O)]+) = 200 μM; c(HQCl-Pro) = 400 μM; pH = 7.40 (20 mM phosphate); c(KCl) = 0.10 M; T = 25.0 °C; = 1 cm}. | ||
Reactions of [Ru(η6-p-cym)(L)(H2O)]+ with various biomolecules were tested as well. Addition of histidine (His), human serum albumin (HSA) and RPMI 1640 medium components resulted in similar spectral changes (Fig. S23a, c and e†). We concluded that no Ru(III) was present in the systems, and most probably a mixed-ligand complex ([Ru(η6-p-cym)(L)(His)]+) is formed in the presence of histidine and the RPMI 1640 medium components (vide infra for NMR spectra). Albumin is also able to bind the complex most probably through its side chains in a monodentate fashion due to the coordination of a histidine nitrogen, or cysteine thiolate or methionine thioether.33,34 The reaction with HSA was somewhat slower under the same experimental setup (Fig. S23b, d and f†).
Based on these findings the following can be concluded: (i) the excess of rigid (O,O) donor bidentate ligands cannot cause the loss of p-cymene; (ii) the excess of the rigid 8-hydroxyquinolate-type (N,O−) and rigid (N,N) donor bidentate ligands can cause liberation of the arene ligand followed by oxidation to Ru(III) at physiological pH in the presence of O2; (iii) the flexible (N,N) donor ethylenediamine and biologically available molecules (like histidine, amino acids of RPMI 1640 or human serum albumin) can readily react with the complex [Ru(η6-p-cym)(L)(H2O)]+ forming mixed-ligand complexes. A previously described class of half-sandwich complexes containing azopyridine ligands also showed arene-loss (even at 1:1 metal-to-ligand ratio), which was explained with the π-acceptor properties of the bidentate ligand35 as the stronger π-acceptor bidentate ligands can compete with the arene.
Deprotonation of the coordinated water molecule and its exchange to the chloride ion in the [M(arene)(L)(H2O)]+ complexes
In the [M(arene)(L)(H2O)]+ complexes (L: the coordinated form of HQCl-Pro) the coordinated aqua ligand can get deprotonated with increasing pH and this process is reported to have an effect on the reactivity of the complex.36 Namely, the hydroxido ligand is considered as a worse leaving group than the water molecule or chloride ion, preventing the complex from interacting with biomolecules. To determine the pKa values 1H NMR spectra of the complexes were recorded in a wide pH range and representative spectra are shown for the titration of the [Ru(η6-p-cym)(L)(H2O)]+ complex in Fig. 8 (and the spectra of the Ru(η6-tol) and Rh(η5-C5Me5) complexes are shown in Fig. S24 and S25†). These spectra reveal that peaks are shifted at pH > 6, and the chemical shifts draw a sigmoid curve as a function of pH (Fig. 8c).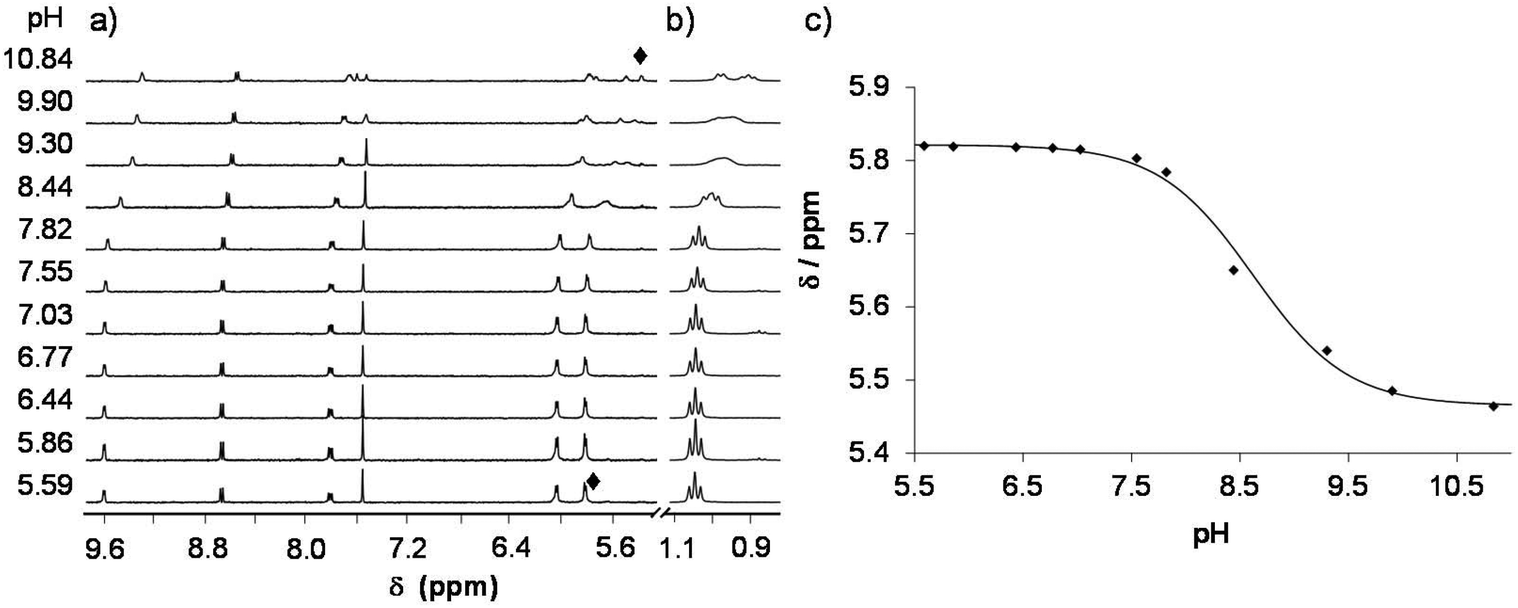 | ||
| Fig. 8
1H NMR spectra of the Ru(η6-p-cym)–HQCl-Pro 1:1 system recorded at pH = 5.6–10.9. (a) Aromatic protons; (b) p-cymene methyl protons. (c) Measured (◆) and fitted (solid line) chemical shift values of one of the protons on the p-cymene ring as a function of pH. {c([Ru(η6-p-cym)(H2O)3]2+) = c(HQCl-Pro) = 300 μM; solvent: 90% H2O/10% D2O; T = 25.0 °C; I = 0.20 M (KNO3)}. | ||
From these sigmoid curves pKa [M(arene)(L)] values were determined (Table 2), which are much lower for the Ru containing complexes than for the Rh-complex. Notably, following the deprotonation the forming hydroxido complex exhibits more peaks than the aqua complex. Most probably the deprotonation leads to the loss of the twofold symmetry of the p-cymene ligand and peaks are doubled. Due to the rigid structure the rotation of the p-cymene ligand is blocked most likely. Earlier, computational studies revealed that the rotation has a very low energy barrier in the ruthenium–arene complexes bearing 1,2-ethylenediamine (RAED), e.g. benzene completes a full rotation in 2 ps.37 In that case, a steric interaction between HQCl-Pro, OH− and p-cymene can increase this rotational barrier.
Using the pKa [M(arene)(L)] values the ratio of the hydroxido form of the complexes was calculated at physiological pH revealing the formation of [M(arene)(L)(OH)] in 6% for Ru(η6-p-cym), 8% for Ru(η6-tol) and less than 1% for the Rh(η5-C5Me5)-complex in the absence of chloride ions. pKa of this type of complexes increases with the chloride ion concentration,24,38 thus these percentages are merely maximum values. When the organometallic fragments are compared with each other, the same trend of the pKa values was observed for complexes of (N,O) ligands such as 2-picolinates, HQ-5-sulfonate (HQS), HQ and 7-(1-piperidinylmethyl)-HQ (PHQ)23,38–40 as Fig. 9 shows.
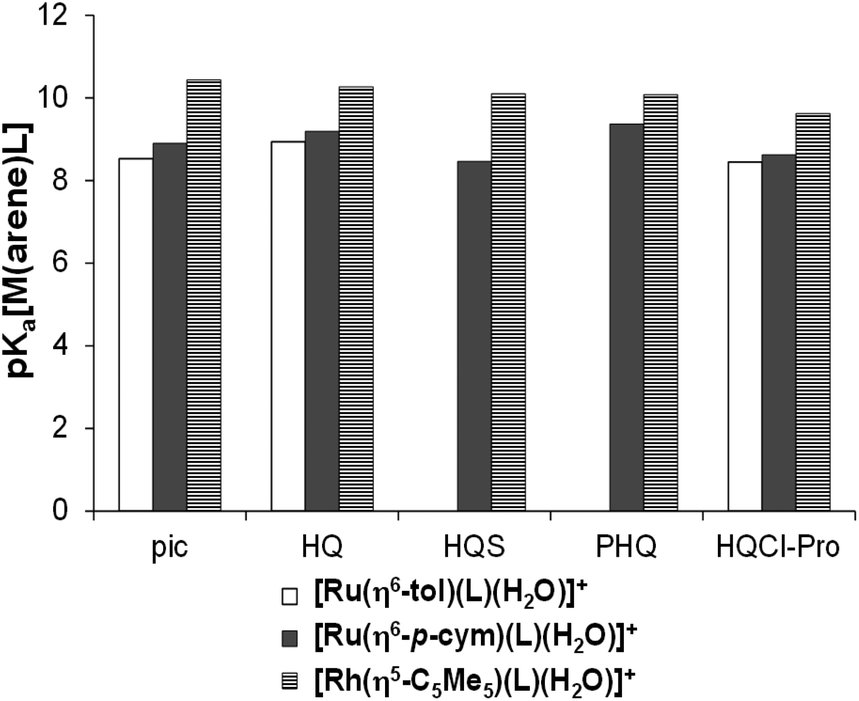 | ||
| Fig. 9 Comparison of pKa[M(arene)(L)] constants for the half-sandwich Ru- and Rh-complexes of (N,O) bidentate ligands (I = 0.20 M KNO3). HL = pic (2-picolinic acid),38–40,a HQ (8-hydroxyquinoline),23,b HQS (8-hydroxyquinoline-5-sulfonate),23 PHQ (7-(1piperidinylmethyl)-8-hydroxyquinoline)23 and HQCl-Pro. aI = 0.20 M KCl; bpKa([Ru(η6-tol)(8-hydroxyquinolinato)(H2O)]+) = 8.94(2) determined by UV-Vis spectrophotometric titration in this work, see Fig. S26.† | ||
Based on the logK and pKa [M(arene)(L)] constants (Table 2) concentration distribution curves were calculated for the Rh(η5-C5Me5) complex of HQCl-Pro (Fig. 10a). A very small amount (<3%) of free organometallic ions appears at pH 2, while the formation of the [Rh(η5-C5Me5)(L)(H2O)]+ complex is predominant at pH 7.4. The stability constants determined for the complexes of HQ derivatives cannot be compared directly due to the different basicity of ligands. For comparison, pM* (the negative logarithm of the unbound metal ion) values were calculated and plotted against the pH (Fig. 10b). (pM* is calculated by taking into consideration the hydrolyzed forms: pM* = −log([M(arene)] + 2×[(M(arene))2(OH)2] + 2×[(M(arene))2(OH)3]). The higher pM* value indicates higher stability of the complex. In these calculations, stability constants of the half-sandwich rhodium complexes with HQ, 8-hydroxyquinoline-5-sulfonate and 7-(1-piperidinyl-methyl)-8-hydroxyquinoline were used.23 Although the HQCl-Pro complex has the highest stability among the others up to pH ∼3.5, at physiological pH the other complexes have somewhat higher stability.
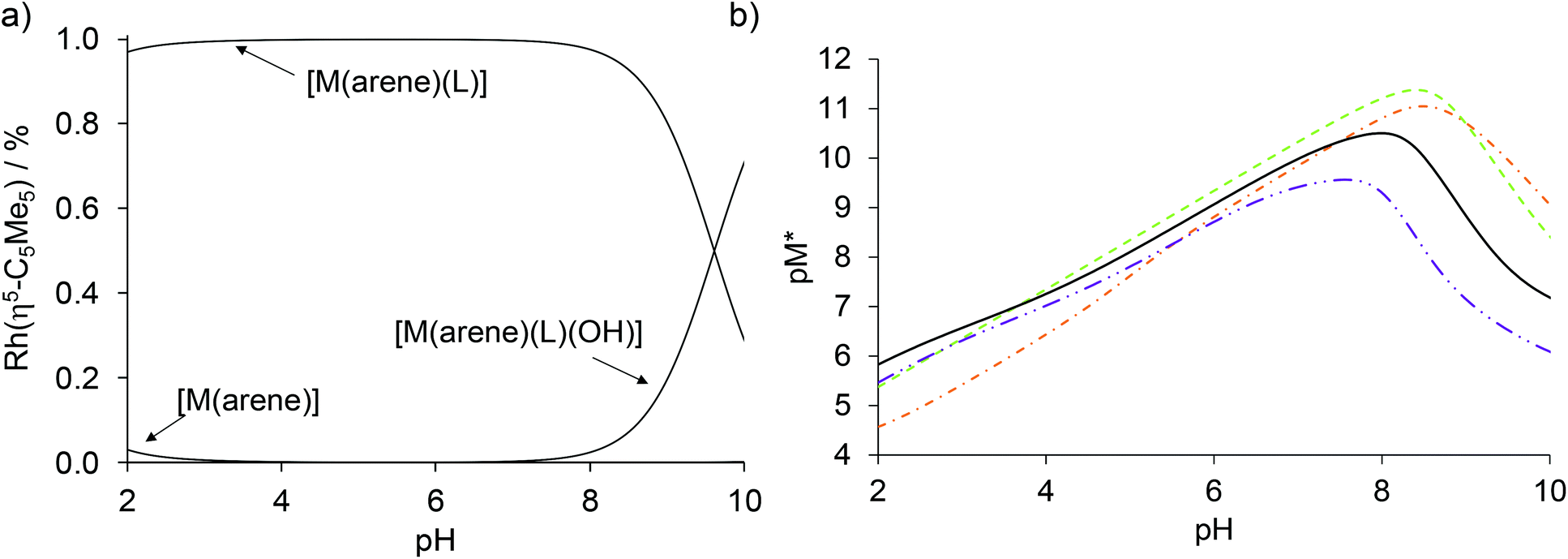 | ||
| Fig. 10 (a) Calculated concentration distribution curves of the [Rh(η5-C5Me5)(H2O)3]2+–HQCl-Pro system based on the stability constants from Table 2. (b) Calculated pM* curves of the Rh(η5-C5Me5)–HQ (-·-),23 8-hydroxyquinoline-5-sulfonate (–),23 7-(1-piperidinylmethyl)-8-hydroxyquinoline (-··-)23 and HQCl-Pro systems (solid line), pM* = −log([M(arene)] + 2 × [(M(arene))2(OH)2] + 2 × [(M(arene))2(OH)3]). {c([Rh(η5-C5Me5)(H2O)3]2+) = c(HQCl-Pro) = 50 μM; T = 25.0 °C; I = 0.20 M (KNO3)}. | ||
Adding chloride ions to the solutions of the complexes causes changes in their 1H NMR and UV-Vis spectra. This is the result of a third equilibrium process, shown in Scheme S1,† which is the exchange of the coordinated water molecule to a chloride ion. Half-sandwich organorhodium and ruthenium complexes have a relatively high chloride ion affinity, and the chlorinated forms of the HQCl-Pro complexes are charge neutral. The chloride content of the medium has an effect on not only the charge, but also on the pKa [M(arene)(L)]24,38 and on the lipophilicity.39,41 This affinity is well described by the logK′ (H2O/Cl−) constant, which is determined from the UV-Vis spectra of the complexes by varying the total concentrations of chloride ions (Fig. S27–S30† and Table 2).
The logK′ (H2O/Cl−) constants show that the Rh(η5-C5Me5) complex has the highest, while [Ru(η6-tol)(L)(H2O)]+ has the lowest value (Table 2). The constants for [Ru(η6-arene)(L)(H2O)]+ complexes have tiny differences, and a similar trend was found previously in the case of 8-hydroxyquinoline, and Fig. S31† shows this tendency.23 The concentration of the chloride ion is around 100 mM in blood 24 mM in the cytoplasm and 4 mM in the nucleus,42 and the actual concentration affects the ratio of the chloride and aqua complexes (Fig. 11). The lower the c(Cl−), the higher fraction of the aqua complex. According to the proposed activation mechanism by aquation42 the complexes are in their neutral (zwitterionic) chlorinated form in the blood serum at 100 mM chloride concentration. The neutral chlorinated form may penetrate more easily through the cell membranes and might be trapped in the cytosol due to the lower chloride concentration and formation of charged aqua forms. While 79% of the rhodium complex is in the neutral form when the c(Cl−) is 100 mM, it drops to 47% and 13% at 24 mM and 4 mM chloride ion concentrations, respectively (Fig. 11). The Ru(η6-arene) complexes show somewhat weaker chloride ion affinity.
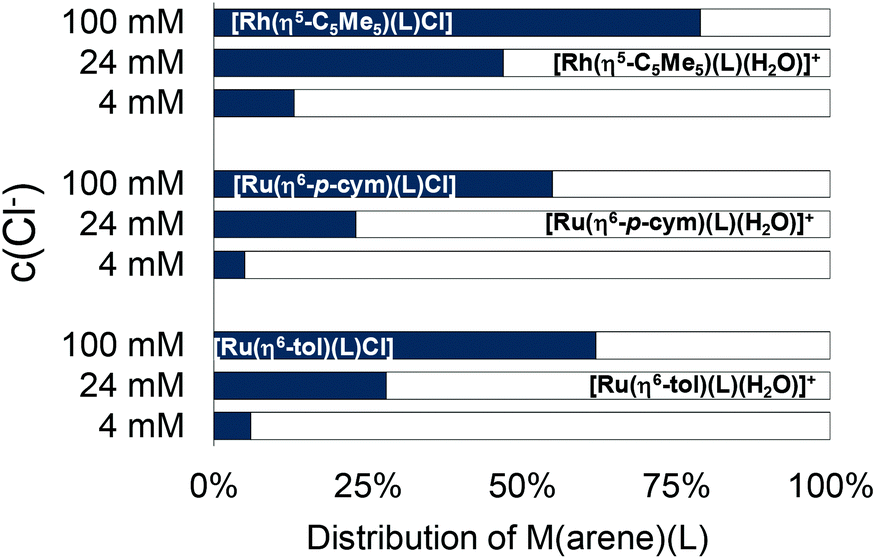 | ||
| Fig. 11 The calculated ratio of aquated ([M(arene)(L)(H2O)]+) (white) and chlorinated ([M(arene)(L)Cl]) (gray) forms of the HQCl-Pro complexes at different chloride concentrations of modelling biofluids, based on the constants in Table 2. {c([M(arene)(H2O)3]2+) = c(HQCl-Pro) = 100 μM; c(Cl−) = 4, 24 and 100 mM; T = 25.0 °C}. | ||
Chloride concentration-dependence of the lipophilicity of HQCl-Pro and its complexes
The conventional shake-flask method was used to determine the chloride-dependent lipophilicity of the ligand and its three organometallic complexes at physiological pH. Compounds were dissolved in 20 mM phosphate buffer (pH = 7.4, saturated with n-octanol), then mixed with n-octanol (saturated with the aqueous buffer). The buffers contained chloride ions in different concentrations related to the different biofluids (4, 24, 100 mM). Distribution coefficients (logD7.4) calculated from UV-Vis quantitative analysis are shown in Fig. 12. The ligand lipophilicity slightly decreases with increasing chloride ion concentration, which is the result of the stronger ionization effect at higher ionic strength.
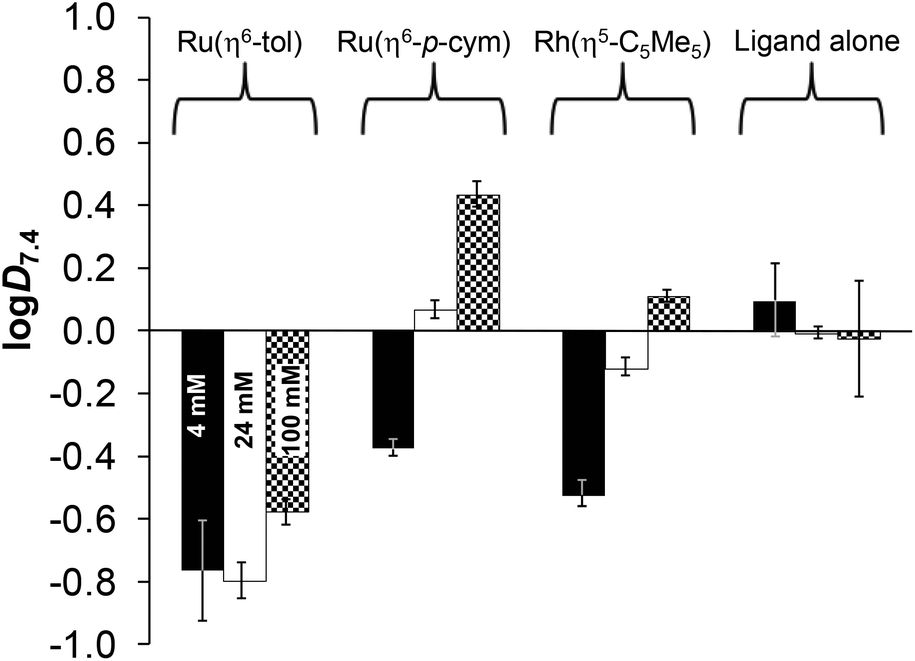 | ||
| Fig. 12
n-Octanol/water distribution coefficients at pH = 7.4 (logD7.4) for complexes [Ru(η6-tol)(L)(H2O)]+, [Ru(η6-p-cym)(L)(H2O)]+, [Rh(η5-C5Me5)(L)(H2O)]+ and the ligand HQCl-Pro at different chloride ion concentrations: 4 mM (black), 24 mM (white) and 100 mM (checkered). {c(compounds) = 200 μM; pH = 7.40 (20 mM phosphate buffer); T = 25.0 °C}. | ||
The actual chemical form of the studied organometallic complexes strongly depends on the chloride ion affinity and concentration, as was described previously. Fig. 11 clarifies the ratios of the neutral chlorinated and the positively charged aquated forms at pH 7.4. Based on the data in Fig. 12 it can be concluded that the most lipophilic complex is [Ru(η6-p-cym)(L)(H2O/Cl)]+/0 at 100 mM of Cl−, and the most hydrophilic is [Ru(η6-tol)(L)(H2O/Cl)]+/0 at 4–24 mM of Cl−. Here the trend of lipophilicity is different from the trend of the logK′ (H2O/Cl−) values: Ru(η6-tol) < Rh(η5-C5Me5) < Ru(η6-p-cym). Although the rhodium complex has the highest chloride ion affinity, other factors also affect the lipophilicity such as the higher charge of the Rh-center (+3) (vs. the +2 charge of Ru) and the negative charge of the C5Me5 ligand (vs. neutral toluene/p-cymene).
In vitro cytotoxicity and antiproliferative activity measurements of HQCl-Pro and its complexes on cancer cell lines
In order to characterize the anticancer effect of HQCl-Pro and to monitor whether the complexation with organometallic triaqua cations can affect the cytotoxic activity of the ligand itself, thiazolyl blue tetrazolium bromide (MTT) assays were performed in two human colonic adenocarcinoma cell lines under two kinds of setup. Colo 205 is a drug sensitive cell line, while Colo 320 is multidrug resistant being primarily mediated by the overexpression of P-gp. Earlier studies showed that the reference compound HQ and its organometallic Ru- and Rh-complexes are cytotoxic in MES-SA and MES-SA/Dx5 cancer cell lines (IC50 = 1–13 μM).23 The structurally related 7-(1-piperidinylmethyl)-8-hydroxyquinoline and its complexes were found to be cytotoxic (IC50 = 0.6–20 μM), however the ligand and its Rh(η5-C5Me5) complex showed collateral sensitivity (with selectivity ratios 5.8 and 5.1, respectively).Inhibiting the proliferation and causing cell death may occur at the same time when the drug is administered to the cells. Using a lower number of cells per well (6 × 103) and a longer incubation time (72 h) the MTT assay provides more information about the activity of the complexes to inhibit cell proliferation rather than growth inhibition. On the other hand, in the case of a higher number of cells per well (6 × 104) and a shorter exposure time (24 h) it is possible to monitor preferably the cytotoxic effect. IC50 values collected for HQCl-Pro in the absence and in the presence of [Rh(η5-C5Me5)(H2O)3]2+, [Ru(η6-p-cym)(H2O)3]2+ and [Ru(η6-tol)(H2O)3]2+ and for the precursor dimers [M(η5/6-arene)(μ-Cl)Cl]2 are shown in Table 3. Doxorubicin and cisplatin were used as positive controls.
| Antiproliferative effect | Cytotoxic effect | |||
|---|---|---|---|---|
| Colo 205 | Colo 320 | Colo 205 | Colo 320 | |
| a 1 × 104 cells were used for cisplatin. | ||||
| HQCl-Pro | 23.4 ± 3.3 | 8.5 ± 1.7 | 42.5 ± 7.4 | 17.4 ± 2.5 |
| [Ru(η6-tol)(L)(H2O)]+ | 25.3 ± 3.1 | 85.0± 7.5 | 72.6 ± 4.8 | 60.9 ± 8.2 |
| [Ru(η6-p-cym)(L)(H2O)]+ | 68.3 ± 10.7 | >100 | >100 | >100 |
| [Rh(η5-C5Me5)(L)(H2O)]+ | 25.8 ± 4.8 | 9.7 ± 1.1 | 81.5 ± 3.3 | 24.1 ± 3.7 |
| [Ru(η6-p-cym)(μ-Cl)Cl]2 | >100 | >100 | >100 | >100 |
| [Ru(η6-tol)(μ-Cl)Cl]2 | >100 | >100 | >100 | >100 |
| [Rh(η5-C5Me5)(μ-Cl)Cl]2 | >100 | >100 | >100 | >100 |
| Doxorubicin | 3.28 ± 0.22 | 3.12 ± 0.27 | 1.56 ± 0.03 | 6.45 ± 0.19 |
| Cisplatin | 10.1 ± 0.3 | 4.78 ± 0.11 | 83.9 ± 3.5a | 18.1 ± 0.3a |
The organometallic precursors have no toxic effect on cancer cell lines (IC50 > 100 μM). HQCl-Pro and its Rh(η5-C5Me5) complex exhibited similar and relatively low IC50 values, while in the presence of Ru(η6-tol) and especially Ru(η6-p-cym) much higher values were obtained. Additionally, HQCl-Pro and its Rh(η5-C5Me5) complex showed higher anticancer activity in Colo 320 than in Colo 205. In contrast the toluene and p-cymene complexes have similar cytotoxicity in both cell lines and the antiproliferative effect is weaker in the Colo 320 cells. These data do not correlate with the lipophilicity of the complexes, suggesting that other factors seem to be more dominant in the bioactivity. Notably, the complexes have similar or weaker cytotoxicity compared to cisplatin.
Reaction with cell culture medium components
To find an answer to the decreased activity of the Ru(η6-p-cym) complex interaction of the complex [Ru(η6-p-cym)(L)(H2O)]+ with the RPMI 1640 cell culture medium was also investigated by 1H NMR spectroscopy (Fig. 13). Previously we described the interaction (vide supra for kinetics), which was followed spectrophotometrically and no oxidation was detected; only mixed ligand complex formation was observed. This medium contains various amino acids and other small biomolecules which may coordinate to the metal center leading to e.g. the (partial) release of the original ligand or the formation of mixed ligand species. According to the recorded 1H NMR spectra the free HQCl-Pro has different spectra in RPMI 1640 medium and in PBS′ (Fig. 13(3 and 4)); namely the shape of doublets are blurry, only the envelope type peak can be seen and peaks are shifted slightly due to the presence of the medium components. The Mg(II) content of the RPMI 1640 medium is relatively high (∼400 μM) and can explain the observed phenomena since 8-hydroxyquinolines are able to form complexes with this metal ion. Using stability constants reported for the Mg(II) complex of 8-hydroxyquinoline-5-sulfonate43 we estimated the amount of the possibly formed Mg(II) complex under the experimental conditions. It is suggested that ∼30% of the ligand can be bound to Mg(II).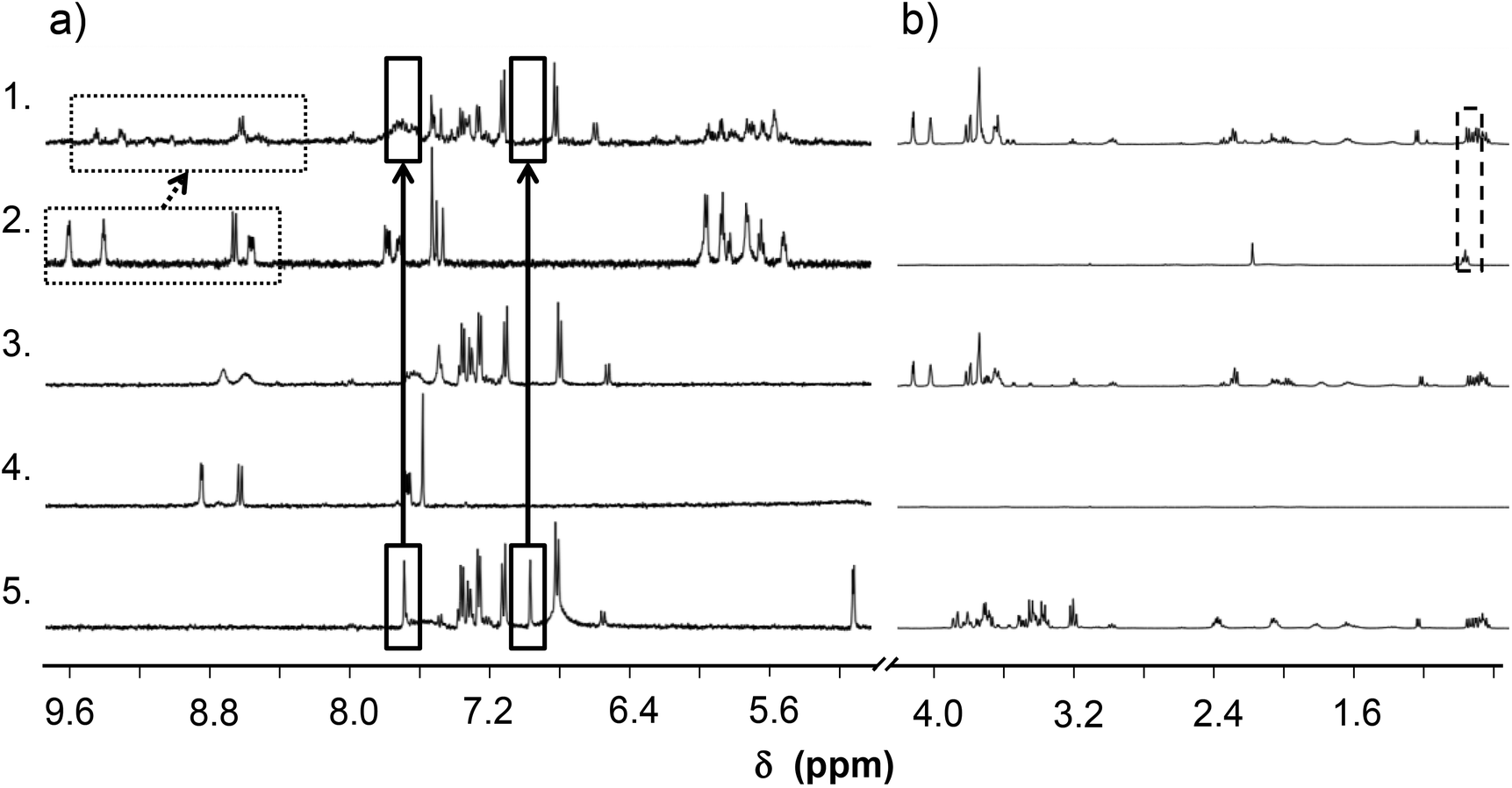 | ||
| Fig. 13 Monitoring of the interaction between HQCl-Pro, [Ru(η6-p-cym)(L)(H2O)]+ (L: coordinated form of HQCl-Pro) and RPMI 1640 medium components by 1H NMR spectroscopy. (a) Aromatic region; (b) aliphatic region. 1: [Ru(η6-p-cym)(L)(H2O)]+ in RPMI 1640; 2: [Ru(η6-p-cym)(L)(H2O)]+ in PBS' buffer; 3: HQCl-Pro in RPMI 1640 medium; 4: HQCl-Pro in PBS' buffer; 5: RPMI 1640 medium. Rectangles show signals due to the interactions: peaks of histidine (solid line), peaks of HQCl-Pro (dotted line), and peaks of p-cymene methyl groups (dashed line). {c([Ru(η6-p-cym)(H2O)3]2+) = c(HQCl-Pro) = 100 μM; solvent: 90% H2O/10% D2O; pH = 7.40 (phosphate buffer); T = 25.0 °C; t = 24 h}. | ||
For the Mg(II) and Ca(II) complexes of the structurally closer HQCl stability constants were determined in a 60% (v/v) dioxane/water mixture,44 and based on these stability constants the bound fraction of the ligand can be ∼60%.
Therefore, peak shifting of the ligand HQCl-Pro can be explained by the fast exchange between the free ligand and the Mg(II)/Ca(II) complexes. However, the competition of Ca2+ and Mg2+ with half-sandwich cations does not occur because the stability of the latter complexes is much higher. Peaks of different amino acid components are shown in the medium (Fig. 13). The singlets of histidine at δ = 7.0 and 7.7 ppm suffer from the most important change, as they disappeared/shifted after reacting with the complex [Ru(η6-p-cym)(L)(H2O)]+. This interaction is most probably the formation of a mixed-ligand species, and can also affect the final biological activity. In all, instead of the ligand displacement only the ternary complex formation with histidine is the most probable interaction, and there is no sign of ligand release (no free HQCl-Pro or in the form of the Mg(II) complex). It can be concluded that the reduced biological activity is most likely not connected to a dissociation process of the original complexes (at least not in the medium).
Cellular uptake of complexes
For the sake of better understanding of the difference in the anticancer activity of the complexes, cellular metal uptake was also measured with total-reflection X-ray fluorescence (TXRF) on the cultures of Colo 205 cancer cells. These cells were incubated for 4 and 24 h with the [Ru(η6-p-cym)(L)(H2O)]+ and [Rh(η5-C5Me5)(L)(H2O)]+ at 200 μM concentration. The effect of using fetal bovine serum (FBS) in the cell culture medium was also investigated. As shown in Fig. 14, both complexes were taken up by cells to a similar extent after 4 h independent of the medium's FBS content. After 24 h, the degree of Rh accumulation in the cells shows a noticeable increase above 400 ng per 106 cells, while the Ru content stays at the same level after 4 h. This result is in contrast to the lipophilicity measurement, where [Ru(η6-p-cym)(L)(H2O)]+ was more lipophilic; active transport might be more important in the transport of these complexes. The higher concentration of the Rh-compound inside the cell may explain the difference in the anticancer activity.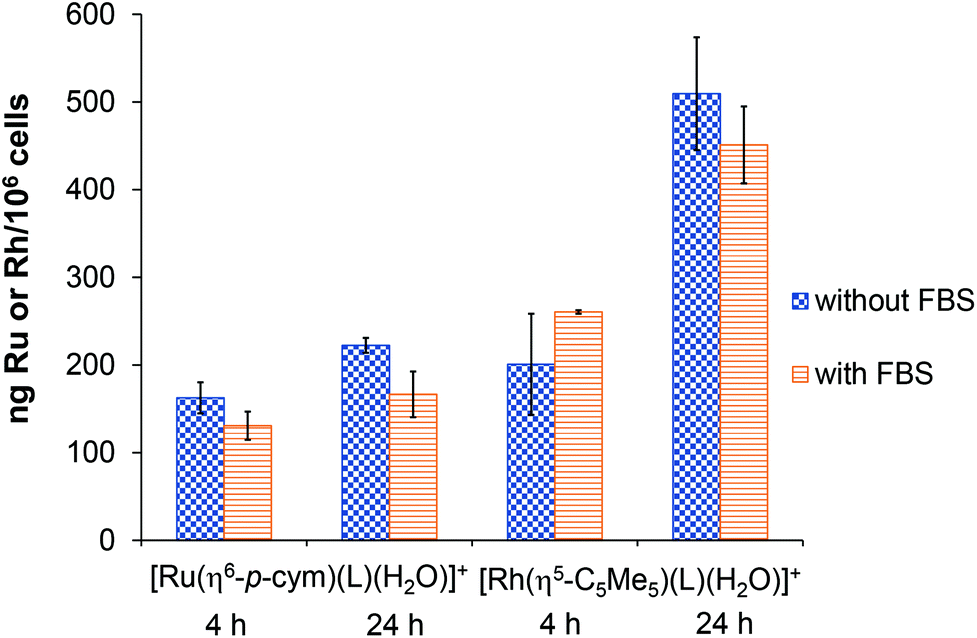 | ||
| Fig. 14 Time-dependent cellular uptake of HQCl-Pro complexes ([Ru(η6-p-cym)(L)(H2O)]+ and [Rh(η5-C5Me5)(L)(H2O)]+) by Colo 205 cancer cells. Checkered bars show samples without fetal bovine serum (FBS) in medium, while striped bars show samples with FBS. Each data bar represents the mean ± standard deviation of the three parallel, separate samples. | ||
Experimental
Chemicals
All solvents were of analytical grade and used without further purification. 8-Hydroxyquinoline, 5-chloro-8-hydroxyquinoline, L-proline, paraformaldehyde, [Rh(η5-C5Me5)(μ-Cl)Cl]2, [Ru(η6-p-cym)(μ-Cl)Cl]2, 1-methyl-1,4-cyclohexadiene, RuCl3 × 3H2O, ethylenediamine, 2-picolylamine, 1,2-dimethyl-3-hydroxy-pyridin-4(1H)-one (deferiprone), 1,10-phenanthroline, doxorubicin, n-octanol, KCl, AgNO3, HNO3, KOH, 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), RPMI 1640, NaH2PO4, Na2HPO4 and KH2PO4 were purchased from Sigma-Aldrich in pure quality, and cisplatin was a TEVA product. Ultrapure Milli-Q water was used for sample preparation. [Ru(II)(η6-tol)(μ-Cl)Cl]2 was prepared according to literature procedures.45The preparation of solutions and the determination of the concentration of stock solutions were performed as in our former works,23,24 see the ESI†/pH-potentiometry part for more information.
The buffered samples were prepared in 20 mM phosphate buffer or in a modified phosphate buffered saline (PBS′) at pH 7.40. PBS′ contains 12 mM Na2HPO4, 3 mM KH2PO4, 1.5 mM KCl and 100.5 mM NaCl; and the concentration of the K+, Na+ and Cl− ions corresponds to that of the human blood serum.
Synthesis and characterization of HQCl-Pro and its complexes
:1 ratio) due to the rigid structure or the existence of diastereomers. For NMR peak-list see the ESI.† ESI-MS: calc. for [M(arene)L]+: 541.0832 (m/z) found: 541.0811 (m/z) and [M(arene)L + H]2+: 271.0455 (m/z) found: 271.0449 (m/z).
:1 ratio) due to the rigid structure or the existence of diastereomers. For NMR peak-list see the ESI.† ESI-MS: calc. for [M(arene)L]+: 499.0363 (m/z) found: 499.0354 (m/z) and [M(arene)L + H]2+: 250.0221 (m/z) found: 250.0214 (m/z).
pH-potentiometric measurements
pH-potentiometric measurements and calculation of the overall stability constants were performed similarly as it was performed in our previous works (see details in the ESI†).23,24,38–40 The computer program Hyperquad201346 was utilized to establish the stoichiometry of the complexes.
UV-Vis spectrophotometric, 1H NMR and distribution coefficient measurements, and ESI-MS
An Agilent Cary 8454 diode array spectrophotometer was used to record the UV-Vis spectra in the interval of 200–800 nm. The path length was 0.5 or 1 cm. Only one of the proton dissociation constants of HQCl-Pro could be determined by spectrophotometric titrations. Samples contained 130 μM HQCl-Pro. UV-Vis spectra were used to investigate the H2O/Cl− exchange processes of complexes at 400 μM concentration, at pH 5.50 (20 mM phosphate buffer) as a function of chloride concentration (0–310 mM).
1H and 13C NMR spectroscopic studies were carried out on a Bruker Avance III HD Ascend 500 Plus instrument. All 1H NMR spectra were recorded with the WATERGATE water suppression pulse scheme using the DSS internal standard, while 13C NMR spectra were recorded with the attached proton test method, which shows CH and CH3 in positive mode, and C and CH2 in negative mode. Samples were made in a 10% (v/v) D2O/H2O mixture to yield a concentration of 300 μM and was titrated at 25 °C, at I = 0.20 M (KNO3) in the absence or presence of [Rh(η5-C5Me5)(H2O)3]2+, [Ru(η6-p-cym)(H2O)3]2+ and [Ru(η6-tol)(H2O)3]2+ at 1:1 metal-to-ligand ratio. Stability constants for the complexes were calculated using the computer program PSEQUAD.47 For characterization, 10 mM CD3OD solutions were used.
Distribution coefficients at physiological pH (D7.4) of the complexes and the ligand were determined by the traditional shake-flask method in n-octanol/buffered aqueous solution at pH 7.40 at various chloride concentrations using UV-Vis detection as described in our former works.23,39
ESI-MS measurements were performed using a Waters Q-TOF Premier (Micromass MS Technologies, Manchester, UK) mass spectrometer equipped with an electrospray ion source. Samples contained 200 μM ligand or complex dissolved in water, and pH was adjusted to ∼7 with a small amount of HCl or KOH.
In vitro cell studies
Samples were analyzed by TXRF spectrometry; a TXRF 8030C spectrometer (Atomika Instruments GmbH, Oberschleissheim, Germany), equipped with a 3 kW fine focus X-ray tube containing a Mo/W alloy anode, a W/C multi-layer monochromator, adjusted to obtain an excitation energy of 33 keV selected out from the Bremsstrahlung was used. A Si(Li) detector with an active area of 80 mm2 was in operation with a resolution of 150 eV at 5.9 keV. 10 μL of 100 mg L−1 Ga was added to the samples prior to the TXRF analysis as the internal standard for the quantification procedure. Rh and Ru were not detected in blank samples. Due to the imprecision of cell counting, the Rh results were normalized to the Zn or S content of the samples.
Conclusions
A novel 8-hydroxyquinoline hybrid, (S)-5-chloro-7-((pro-line-1-yl)methyl)8-hydroxyquinoline (HQCl-Pro) was designed and prepared possessing excellent water solubility compared to the poorly soluble 8-hydroxyquinoline. HQCl-Pro harbors a CH2–N building block at position 7 that often has a role in MDR reversal activity, and as a bidentate ligand bearing an (N,O) donor it is an efficient chelator for half-sandwich organometallic Ru(II) and Rh(III) triaqua cations. Herein complex formation equilibria of HQCl-Pro with [Rh(η5-C5Me5)(H2O)3]2+, [Ru(η6-p-cym)(H2O)3]2+ and [Ru(η6-tol)(H2O)3]2+ were investigated in chloride-free aqueous solutions by pH-potentiometry, UV-visible spectrometry and 1H NMR spectroscopy. Stability and proton dissociation constants of the [M(arene)(L)(H2O)]+ complexes in addition to H2O/Cl− co-ligand exchange constants were determined. HQCl-Pro forms complexes with high but slightly lower stability compared with the unsubstituted 8-hydroxyquinoline. Arene loss and the oxidation of Ru(II) were found as side reactions with ligand excess at physiological pH. The excess of HQCl-Pro or rigid aromatic (N,N) bidentate ligands reacted with [Ru(η6-p-cym)(L)(H2O)]+ causing loss of p-cymene followed by oxidation or binding of a third bidentate ligand. Reaction did not occur with deferiprone containing oxygen donor atoms. The reactions with ethylenediamine, HSA, histidine and RPMI 1640 components were similar: most probably the formation of the mixed-ligand complex occurred without oxidation or p-cymene loss.Formation of mixed hydroxido [M(arene)(L)(OH)] complexes is less than 10% at physiological pH in the absence of chloride ions, and it is assumed to be an even lower fraction in the presence of this coordinating co-ligand. Substitution of the coordinated water molecule in the complex [M(arene)(L)(H2O)]+ by chloride ions results in neutral complexes, which have much higher lipophilicity than the aqua form and the ligand itself. The Rh(η5-C5Me5) complex has stronger chloride ion affinity than the Ru(η6-arene) containing ones, and the Ru(η6-tol) complex was found to be the most hydrophilic among the studied compounds.
The in vitro cytotoxic and antiproliferative activity of HQCl-Pro and its half-sandwich organometallic complexes were studied in Colo 205 drug sensitive and Colo 320 multidrug resistant cancer cell lines by the MTT assay in addition to cellular metal uptake studies. HQCl-Pro and its [Rh(η5-C5Me5)(L)(H2O)]+ complex showed relatively strong anticancer activity and moderate MDR selectivity, while complexation with ruthenium–arene species results in a lower or similar activity in the Colo 320 cell lines compared to that obtained in Colo 205 cells. Based on the 1H NMR spectra recorded for the Ru(η6-p-cym) complex of HQCl-Pro in the cell medium RPMI 1640 it was concluded that only mixed-ligand complex formation occurred with histidine, and the bidentate ligand is not dissociated from the complex. While a similar metal uptake level was found for both the Rh(η5-C5Me5) and Ru(η6-p-cymene) complexes using a 4 h incubation period, the longer incubation led to a higher intracellular Rh content which might contribute to the lower IC50 values of the Rh(η5-C5Me5) complex.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research, Development and Innovation Office-NKFIA through projects GINOP-2.3.2-15-2016-00038, FK 124240 and FIKP program TUDFO/47138-1/2019-ITM. J. P. M. acknowledges the support of the ÚNKP-19-3 National Excellence Program of the Ministry for Innovation and Technology.References
- R. Callaghan, F. Luk and M. Bebawy, Drug Metab. Dispos., 2014, 42, 623, DOI:10.1124/dmd.113.056176.
- A. Füredi, S. Tóth, K. Szebényi, V. F. S. Pape, D. Türk, N. Kucsma, L. Cervenak, J. Tóvári and G. Szakács, Mol. Cancer Ther., 2017, 16, 45, DOI:10.1158/1535-7163.MCT-16-0333-T.
- Y. Song, H. Xu, W. Chen, P. Zhan and X. Liu, MedChemComm, 2015, 6, 61, 10.1039/c4md00284a.
- V. Prachayasittikul, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Drug Des., Dev. Ther., 2013, 7, 1157, DOI:10.2147/DDDT.S49763.
- V. Oliveri and G. Vecchio, Eur. J. Med. Chem., 2016, 120, 252, DOI:10.1016/j.ejmech.2016.05.007.
- M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658, DOI:10.1021/acs.organomet.5b00868.
- A. R. Timerbaev, Metallomics, 2009, 1, 193, 10.1039/b902861g.
- R. Gogna, E. Madan, B. Keppler and U. Pati, Br. J. Pharmacol., 2012, 166, 617, DOI:10.1111/j.1476-5381.2011.01780.x.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219, 10.1039/c2cc30678f.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805, DOI:10.1021/acs.jmedchem.7b01689.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86, DOI:10.1016/j.ccr.2015.06.014.
- C. Artner, H. U. Holtkamp, W. Kandioller, C. G. Hartinger, S. M. Meier-Menches and B. K. Keppler, Chem. Commun., 2017, 53, 8002, 10.1039/c7cc04582d.
- R. E. Aird, J. Cummings, A. A. Titchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652, DOI:10.1038/sj/bjc/6600290.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909, 10.1039/c7cs00332c.
- A. Habtemariam, M. Melchart, R. Fernandez, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858, DOI:10.1021/jm060596m.
- A. Mitrović, J. Kljun, I. Sosič, S. Gobec, I. Turel and J. Kos, Dalton Trans., 2016, 45, 16913, 10.1039/c6dt02369j.
- M. C. Ruiz, J. Kljun, I. Turel, A. L. Di Virgilio and I. E. León, Metallomics, 2019, 11, 666, 10.1039/c8mt00369f.
- M. Gobec, J. Kljun, I. Sosič, I. Mlinarič-Raščan, M. Ursič, S. Gobec and I. Turel, Dalton Trans., 2014, 43, 9045, 10.1039/c4dt00463a.
- J. Kljun, I. E. León, S. Peršič, J. F. Cadavid-Vargas, S. B. Etcheverry, W. He, Y. Bai and I. Turel, J. Inorg. Biochem., 2018, 186, 187, DOI:10.1016/j.jinorgbio.2018.05.009.
- A. Mitrović, J. Kljun, I. Sosič, M. Ursič, A. Meden, S. Gobec, J. Kos and I. Turel, Inorg. Chem., 2019, 58, 12334, DOI:10.1021/acs.inorgchem.9b01882.
- U. Sliwinska, F. P. Pruchnik, S. Ułaszewski, M. Latocha and D. Nawrocka-Musiał, Polyhedron, 2010, 29, 1653, DOI:10.1016/j.poly.2010.02.013.
- S. Movassaghi, M. Hanif, H. U. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Dalton Trans., 2018, 47, 2192, 10.1039/c7dt04451h.
- O. Dömötör, V. F. S. Pape, N. V. May, G. Szakács and É. A. Enyedy, Dalton Trans., 2017, 46, 4382, 10.1039/c7dt00439g.
- O. Dömötör, S. Aicher, M. Schmidlehner, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and É. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57, DOI:10.1016/j.jinorgbio.2014.01.020.
- L. Bíró, A. J. Godó, Z. Bihari, E. Garribba and P. Buglyó, Eur. J. Inorg. Chem., 2013, 2013, 3090, DOI:10.1002/ejic.201201527.
- A. A. Kurganov, V. A. Davankov, Y. D. Koreshkov and S. V. Rogozhin, Russ. J. Coord. Chem., 1977, 3, 667 CAS.
- É. A. Enyedy, O. Dömötör, E. Varga, T. Kiss, R. Trondl, C. G. Hartinger and B. K. Keppler, J. Inorg. Biochem., 2012, 117, 189, DOI:10.1016/j.jinorgbio.2012.08.005.
- Marvin, version 18.23.0, ChemAxon, Budapest, Hungary, 2018, https://chemaxon.com/ Search PubMed.
- V. F. S. Pape, N. V. May, G. T. Gál, I. Szatmári, F. Szeri, F. Fülöp, G. Szakács and É. A. Enyedy, Dalton Trans., 2018, 47, 17032, 10.1039/c8dt03088j.
- M. Albrecht, K. Witt, P. Weis, E. Wegelius and R. Fröhlich, Inorg. Chim. Acta, 2002, 341, 25, DOI:10.1016/S0020-1693(02)01201-X.
- J. P. Mészáros, O. Dömötör, C. M. Hackl, A. Roller, B. K. Keppler, W. Kandioller and É. A. Enyedy, New J. Chem., 2018, 42, 11174, 10.1039/c8nj01681j.
- J. G. Malecki, M. Jaworska, R. Kruszynski and J. Klak, Polyhedron, 2005, 24, 3012, DOI:10.1016/j.poly.2005.06.053.
- W. Hu, Q. Luo, X. Ma, K. Wu, J. Liu, Y. Chen, S. Xiong, J. Wang, P. J. Sadler and F. Wang, Chem. – Eur. J., 2009, 15, 6586, DOI:10.1002/chem.200900699.
- A. Merlino, Coord. Chem. Rev., 2016, 326, 111, DOI:10.1016/j.ccr.2016.08.001.
- S. J. Dougan, M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, Inorg. Chem., 2006, 45, 10882, DOI:10.1021/ic061460h.
- H. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 173, DOI:10.1021/ja027719m.
- C. Gossens, I. Tavernelli and U. Rothlisberger, J. Phys. Chem. A, 2009, 113, 11888, DOI:10.1021/ct6003577.
- É. Sija, C. G. Hartinger, B. K. Keppler, T. Kiss and É. A. Enyedy, Polyhedron, 2014, 67, 51, DOI:10.1016/j.poly.2013.08.057.
- J. M. Poljarević, G. T. Gál, N. V. May, G. Spengler, O. Dömötör, A. R. Savić, S. Grgurić-Šipka and É. A. Enyedy, J. Inorg. Biochem., 2018, 181, 74, DOI:10.1016/j.jinorgbio.2017.12.017.
- É. A. Enyedy, O. Dömötör, C. M. Hackl, A. Roller, M. S. Novak, M. A. Jakupec, B. K. Keppler and W. Kandioller, J. Coord. Chem., 2015, 68, 1583, DOI:10.1080/00958972.2015.1023195.
- O. Dömötör, C. M. Hackl, K. Bali, A. Roller, M. Hejl, M. A. Jakupec, B. K. Keppler, W. Kandioller and É. A. Enyedy, J. Organomet. Chem., 2017, 846, 287, DOI:10.1016/j.jorganchem.2017.06.027.
- A. M. Pizarro, A. Habtemariam and P. J. Sadler, Top. Organomet. Chem., 2010, 32, 21, DOI:10.1007/978-3-642-13185-1_2.
- C. F. Richard, R. L. Gustafson and A. E. Martell, J. Am. Chem. Soc., 1959, 81, 1033, DOI:10.1021/ja01514a006.
- H. F. Steger and A. Corsini, J. Inorg. Nucl. Chem., 1973, 35, 1621, DOI:10.1016/0022-1902(73)80253-2.
- R. A. Zelonka and M. C. Baird, Can. J. Chem., 1972, 50, 3063, DOI:10.1139/v72-486.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739, DOI:10.1016/0039-9140(96)01958-3.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Stability Constants, ed. D. L. Leggett, Plenum Press, New York, 1985, pp. 291–353 Search PubMed.
- GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla, California, USA, 2018, http://www.graphpad.com Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization, additional UV-Vis spectra of kinetic measurements and titrations, and pH-dependent 1H NMR spectra of compounds. See DOI: 10.1039/D0DT01256D |
| This journal is © The Royal Society of Chemistry 2020 |