
Structural analysis of and selective CO2 adsorption in mixed-ligand hydroxamate-based metal–organic frameworks†
Koh
Sugamata,  *a
Mao
Minoura
*a
*a
Mao
Minoura
*a
*a
Mao
Minoura
*a
Abstract
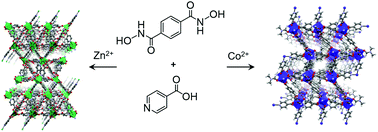
Two mixed-ligand metal–organic frameworks, [Zn2(BDHA)0.5(INA)3] (MOF-1: H2BDHA = benzene-1,4-dihydroxamic acid; HINA = isonicotinic acid) and [Co2(BDHA)0.5(INA)3(DMF)] (MOF-2), were solvothermally synthesized and fully characterized by single-crystal X-ray crystallography as well as N2, H2, and CO2 gas-sorption measurements. The results constitute the first detailed analysis of the bonding environment around the hydroxamates in such MOFs, which are simultaneously decorated with Lewis-basic sites from the hydroxamate moieties and metal sites predisposed for coordinative unsaturation. MOF-2 shows a desirably selective adsorption of CO2 relative to N2.
 Please wait while we load your content...
Please wait while we load your content...

