
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface modification of aqueous miscible organic layered double hydroxides (AMO-LDHs)†
Chunping
Chen
,
Jean-Charles
Buffet
 and
Dermot
O'Hare
*
and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: dermot.ohare@chem.ox.ac.uk
First published on 10th June 2020
Abstract
Silane modification of layered double hydroxides (LDHs) plays an important role in controlling the surface hydrophobicity and improving the compatibility of LDHs dispersed in non-polar materials. However, the surface modification of conventional LDHs in hydrous conditions typically results in aggregated particle morphologies, low surface areas and accessible pore volumes. In this study, well dispersed and high surface area silane grafted AMO-Zn2MgAl-CO3 LDH were prepared using the silane coupling agents (triethoxyvinylsilane (TEVS), triethoxyoctylsilane (TEOS) and (3-glycidyloxypropyl)trimethoxysilane (TMGPS)) in anhydrous acetone. Solution 1H NMR spectroscopy was initially used to study the rate and extent of silane reactivity with AMO-Zn2MgAl-CO3 LDH. Powder XRD, TEM, N2 BET specific surface area and total pore volume measurements showed that the structure and morphology of silane-treated AMO-Zn2MgAl-CO3 LDHs remained largely unchanged. Solid state 13C CP-MAS, 27Al DP-MAS and 29Si CP-MAS NMR spectroscopy indicates that the silanes have been successfully grafted onto the surface of the LDH. In addition to maintaining their structure, morphology, high surface area and total pore volume, these surface-functionalised LDHs are now more hydrophobic, displaying a saturation water vapour uptake (<4 wt%) that is ca. 60% lower than the untreated AMO-LDH.
Introduction
Layered double hydroxides (LDHs) comprise a large family of anion exchangeable layered solids, they may be represented by the general formula [Mz+1−xM′y+x(OH)2](Xn−)a/n·bH2O, wherein M and M′ are metal cations, most commonly z = 2 (e.g. Mg2+); y = 3 (e.g. Al3+), 0 < x < 1, 0 < b < 5, giving a = z(1 − x) + xy − 2; Xn− is a charge compensating anionic moiety either organic, inorganic or biological.1–3 Owing to their excellent anionic exchangeability and good biocompatibility, LDHs have been widely used in wastewater treatment and biomolecular delivery.4–6 The possibility of a high aspect 2D structure with a controllable layer thickness gives LDHs a promising application in gas and moisture barrier coatings on flexible polymeric films.7–10 With the advantages of composition flexibility, homogeneous distribution of the two or more metal cations within the layers and the tunability of the number and strength of basic/acidic sites, LDHs have received considerable interest as catalysts or precursors to mixed metal oxide catalysts.11–15Recently, LDHs have attracted increasing attention as a new generation of inorganic additives in polymers.16–22 LDHs were reported to be an effective thermal and UV stabiliser in poly(vinyl chloride) (PVC). Since LDHs are intrinsically basic, they can act as acid scavenger or radical trap; both features are key factors for the thermal and UV stabilisation of PVC.22 Dispersions of LDHs in polymers also impart excellent flame retardancy and smoke suppression properties. During combustion, LDHs undergo endothermic decomposition, absorbing heat and releasing water and CO2 in the case of a carbonate LDHs. This LDHs facilitate absorption of heat and the formation of an expanded carbonaceous coating or char on the polymer.17 LDHs may be intercalated with functional anions such as borate, phosphate, leading to new generations of LDHs based flame retardant materials with enhanced performance and environmentally-friendly (halogen free) character.23–26 However, LDHs are inherently hydrophilic materials that can display very rapid moisture uptake leading to a material with a very high moisture content. This seriously affects storage, transport and the compounding of LDHs with non-polar polymers (LDHs can release the surface bound water resulting in bubble formation).
To facilitate formation of effective LDH-organic composites, a number of strategies have been previously explored to obtain LDHs containing less water and possessing hydrophobic surface character. Intercalation or surface adsorption of anionic surfactants such as sulfonates, carboxylates, phosphates and sulphates.27–29 The interaction between these surfactants and the LDHs is mainly via electrostatic interactions and hydrogen bonding. Silane modification is another promising strategy, forming chemical bonding via reaction of a silane with the hydroxyl groups on LDHs surface. Silanes may contain one or more functional groups enabling chemical linkages with various polymers improving their compatibility. In 2005, Park et al. reported that dodecylsulfate intercalated LDHs can be surface modified with silane in the presence of a cationic surfactant (N-cetyl-N,N,N-trimethylammonium bromide).30 The formation of covalent bonding was demonstrated using solid-state 29Si DP-MAS NMR spectroscopy. Wypych et al. studied the surface modification of LDH single sheets with (3-aminopropyl)triethoxysilane (TEAPS) in toluene.31 However, the synthetic method required at least two steps. Subsequently, Qi et al. investigated a series of synthetic methods including induced hydrolysis, surfactant assisted in situ coprecipitation, calcination-reconstruction in Na2CO3 solution with direct silylation.32–36 They found that in an aqueous environment, the silylation process is induced by hydrolysis and condensation of TEAPS on LDH surface even without any surfactant, whilst the direct silylation reaction cannot occur between the LDHs and TEAPS. The condensation reaction can only takes place between the adsorbed TEAPS and LDHs under thermal treatment. Recently, Guo et al. found that an LDH can be directly silylated with phenyltriethoxysilane upon reflux in toluene for 12 h.37 However, the direct observation of the reaction between a silane and an LDH has not yet been reported. The silylation of LDHs under anhydrous conditions is still at an early stage, the challenge is to maintain particle properties such as morphology, surface area and porosity after silane modification.
In this work, we present the silane modification of Aqueous Miscible Organic Solvent Treated (AMOST) [Zn0.47Mg0.24Al0.25(OH)2](CO3)0.125 (AMO-Zn2MgAl-CO3 LDH) under anhydrous conditions. Three silane coupling agents, triethoxyvinylsilane (TEVS), triethoxyoctylsilane (TEOS), and (3-glycidyloxypropyl) trimethoxysilane (TMGPS) were used in this study. In situ solution 1H NMR spectroscopy was used to monitor the rate and extent of reaction by observing the soluble products produced during the reaction between silanes and AMO-Zn2MgAl-CO3 LDH. The silane-modified LDH products were characterised by X-ray powder diffraction (XRD), solid-state 13C CP-MAS, 27Al DP-MAS and 29Si CP-MAS nuclear magnetic resonance (NMR) spectroscopy, thermogravimetric analysis (TGA), transmission electron microscopy (TEM), N2 Brauner–Emmett–Teller (BET) specific surface measurements and Barrett–Joyner–Halenda (BJH) total pore volume measurements. The hydrophobicity of the modified LDHs was investigated by monitoring the rate and quantity of water vapour adsorption at a range of relative humidities.
Results and discussion
The synthesis of [Zn0.47Mg0.24Al0.25(OH)2](CO3)0.125·0.06(H2O)·0.03(ethanol), AMO-Zn2MgAl-CO3 LDH, was performed in two steps. Firstly, Zn2MgAl-CO3 was prepared using conventional co-precipitation at pH 10 followed by ageing for 16 h. Then, AMO solvent treatment was performed by dispersion of the aqueous wet solid in ethanol for 4 h. The XRD, TGA, and N2 BET data for AMO-Zn2MgAl-CO3 LDH are shown in Fig. S4a, S7a and S9a† respectively. The chemical composition of the AMO-Zn2MgAl-CO3 LDH was determined by TGA, ICP and CHN combustion microanalysis. As reported previously, AMO-LDHs have enhanced surface areas and particle dispersion compared to conventionally prepared materials.38–40 The TEM image of AMO-Zn2MgAl-CO3 LDH shows well dispersed LDH platelets with primary particle sizes in the range 10–250 nm (Fig. 7a). AMO-LDHs exhibit very high-water vapour uptake responses. Fig. 1 shows the time dependence of the water vapour adsorption of AMO-Zn2MgAl-CO3 LDH under different relative humidities at 20 °C. In a RH60 relative humidity environment, AMO-Zn2MgAl-CO3 LDH can adsorb up to 10 wt% water. The total water vapour uptake capacity increases with increasing relative humidity, reaching maximum of 21 wt% moisture uptake in an environment with a relative humidity of RH99. Rapid water vapour uptake is a major impediment to the usage of AMO-LDHs in moisture sensitive applications and in the applications requiring the compounding of LDHs with hydrophobic, non-polar polymers to prepare LDH-polymer nanocomposites. Hence, a silane treatment approach was developed using anhydrous conditions in order to modify the hydrophobicity of the AMO-LDH surfaces. | ||
| Fig. 1 Time dependence of water vapour uptake of AMO-Zn2MgAl-CO3 LDH in different humidity atmospheres (a) RH99, (b) RH70 and (c) RH60 at 20 °C. | ||
The AMO-LDHs were initially thermally treated to remove any surface-bound and co-intercalated water located in the interlayer galleries (maximising the available surface hydroxyl concentration). Under anhydrous conditions, the alkoxy groups of the triethoxy- or trimethoxysilanes should react with chemically accessible surface hydroxyl groups on the LDH via direct condensation as shown schematically in Fig. 2(a), forming new Si–O−M bonds (M = Mg, Zn, Al) to the LDH surface.41,42 Although, it is challenging to directly observe this type of heterogeneous reaction, solution 1H NMR spectroscopy was successfully used to follow the course of the reaction between the trialkoxysilanes and the LDH.
 | ||
| Fig. 2 (a) Proposed reaction of a trialkoxysilane with LDH surface hydroxyl groups; (b) solution 1H NMR spectra of (i) methanol, (ii) TMGPS, a suspension of AMO-Zn2MgAl-CO3 LDH with TMGPS at 60 °C after (iii) 3 h, (iv) 33 h and (v) 68 h; (c) time dependence of the integrated intensity of the 1H resonances for (MeO)3Si- (P1) and MeOH (P2). | ||
The 1H solution NMR spectra of TMGPS in the presence of a dispersion of AMO-Zn2MgAl-CO3 LDH (acetone-d6) are shown in Fig. 2(b) and Fig. S1.† The proton resonances at 3.53 ppm (labelled as P1) and 3.30 ppm (labelled as P2) (Fig. 2(b)) can be assigned to the (MeO)3Si-protons in TMGPS and MeOH respectively. After heating the mixture at 60 °C for 3 h, the resonance at 3.30 ppm appears indicating the formation of methanol. The resonance related to methanol dramatically increased while the (MeO)3Si-resonance in TMGPS decreases significantly within the first 24 h, indicating that the condensation happened quickly at 60 °C (Fig. 2b). By way of control, no change was observed when TMGPS was heated in acetone-d6 without LDH (Fig. S2†). When TEVS was added to a dispersion of AMO-Mg3Al-CO3 LDH, the formation of ethanol was observed, indicating the reaction between TEVS and LDH surface hydroxyl groups has occurred (Fig. S3†). Ethanol was generated almost immediately at room temperature.
Bulk samples (2 g) of TMGPS, TEVS and TEOS modified AMO-Zn2MgAl-CO3 LDH were prepared in acetone at 60 °C. The XRD data indicate the basic layer structure and crystallinity of silane-modified AMO-Zn2MgAl-CO3 LDH remained unchanged compared to pristine AMO-Zn2MgAl-CO3 LDH (Fig. S4 and Table S1†). In the XRD, a small increase was observed in the c-lattice parameter from 20.4 to 21.1 and 21.7 Å for TEOS, TEVS and TMGPS treated samples respectively. The interlayer separation (6.8 Å) of pristine AMO-Zn2MgAl-CO3 LDH is slightly lower than that of normal carbonated containing LDH due to liberation of interlayer water molecules after thermally pre-treated at 180 °C.43 There was no change observed in the a-lattice parameter. The small expansion of the interlayer separation may result from some of silane modifying the internal surface of the LDH.
The solid-state 13C CP-MAS NMR spectrum of pristine AMO-Zn2MgAl-CO3 LDH (Fig. 3a) exhibits one resonance at around 170 ppm, which is due to the presence of interlayer carbonate ions. After silane reaction with TMGPS, additional resonances in the range of 0–100 ppm can be observed, which correspond to the 13C resonances of the 3-glycidyloxypropyl group (Fig. 3b). No resonances from the methoxy groups (marked as 13, 14, and 15 in Fig. 3) of the TMGPS can be observed, supporting the hypothesis that silane binding involves the loss of most of the methoxy groups.
 | ||
| Fig. 3 13C CPMAS NMR spectra of (a) AMO-Zn2MgAl-CO3 LDH and (b) TMGPS modified AMO-Zn2MgAl-CO3 LDH. | ||
The solid-state 27Al NMR spectrum of the pristine AMO-Zn2MgAl-CO3 LDH shows a single resonance at −15 ppm, owing to the presence of octahedral Al(Oh) in the metal hydroxide structure of LDH (Fig. 4a). After silane modification, a broad, weak resonance attributed to a tetrahedral Al(Td) environment can be observed between 20–80 ppm (Fig. 4b inset), which can be ascribed to the formation of Si–O−Al(O)3 moiety on the surface of the LDH.44,45 This resonance is broad and asymmetric, probably because the tetrahedral surface Al sites on the LDH bond to different degrees with the silane. Both TEVS and TEOS also react in a similar fashion (Fig. S5†).
 | ||
| Fig. 4 27Al DP-MAS NMR spectra of (a) AMO-Zn2MgAl-CO3 LDH and (b) TMGPS modified AMO-Zn2MgAl-CO3 LDH. Inset is the enlarge range (20–80 ppm) of TMGPS modified AMO-Zn2MgAl-CO3 LDH at Td peak. | ||
The solid-state 29Si CP-MAS NMR spectrum of TMGPS modified AMO-Zn2MgAl-CO3 LDH shows resonances between −25 and −75 ppm (Fig. S6†), which can be assigned to Tn sites of silicon (where n represents the number of Si–(O–M)n bonds formed between silane and LDH or a silicon neighbour).30,36,46 Deconvolution of the 29Si CP-MAS NMR spectrum (Table S2†) reveals that the main 29Si resonance in TMGPS modified LDH is a monodentate T1 site, including 1st hydrolysed monodentate at −47 ppm (49%), 2nd hydrolysed monodentate at −52 ppm (8%) and monodentate at −54 ppm (14%). In addition, 29% of total surface bound silicon population is due to a T2 site, i.e. Si–(O–M)2 involving the AMO-Zn2MgAl-CO3 LDH surface and/or a neighbouring silicon.
Thermogravimetric analysis (TGA) of AMO-Zn2MgAl-CO3 LDH and TMGPS modified AMO-Zn2MgAl-CO3 LDH present characteristic thermal events of an LDH (Fig. 5). The weight loss up to 200 °C is due to the release of water which includes both physiosorbed and co-intercalated interlayer water. All silane modified AMO-Zn2MgAl-CO3 LDHs still show some water loss at 200 °C but much less when compared with the pristine LDH (14 wt%) at 200 °C (Fig. S7, Table S3†). For example, the TMGPS modified LDH exhibits a water loss of only 4 wt% compared with that of pristine LDH (14 wt%) at 200 °C. We presume this small amount of water resides within the interlayer galleries and so was not accessible to react with the silane reagents.
 | ||
| Fig. 5 TGA curves of (a) AMO-Zn2MgAl-CO3 LDH and (b) TMGPS modified AMO-Zn2MgAl-CO3 LDH. | ||
The water vapour adsorption of AMO-Zn2MgAl-CO3 LDH and TMGPS-modified AMO-Zn2MgAl-CO3 LDH was evaluated at RH70 and 20 °C (Fig. 6). Pristine AMO-Zn2MgAl-CO3 LDH exhibits rapid water vapour adsorption, weight gain of 12 wt% compared to the dry LDH. After TMGPS surface modification, the hydrophobicity of the materials is evidenced by a significantly reduced saturation water vapour adsorption capacity (below 5 wt% after 13 days). The reduced water vapour adsorption behaviour of the other silane modified LDH are shown in Fig. S8 and Table S3.† Surface modification using vinyl silane produces a similar saturation water vapour adsorption capacity (5 wt%).
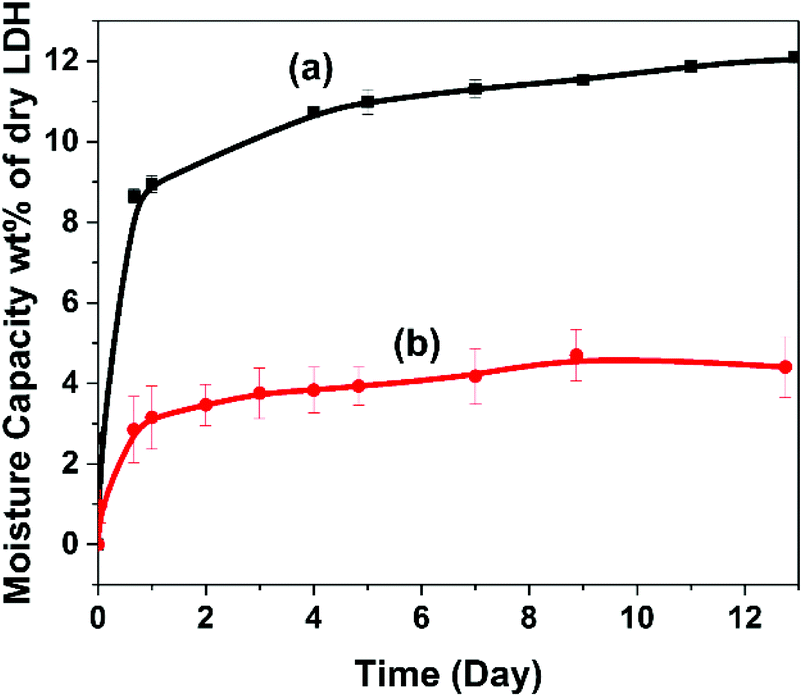 | ||
| Fig. 6 Water vapour uptake of (a) AMO-Zn2MgAl-CO3 LDH and (b) TMGPS modified AMO-Zn2MgAl-CO3 LDH in RH 70 at 20 °C. | ||
However, alkyl silane treated LDHs showed a slightly lower moisture sorption inhibition (6.5 wt% after 13 days). This could be due to a less efficient surface functionalisation by alkyl silane.
The morphology, surface area and total pore size of the AMO-Zn2MgAl-CO3 LDH was compared before and after surface modification. AMO-Zn2MgAl-CO3 LDH adopts a typical LDH platelet morphology, the as-prepared sample exhibits a particle size range of 10–250 nm (Fig. 7a). After reaction with silanes (TEVS, TEOS and TMGPS) in acetone at 60 °C, the particle size distribution remains unchanged. No significant agglomerates can be observed in the modified samples. The N2 BET specific surface areas of the modified LDHs are also unaffected (Fig. S9†) while the total pore volumes are slightly larger than the pristine LDH (Fig. S10†). Silane treatment using acetone seems to be effective in achieving a high degree of surface treatment, it prevents agglomeration of the individual particles during the reaction leading to unchanged morphology, specific surface area and porosity after silane modification.
 | ||
| Fig. 7 TEM images of (a) AMO-Zn2MgAl-CO3 LDH, (b) TEVS modified AMO-Zn2MgAl-CO3 LDH, (c) TEOS modified AMO-Zn2MgAl-CO3 LDH and (d) TMGPS modified AMO-Zn2MgAl-CO3 LDH. | ||
Conclusions
Trialkoxysilanes react efficiently with a dispersion of an AMO-LDH in acetone via elimination of the equivalent alcohol. The elimination of ethanol or methanol using either triethoxyvinylsilane (TEVS), triethoxyoctylsilane (TEOS), or (3-glycidyloxypropyl) trimethoxysilane (TMGPS) can be observed by solution 1H NMR spectroscopy. Solid state 13C CP-MAS, 27Al DP-MAS and 29Si CP-MAS NMR spectroscopy confirms the surface functionalisation.In addition to maintaining their structure, morphology, high surface area and total pore volume, these LDHs are now more hydrophobic, displaying a saturation water vapour uptake (<4 wt%) that is ca. 60% lower than the untreated AMO-LDH.
Experimental
Synthesis of AMO-Zn2MgAl-CO3 LDH
The metal precursor solution containing 25 mmol Mg(NO3)2·6H2O, 50 mmol Zn(NO3)2·6H2O, 25 mmol Al(NO3)3·9H2O and 100 mL water was added dropwise into Na2CO3 base solution (100 mL water with 12.5 mmol Na2CO3) within 1 h. The pH was kept constant around 10.0 by dropwise addition of a 4.0 M NaOH solution. After stirring for 16 h at room temperature, the mixture was filtered and washed with deionised water until pH 7. The wet LDH solid was dispersed with ethanol according to the AMOST method.38,39 The wet LDH solid was washed with ethanol (1000 mL) by filtration and then re-dispersed in fresh ethanol (600 mL) under stirring for 4 h. The LDH solid was then filtered and dispersed in fresh ethanol (400 mL). The final AMO-LDH solid was dried under vacuum overnight. AMO-Zn2MgAl-CO3 LDH has the chemical composition [Zn0.47Mg0.24Al0.25(OH)2](CO3)0.125·0.06(H2O)·0.03(ethanol).Monitoring silane modification of AMO-Zn2MgAl-CO3 LDH
AMO-Zn2MgAl-CO3 LDH (20 mg) was loaded in a J. Young NMR tube followed by heating in oven at 180 °C. After 6 h, the NMR tube was closed and cooled in a desiccator under vacuum. 700 μL of a deuterated acetone (acetone-d6) solution containing 2.8 mmol per g-LDH of the appropriate silane was introduced into the tube, then sealed and heated at 60 °C. The reaction was followed by 1H NMR spectroscopy.Bulk synthesis of silane modified AMO-Zn2MgAl-CO3 LDH
AMO-Zn2MgAl-CO3 LDH (2 g) was dried in an oven at 180 °C. After 6 h, the solid was cooled in a desiccator under vacuum followed by addition of 100 mL of acetone. Silanes (2.8 mmol per g-LDH) were introduced by slow injection under a N2 flow while stirring. The slurry was refluxed at 60 °C for 18 h. The obtained solid was collected and washed with acetone. The final solid was dried in an oven at 80 °C for 18 h.Water vapour uptake measurements
The water vapour uptake measurement was tested according to a modified Callahan's method.47 The water vapour uptake was tested in a sealed box at room temperature (20 °C) and repeated at least three times. The relative humidity of RH99, RH70 and RH60 were generated by saturated solution of KNO3, NaCl and Mg(NO3)2, respectively, monitored by an electrical moisture meter. Detailed measurement procedure is in shown in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C. and J.-C. B. would like to thank SCG Chemicals Co., Ltd (Thailand) for funding, surface analysis facility (University of Oxford) for use of the TGA and FTIR instruments and Dr Nicholas H. Rees (University of Oxford) for solid state NMR spectroscopy.Notes and references
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- V. Rives, Layered double hydroxides: present and future, Nova Publishers, 2001 Search PubMed.
- D. G. Evans and R. C. Slade, in Layered double hydroxides, Springer, 2006, pp. 1–87 Search PubMed.
- C. Chen, P. Wang, T.-T. Lim, L. Liu, S. Liu and R. Xu, J. Mater. Chem. A, 2013, 1, 3877–3880 RSC.
- C. Chen, K. Y. Lee, H. Gong, Y. Zhang and R. Xu, Nanoscale, 2013, 5, 4314–4320 RSC.
- J.-M. Oh, D.-H. Park and J.-H. Choy, Chem. Soc. Rev., 2011, 40, 583–595 RSC.
- Y. Dou, S. Xu, X. Liu, J. Han, H. Yan, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2014, 24, 514–521 CrossRef CAS.
- F. Zhang, L. Zhao, H. Chen, S. Xu, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2008, 47, 2466–2469 CrossRef CAS PubMed.
- Y. Dou, A. Zhou, T. Pan, J. Han, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2014, 50, 7136–7138 RSC.
- J. Yu, K. Ruengkajorn, D.-G. Crivoi, C. Chen, J.-C. Buffet and D. O'Hare, Nat. Commun., 2019, 10, 2398 CrossRef PubMed.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- T. Baskaran, J. Christopher and A. Sakthivel, RSC Adv., 2015, 5, 98853–98875 RSC.
- V. R. L. Constantino and T. J. Pinnavaia, Catal. Lett., 1994, 23, 361–367 CrossRef CAS.
- V. R. L. Constantino and T. J. Pinnavaia, Inorg. Chem., 1995, 34, 883–892 CrossRef CAS.
- B. Sels, D. D. Vos, M. Buntinx, F. Pierard, A. Kirsch-De Mesmaeker and P. Jacobs, Nature, 1999, 400, 855–857 CrossRef CAS.
- Y. S. Gao, Q. Wang, J. Y. Wang, L. Huang, X. R. Yan, X. Zhang, Q. L. He, Z. P. Xing and Z. H. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 5094–5104 CrossRef CAS PubMed.
- Y. S. Gao, J. W. Wu, Q. Wang, C. A. Wilkie and D. O'Hare, J. Mater. Chem. A, 2014, 2, 10996–11016 RSC.
- Y. Z. Bao, Z. M. Huang, S. X. Li and Z. X. Weng, Polym. Degrad. Stab., 2008, 93, 448–455 CrossRef CAS.
- C. X. Zhao, Y. Liu, D. Y. Wang, D. L. Wang and Y. Z. Wang, Polym. Degrad. Stab., 2008, 93, 1323–1331 CrossRef CAS.
- X. D. Wang and Q. Zhang, Polym. Int., 2004, 53, 698–707 CrossRef CAS.
- D. G. Evans and X. Duan, Chem. Commun., 2006, 485–496 RSC.
- F. J. Labuschagne, D. M. Molefe, W. W. Focke, I. van der Westhuizen, H. C. Wright and M. D. Royeppen, Polym. Degrad. Stab., 2015, 113, 46–54 CrossRef CAS.
- A. Shimamura, E. Kanezaki, M. I. Jones and J. B. Metson, J. Solid State Chem., 2012, 186, 116–123 CrossRef CAS.
- J. Qin, P. Xie, Y. Z. Tian, H. Zhang and J. Yu, J. Therm. Anal. Calorim., 2012, 110, 1193–1198 CrossRef CAS.
- M. Badreddine, A. Legrouri, A. Barroug, A. De Roy and J. P. Besse, Mater. Lett., 1999, 38, 391–395 CrossRef CAS.
- L. Wang, S. Su, D. Chen and C. A. Wilkie, Polym. Degrad. Stab., 2009, 94, 770–781 CrossRef CAS.
- F. R. Costa, A. Leuteritz, U. Wagenknecht, D. Jehnichen, L. Häußler and G. Heinrich, Appl. Clay Sci., 2008, 38, 153–164 CrossRef CAS.
- Z. P. Xu and P. S. Braterman, J. Mater. Chem., 2003, 13, 268–273 RSC.
- B. Wang, H. Zhang, D. G. Evans and X. Duan, Mater. Chem. Phys., 2005, 92, 190–196 CrossRef CAS.
- A. Y. Park, H. Kwon, A. J. Woo and S. J. Kim, Adv. Mater., 2005, 17, 106–109 CrossRef CAS.
- F. Wypych, A. Bail, M. Halma and S. Nakagaki, J. Catal., 2005, 234, 431–437 CrossRef CAS.
- Q. Tao, H. He, R. L. Frost, P. Yuan and J. Zhu, Appl. Surf. Sci., 2009, 255, 4334–4340 CrossRef CAS.
- Q. Tao, H. He, R. L. Frost, P. Yuan and J. Zhu, J. Therm. Anal. Calorim., 2010, 101, 153–159 CrossRef CAS.
- Q. Tao, J. Zhu, R. L. Frost, T. E. Bostrom, R. M. Wellard, J. Wei, P. Yuan and H. He, Langmuir, 2010, 26, 2769–2773 CrossRef CAS PubMed.
- Q. Tao, J. Zhu, R. M. Wellard, T. E. Bostrom, R. L. Frost, P. Yuan and H. He, J. Mater. Chem., 2011, 21, 10711–10719 RSC.
- Q. Tao, H. He, T. Li, R. L. Frost, D. Zhang and Z. He, J. Solid State Chem., 2014, 213, 176–181 CrossRef CAS.
- W. Guo, Y. Zhao, F. Zhou, X. Yan, B. Fan and R. Li, Appl. Catal., A, 2016, 522, 101–108 CrossRef CAS.
- C. Chen, A. Wangriya, J.-C. Buffet and D. O'Hare, Dalton Trans., 2015, 44, 16392–16398 RSC.
- C. Chen, M. Yang, Q. Wang, J.-C. Buffet and D. O'Hare, J. Mater. Chem. A, 2014, 2, 15102–15110 RSC.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301–6303 RSC.
- K. L. Mittal, Silanes and other coupling agents, CRC Press, 2007 Search PubMed.
- A. Michelot, S. Sarda, C. Audin, E. Deydier, E. Manoury, R. Poli and C. Rey, J. Mater. Sci., 2015, 50, 5746–5757 CrossRef CAS.
- V. R. L. Constantino and T. J. Pinnavaia, Inorg. Chem., 1995, 34, 883–892 CrossRef CAS.
- C. Chen, R. Felton, J.-C. Buffet and D. O'Hare, Chem. Commun., 2015, 51, 3462–3465 RSC.
- C. Chen, C. F. Byles, J.-C. Buffet, N. H. Rees, Y. Wu and D. O'Hare, Chem. Sci., 2016, 7, 1457–1461 RSC.
- Q. Tao, J. Zhu, R. M. Wellard, T. E. Bostrom, R. L. Frost, P. Yuan and H. He, J. Mater. Chem., 2011, 21, 10711–10719 RSC.
- J. C. Callahan, G. W. Cleary, M. Elefant, G. Kaplan, T. Kensler and R. A. Nash, Drug Dev. Ind. Pharm., 1982, 8, 355–369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, PXRD, BET surface area and Pore volume, moisture uptake, NMR spectroscopy, 13C CP-MAS, 27Al DP-MAS and 29Si CP-MAS NMR spectroscopy. See DOI: 10.1039/d0dt01213k |
| This journal is © The Royal Society of Chemistry 2020 |