
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Accurate computed spin-state energetics for Co(III) complexes: implications for modelling homogeneous catalysis†
Samuel E.
Neale‡
 a,
Dimitrios A.
Pantazis
*b and
Stuart A.
Macgregor
*a
a,
Dimitrios A.
Pantazis
*b and
Stuart A.
Macgregor
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Edinburgh, United Kingdom EH14 4AS. E-mail: s.a.macgregor@hw.ac.uk
bMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: dimitrios.pantazis@kofo.mpg.de
First published on 21st April 2020
Abstract
Co(III) complexes are increasingly prevalent in homogeneous catalysis. Catalytic cycles involve multiple intermediates, many of which will feature unsaturated metal centres. This raises the possibility of multi-state character along reaction pathways and so requires an accurate approach to calculating spin-state energetics. Here we report an assessment of the performance of DLPNO-CCSD(T) (domain-based local pair natural orbital approximation to coupled cluster theory) against experimental 1Co to 3Co spin splitting energies for a series of pseudo-octahedral Co(III) complexes. The alternative NEVPT2 (strongly-contracted n-electron valence perturbation theory) and a range of density functionals are also assessed. DLPNO-CCSD(T) is identified as a highly promising method, with mean absolute deviations (MADs) as small as 1.3 kcal mol−1 when Kohn–Sham reference orbitals are used. DLPNO-CCSD(T) out-performs NEVPT2 for which a MAD of 3.5 kcal mol−1 can be achieved when a (10,12) active space is employed. Of the nine DFT methods investigated TPSS is the leading functional, with a MAD of 1.9 kcal mol−1. Our results show how DLPNO-CCSD(T) can provide accurate spin state energetics for Co(III) species in particular and first row transition metal systems in general. DLPNO-CCSD(T) is therefore a promising method for applications in the burgeoning field of homogeneous catalysis based on Co(III) species.
1. Introduction
The spin-state energetics of transition-metal (TM) complexes are of great interest in bioinorganic, catalytic and materials chemistry, as they are strong indicators of spin-forbidden reactivity1,2 (also known as two-state reactivity3) and spin-crossover (SCO). The term ‘spin-accelerated reactivity’ has also been used when changes in spin-state enhance a reaction kinetically.4 Identifying these phenomena can lead to improved rationalizations and predictions of catalytic reactivity that can then inform new routes of chemical synthesis. Moreover, the identification of SCO behaviour in first-row TM systems has important implications for the development of new display, sensor and data-storage technologies.5,6While concepts such as the spectrochemical series of ligands7–9 and first-row TM ions Racah parameters10 can often serve to predict the ground-state of a particular complex, quantifying the relative spin-state energetics of first-row TM complexes is a challenge for quantum chemical methods. Indeed, in some biologically relevant systems, and other particularly challenging cases, even gaining a qualitative description of the correct ground spin-state can be problematic.11–14 Extensive effort has therefore been devoted to evaluate different quantum chemical approaches for spin-state energetics and numerous studies on Fe(II), Fe(III) and Co(II) species have been reported.15–25 Recent examples include the 2017 study by Pierloot et al. into the performance of CASPT2 and NEVPT2 to describe the spin-state energetics of TM ions, TM ions surrounded by point-charges, and a range of first-row TM complexes,16 and Radoń's 2019 assessment of CCSD(T), CASPT2, NEVPT2, MRCI and DFT for the calculation of experimental spin-state data for four octahedral Fe(II) and Fe(III) complexes.20 Additionally, density matrix renormalization group (DMRG) methods have found increasing use in the study of spin-state energetics of first-row TM complexes, particularly for systems where the TM centres are situated within extensively conjugated ligands (e.g. porphyrin rings), or for complexes featuring multiple TM centres.26
In contrast, Co(III) complexes have generally been overlooked in this regard. Co(III) complexes have a d6 valence electron configuration and are typically low-spin (S = 0) and exhibit a saturated octahedral coordination geometry. As a result, only a few cases of intermediate-spin (S = 1) or high-spin (S = 2) ground states, or observable spin-crossover, have been reported.27 Early examples include the [CoF6]3− anion28 and [CoF3(H2O)3] complex,29 which neatly demonstrate how weak field ligands generate a minimal ligand field splitting term Δo, and thus provide a high-spin quintet (S = 2) ground-state. The first examples of spin-crossover in Co(III) complexes were reported by Kläui, beginning in 1979 (Fig. 1a),30–34 and these were added to more recently by Theopold et al.35,36 and by Betley et al. (Fig. 1b).37 The latter study also characterized a Co(III) arylimido complex with a triplet ground state (Fig. 1c). The formal metal unsaturation present in these Co-imido systems is a key factor in making the higher spin states accessible.
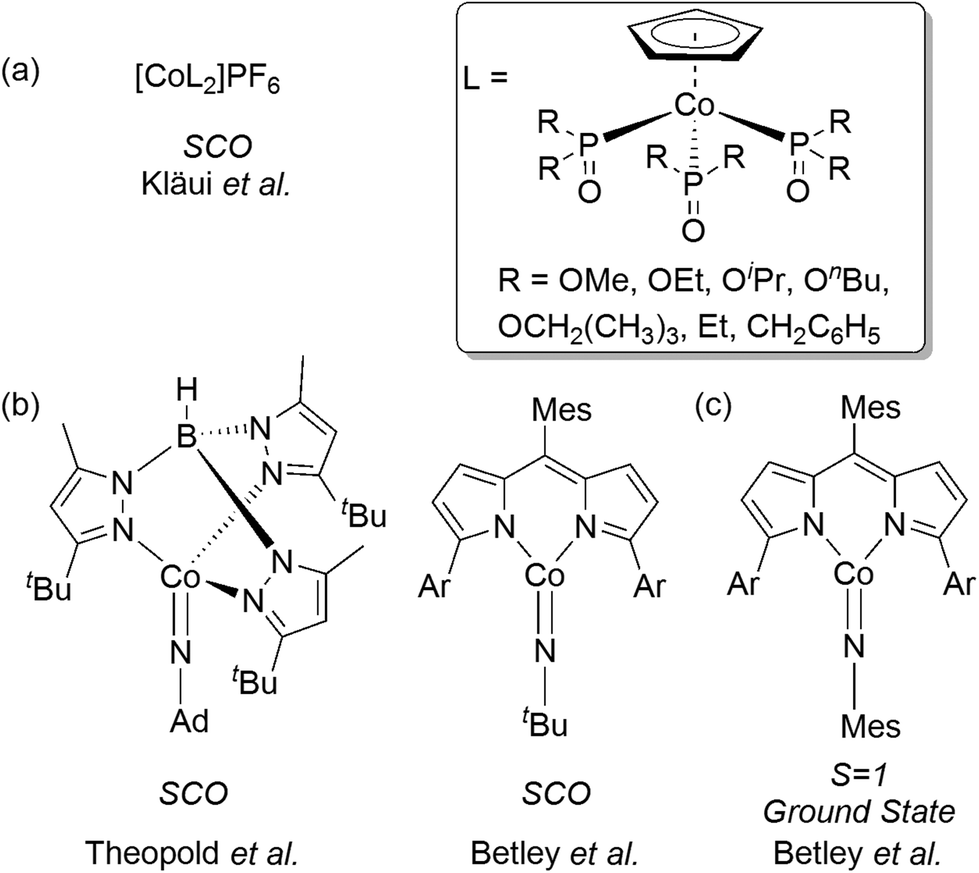 | ||
| Fig. 1 (a) Kläui's trinuclear spin crossover Co(III) complexes. (b) Co(III) imido complexes exhibiting SCO character. (c) A Co-arylimido complex with a triplet ground state (Ar = (2,4,6-triphenyl)phenyl; Mes = 2,4,6-trimethylphenyl; Ad = adamantyl). | ||
Recent years have seen significant growth in the use of organometallic Co(III) complexes in homogeneous catalysis, particularly in the area of carboxylate-assisted C–H functionalization.38–40 This has been accompanied by experimental mechanistic studies, often supported by computation to yield further insight.41–54 Density functional theory (DFT) is currently the most common method for the computation of reaction profiles, however, there is currently no clear consensus on which functional yields the most reliable results. As the choice of functional can significantly affect the computed energies of different spin-states in 1st row TM complexes,21,22 there is potential ambiguity in tackling the issue of multi-state reactivity.
An alternative strategy would be to move away from DFT and utilize linear-scaling local coupled cluster methods. The domain-based local pair natural orbital approximation to coupled cluster theory with single, double and perturbative triple excitations, DLPNO-CCSD(T),55–58 has emerged as a promising and significantly cheaper alternative to canonical CCSD(T), where much larger systems can be computed that were previously challenging and/or intractable. Since the release of the open-shell formalism of DLPNO-CCSD(T) in ORCA,58 a handful of studies have investigated its use in modelling first-row TM-mediated reactivity. Feldt, Harvey et al. probed the use of DLPNO-CCSD(T) and local unrestricted coupled-cluster theory (LUCCSD(T)) as implemented in Molpro59–62 in describing two-state reactivity of H-atom abstraction by non-heme iron complexes, where canonical CCSD(T) energies were used as references.63,64 Chen et al. employed a DLPNO-CCSD(T)/CBS//B3LYP/TZVP protocol to study Co(III)-catalysed carboxylate-assisted C–H activation reactions, with reference unrestricted Kohn–Sham B3LYP orbitals used in the DLPNO-CCSD(T) calculations.65 Within this study, calculations on the C–H activation of 1-phenylpyrazole at a [Co(OAc)Cp*]+ moiety (I → II, Fig. 2) suggested that reaction remains on the singlet surface, i.e. with no spin-crossover. However, the equivalent process at an amidoquinolinylcobalt(III) species identified a 5Co precursor (III, Fig. 2) that formed a 3Co product (IV, Fig. 2) via a 1Co transition state.
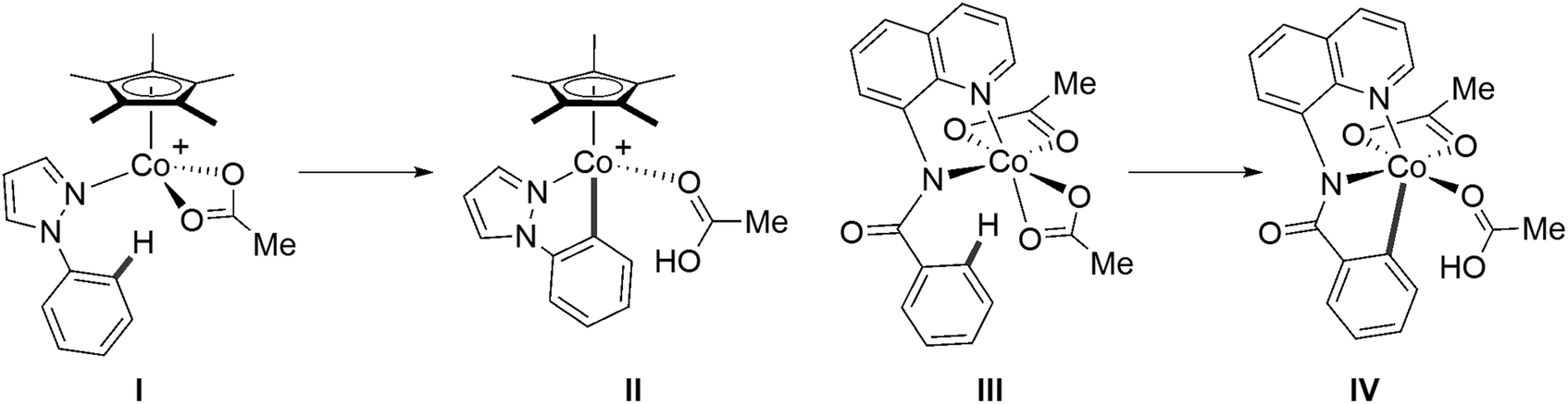 | ||
| Fig. 2 Species featured in Chen et al.'s DLPNO-CCSD(T) investigation of Co(III) C–H activation.65 | ||
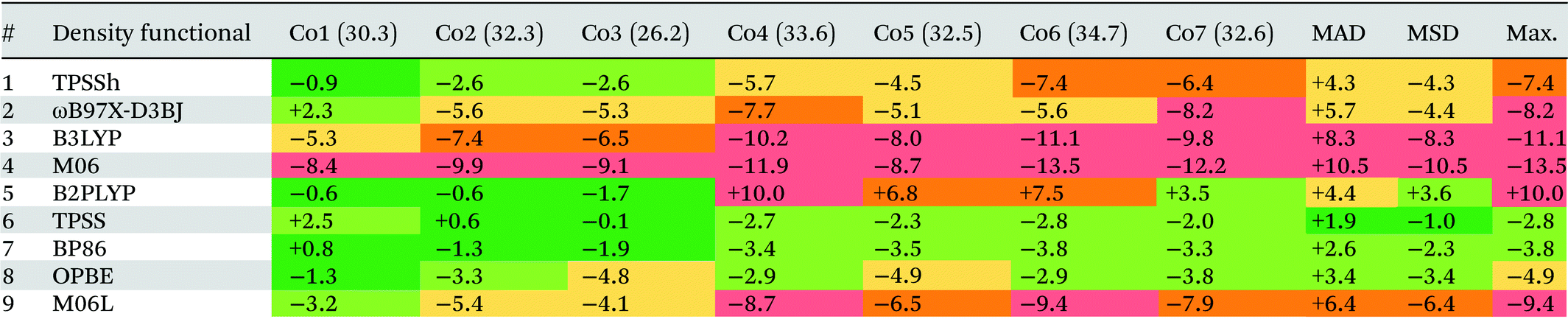
We have an ongoing interest in modelling 1st row TM organometallic catalysis using DFT and have noted the extreme sensitivity of computed energetics on the functional employed.66 In targeting new studies on Co(III) catalysis we wished to investigate open-shell DLPNO-CCSD(T) as an alternative approach. The present work therefore presents an initial assessment of Co(III) spin-state energetics using DLPNO-CCSD(T) against experimental 1Co → 3Co spin-splitting energy data derived by Hughes and Friesner.67 This study provided the seven octahedral Co(III) complexes shown in Fig. 3: [Co(NH3)6]3+ (herein referred to as Co1), [Co(en)3]3+ (Co2), [Co(NH3)5(Me2SO)]3+ (Co3), [Co(AMN3S3sarH)]4+ (Co4), [Co(AMN5SsarH)]4+ (Co5), [Co(N3S3)]3+ (Co6), and [Co(EtN4S2amp)]3+ (Co7).
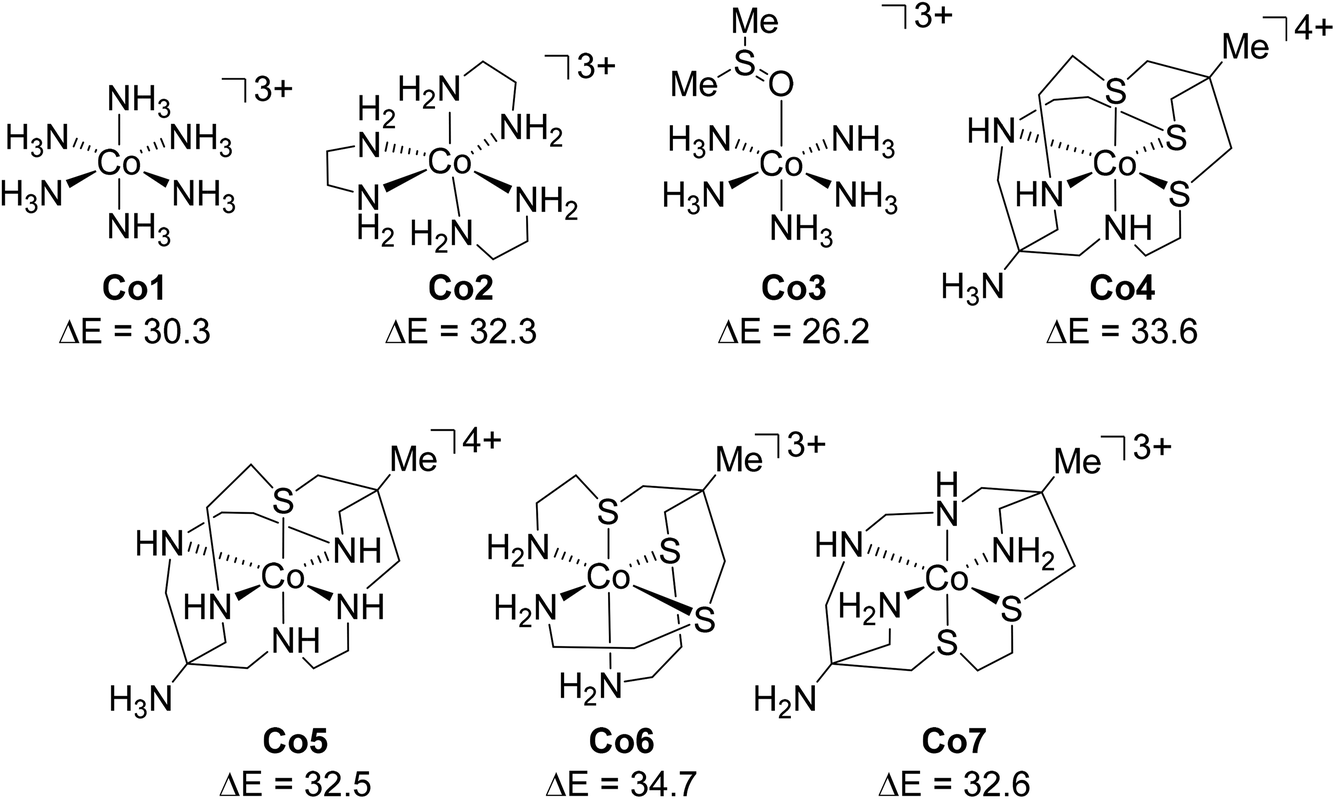 | ||
| Fig. 3 Structures and labels of the Co(III) complexes studied here, with the experimental 1Co → 3Co spin-splitting energies shown in kcal mol−1. | ||
In addition to DLPNO-CCSD(T), another promising alternative to DFT for modelling spin-state energetics is multi-reference perturbation theory (MRPT), and, in particular, strongly-contracted n-electron valence perturbation theory (NEVPT2).68–70 Apart from the previously mentioned studies on the performance of NEVPT2 for 1st row metals by Pierloot16 and Radoń20 no study at this particular level of theory on spin-state energetics of Co(III) complexes has so far been published. We have therefore extended our study to include NEVPT2 calculations on the Co(III) complexes in Fig. 3. Finally, the performance of a range of DFT methods will also be assessed.
2. Computational details
All calculations were carried out using ORCA 4.0 (version 4.0.1) and 4.1 (versions 4.1.0, 4.1.1 and 4.1.2).71,72 Further details on which version was used for each type of calculation can be found in the relevant sections below.2.1. Geometries and frequencies
Geometry optimization and frequency calculations were performed with ORCA 4.0.1 at the BP86/def2-TZVP level,73–75 where the resolution of identity (RI) approximation was employed76 with the “def2/J” general Weigend auxiliary basis sets.77 Structures were optimized separately in the singlet and triplet spin-states. Analytical frequency calculations were also performed to confirm minima and afford thermodynamic corrections, where the quasi-rigid rotor-harmonic oscillator approximation was employed for vibrational frequencies under 35 cm−1.782.2. DLPNO-CCSD(T) and canonical CCSD(T) calculations
DLPNO-CCSD(T) calculations were carried out both with Hartree–Fock (HF) orbitals and Kohn–Sham orbitals as references, where the latter used the BP86 level (BP), where in all cases non-iterative “(T0)” corrections were employed. Calculations tested both the “NormalPNO” and “TightPNO” thresholds and will be referred to as “N-PNO(HF/BP)” and “T-PNO(HF/BP)”, respectively. N-PNO(HF) calculations were performed using cc-pVTZ (cc-TZ) and cc-pVQZ (cc-QZ) basis sets,79–82 while for T-PNO(HF) calculations only the cc-TZ basis set was employed.83 N-PNO(BP) and T-PNO(BP) calculations were run with cc-DZ, cc-TZ, and def2-TZVPP (def2-TZ)75 basis sets, where the respective auxiliary basis sets cc-pVTZ/C,84,85 cc-pVQZ/C, and def2-TZVPP/C86 were used for the correlation calculations. In the absence of the cc-pVDZ/C auxiliary basis set for cobalt, the “AutoAux” automatic generation procedure was employed.87Extrapolation to the complete basis-set (CBS) limit was carried out at the T-PNO(BP) level, with cc-DZ and cc-TZ as the two basis-sets (i.e. CBS[2:3]). The following scheme for the SCF energy developed by Karton and Martin, was employed (eqn (1)):88
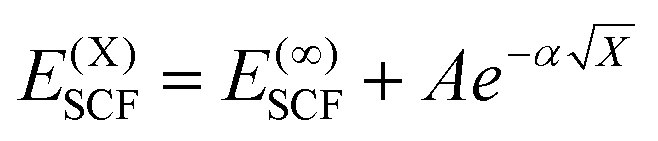 | (1) |
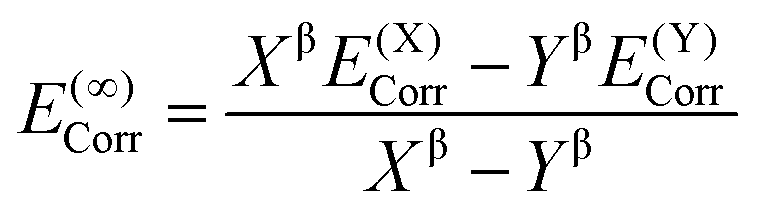 | (2) |
T-PNO(BP)/def2-SVP and CCSD(T)(BP)/def2-SVP calculations were also performed on the three smallest complexes, Co1, Co2 and Co3, to compare spin-state energetics obtained with DLPNO-CCSD(T), and those with canonical CCSD(T). In the canonical open-shell CCSD(T) calculations, quasi-restricted orbitals (QROs)91 at the BP86 level were used as references, while the standard converged BP86 wavefunction was used as a reference for the closed-shell calculations.
All DLPNO-CCSD(T) and canonical CCSD(T) calculations were performed with ORCA versions 4.1.0 and 4.1.2.
2.4. CASSCF and NEVPT2 calculations
In all CASSCF calculations, localized Kohn–Sham orbitals at the BP86/def2-TZVPP level were used as reference orbitals. Based on the general rules for active space selection outlined by Roos92 and Pierloot,93 both CASSCF and strongly contracted NEVPT2 calculations employed on a (10,7) active space, in which the 7 active orbitals are the five orbitals of 3d character, and the 2 ligand σ-bonding orbitals. Subsequent calculations were performed with a (10,12) active space to include the five double-shell 4d orbitals. Both sets of calculations employed the def2-QZVPP (def2-QZ) basis set. A detailed description of the methodology in setting up the orbitals prior to the CASSCF/NEVPT2 calculations is presented in the ESI.† All CASSCF/NEVPT2 calculations were performed with ORCA versions 4.1.0 and 4.1.2.2.3. DFT calculations
A selection of nine functionals were chosen such that all rungs of “Jacob's Ladder” were represented:94 B2PLYP,95 B3LYP,96,97 BP86,73,74 M06L,98 M06,98 OPBE,11,99,100 TPSS,101 TPSSh,101,102 and ωB97X-D3BJ.103,104 With the exception of the Minnesota functionals and ωB97X-D3BJ, an additional D3 dispersion correction with Becke–Johnson damping was included.105,106 The def2-QZ basis set was employed for each functional, and in the case of GGA and meta-GGA functionals, the RI-J approximation was employed with the general Weigend “def2/J” auxiliary basis sets, while with the double-hybrid functional B2PLYP the RI-MP2 approximation was employed. Calculations featuring an admixture of HF exchange additionally used the chain-of-spheres (COSX) approximation to exact exchange for computational speed up of the SCF step.107 These calculations were performed with ORCA version 4.1.2.2.5. Solvation
At all levels of theory the CPCM model was employed to account for environment effects.108–111 For the DLPNO-CCSD(T) and CCSD(T) calculations, the solvation correction was obtained by performing a single-point calculation at the BP86(CPCM)/def2-TZVP level of theory and subtracting the energy at the BP86/def2-TZVP level. For NEVPT2 the CPCM correction was included in the CASSCF wavefunction, while for DFT energies this was applied as a single-point correction on the BP86-optimized structure using the relevant functional. The overall free energies at each level of theory were thus generally calculated as follows: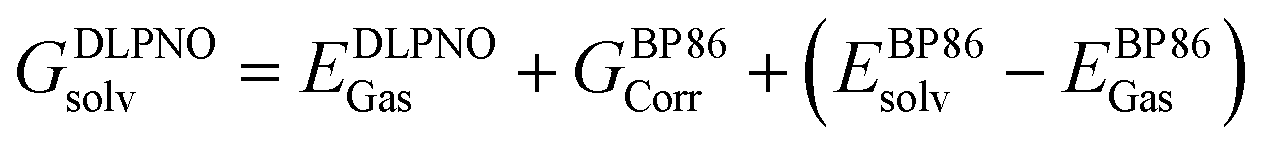 | (3) |
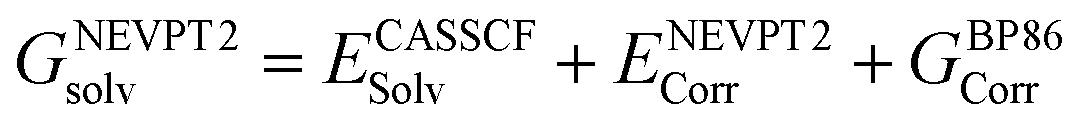 | (4) |
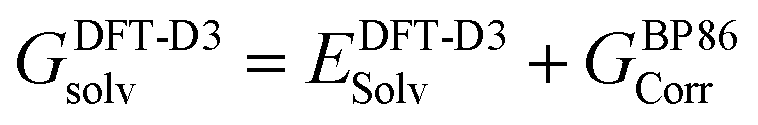 | (5) |
For complexes Co1, Co2 and Co3, the absorption spectra utilized in ref. 67 were measured in water, so the default solvation parameters for water were used (e.g. ε = 80.4). For complexes Co4, Co5, Co6 and Co7, spectra were obtained in Nafion® film and so a dielectric constant of 20 was employed based on work reported by Paddison et al. on Nafion® 117 at various states of hydration.112,113 All solvation corrections for use with DLPNO-CCSD(T) were performed with ORCA 4.0.1.
3. Results and discussion
3.1 DLPNO-CCSD(T) calculations
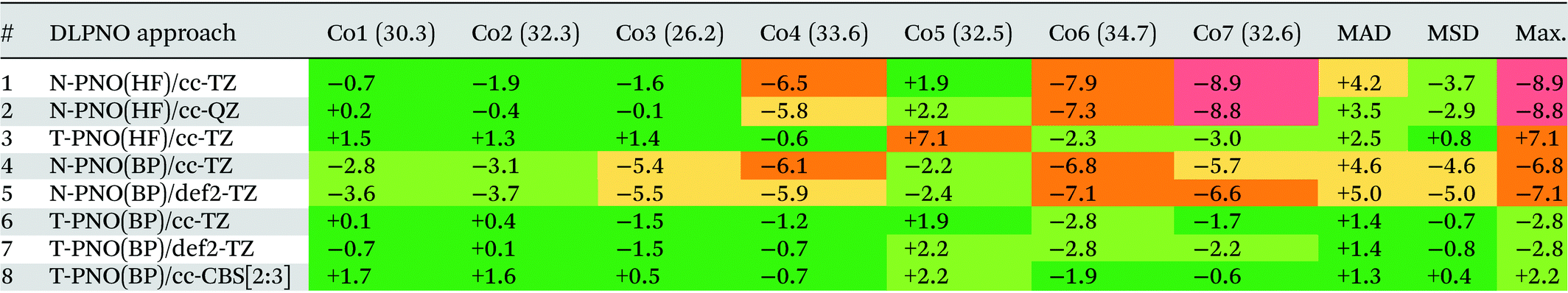
Deviations of the computed spin-splitting energies from the experimental values for the seven Co(III) complexes are shown in Table 1. This features results from the eight different DLPNO-CCSD(T) protocols employed, along with the mean absolute deviations (MAD), mean signed deviation (MSD) and the maximum deviation (Max.) in each case. These statistical deviations are also shown on Fig. 4.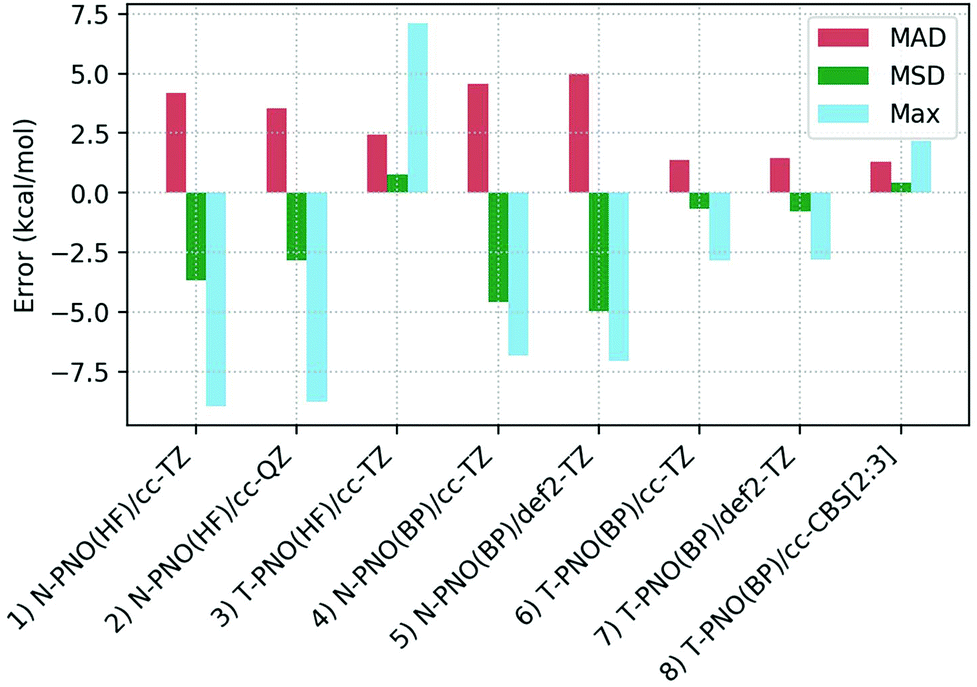 | ||
| Fig. 4 Mean absolute deviations (MAD, red), mean signed deviations (MSD, green) and Maximum deviations (Max, blue) for the 8 DLPNO-CCSD(T) protocols studied. | ||
|
While the N-PNO(HF)/cc-TZ approach (entry 1) yields reasonable 1Co → 3Co spin-splittings for four of the seven complexes, the performance for the larger complexes Co4, Co6 and Co7 is poor, with deviations of up to −8.9 kcal mol−1 (Co7), resulting in a MAD of 4.2 kcal mol−1. Moving to a larger cc-QZ basis set (entry 2) does not resolve the large deviations for Co4, Co6 and Co7, with only a small improvement to the MAD (3.5 kcal mol−1).
Switching to the T-PNO settings yields a better agreement with the experimental data, with an MAD at the T-PNO(HF)/cc-TZ level of 2.5 kcal mol−1 (entry 3). The tighter settings also provide significant improvement for complexes Co4, Co6 and Co7; however, an anomalously large deviation is now seen for Co5 (+7.1 kcal mol−1), which contributes significantly to the MAD of 2.5 kcal mol−1. In the absence of Co5, the MAD lowers to 1.7 kcal mol−1.
For the calculations employing BP86 reference orbitals (entries 4–8), those using N-PNO settings provide a similar performance, both to each other, (N-PNO(BP)/cc-TZ: MAD = 4.6 kcal mol−1, entry 4; N-PNO(BP)/def2-TZ: MAD = 5.0 kcal mol−1, entry 5) and to N-PNO(HF)/cc-TZ (MAD = +4.2 kcal mol−1, entry 1). With the BP86 reference orbitals, however, use of the T-PNO settings now provides significant improvement, with MADs of 1.4 kcal mol−1 for both T-PNO(BP)/cc-TZ (entry 6) and T-PNO(BP)/def2-TZ (entry 7). Much of this improvement again stems from a better description of the larger thioether-containing complexes, Co4–Co7. Furthermore, the problems associated with Co5 seen at the T-PNO(HF)/cc-TZ level are now resolved, the error reducing from +7.1 kcal mol−1 to only +1.9 kcal mol−1 and +2.2 kcal mol−1 for T-PNO(BP)/cc-TZ and T-PNO(BP)/def2-TZ respectively. This improvement in the performance of coupled-cluster energies when using Kohn–Sham reference orbitals was also seen by Radoń in his 2019 study against spin-states of octahedral Fe(II)/Fe(III) complexes.20 Harvey and Tew also identified that using Kohn–Sham orbitals in CCSD(T) gave more promising energetics (comparable to those obtained with Brueckner orbitals) for the spin-forbidden reactions of FeO+ with H2.114 In contrast, the choice of basis sets used in this study has less impact, irrespective of the DLPNO threshold or reference orbitals selected.
Based on the data in Table 1 the T-PNO(BP)/cc-TZ level was selected as the leading performer for further investigation. For all complexes the computed T1 diagnostic for multi-reference character was below the 0.05 criterion suggested for TM complexes,115 and the largest DLPNO-CCSD amplitudes were all found to be small (<0.11). Moreover, the largest doubles amplitude (i.e. |t2,max|) for each species was under 0.06, further demonstrating the lack of multireference character (see Table S2b†). A single-determinant approach therefore appears to be suitable for these pseudo-octahedral Co(III) complexes. A complete basis-set extrapolation using the cc-DZ and cc-TZ basis sets was also carried out, and the MAD obtained, 1.3 kcal mol−1 (entry 8), was similar to that from obtained at the T-PNO(BP)/cc-TZ level.
The DLPNO-CCSD(T) approach was also compared with canonical CCSD(T) theory. Due to the higher cost, the CCSD(T) calculations could only be run on the 3 smallest complexes, Co1, Co2 and Co3, using a smaller double-ζ def2-SVP basis-set. The DLPNO-CCSD(T) results were therefore repeated with this smaller basis set to allow direct comparison (see Fig. 5). Although both DLPNO-CCSD(T)/def2-SVP and CCSD(T)/def2-SVP over-stabilize the triplet state (by between 4–8 kcal mol−1), it is the comparison of these two methods (and the individual energy contributions) that is the focus here, rather than the absolute agreement with experiment.
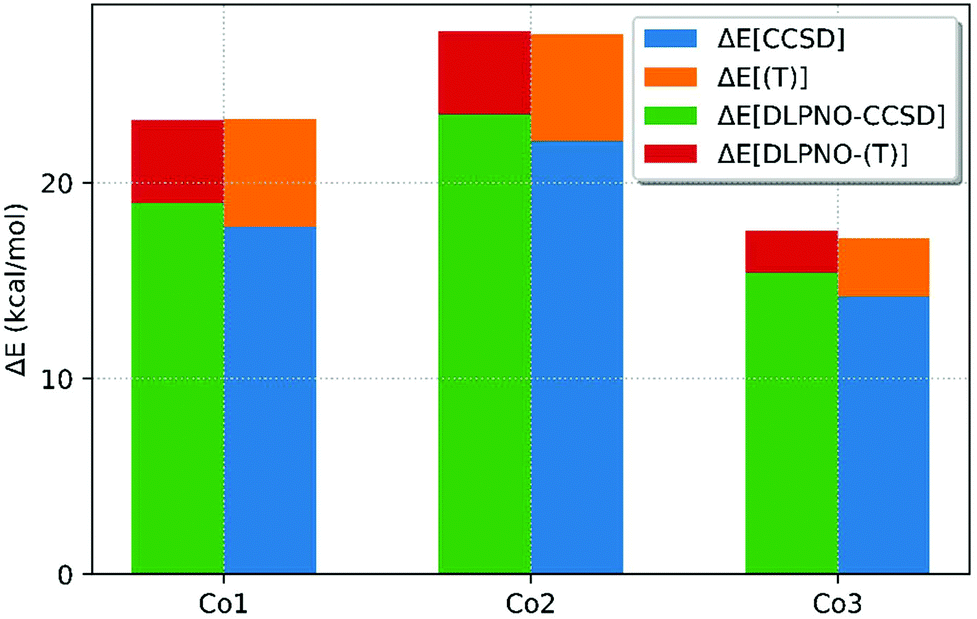 | ||
| Fig. 5 Energy differences for the 1Co → 3Co spin splitting energies of complexes Co1, Co2 and Co3 at the DLPNO-CCSD(T)/def2-SVP and CCSD(T)/def2-SVP levels with the contributions from singles and doubles (ΔE[CCSD], ΔE[DLPNO-CCSD] and triples (ΔE[(T)] and ΔE[DLPNO-(T)] highlighted. Energies are in kcal mol−1. | ||
Fig. 5 shows that both CCSD(T)/def2-SVP and DLPNO-CCSD(T)/def2-SVP give very similar spin-state splittings, with differences of −0.04, +0.35 and +0.16 kcal mol−1 for Co1, Co2 and Co3, respectively. Consideration of the different energy contributions within these overall outcomes revealed slightly larger differences. For example, with Co1 the spin-splitting energy at the CCSD level is +1.2 kcal mol−1 when computed within the DLPNO algorithm, but this is counterbalanced by the perturbative triples correction which is now 1.3 kcal mol−1 lower. The cumulative effect yields the remarkably small deviation of only −0.04 kcal mol−1.
3.2. NEVPT2 calculations
Table 2 shows the spin-splitting deviations computed with various NEVPT2 protocols, with the associated statistical deviations shown in Fig. 6.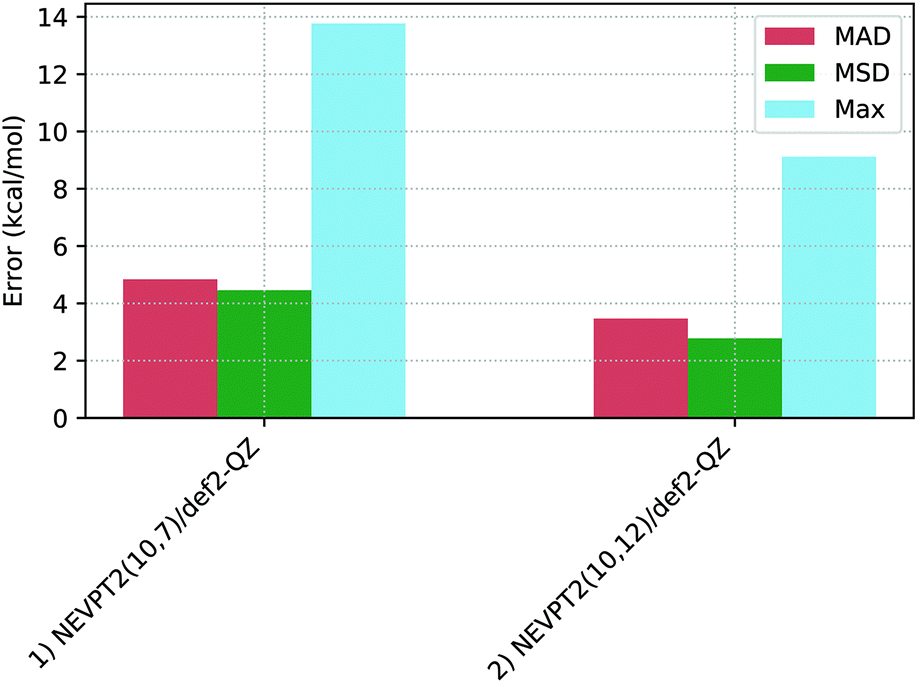 | ||
| Fig. 6 Mean absolute deviations (MAD, red), mean signed deviations (MSD, green) and Maximum deviations (Max, blue) for the two NEVPT2 protocols studied. | ||

|
For the NEVPT2(10,7)/def2-QZ calculations (entry 1) the spin-splitting energies of Co1–Co3 and Co7 are all well described with this active space, with deviations between −0.1 kcal mol−1 and −2.6 kcal mol−1. In contrast, Co4, Co5, and Co6, show large positive deviations (+7.8, +13.8 and +10.4 kcal mol−1, respectively), leading to a moderate MAD of 4.8 kcal mol−1.
Increasing the active space in the NEVPT2(10,12)/def2-QZ approach (in which the 3d′ orbitals are included in the active space, entry 2) gives only a small overall improvement in performance (MAD = 3.5 kcal mol−1) that does, however, mask some non-systematic behaviour. Thus while only small changes below 1 kcal mol−1 are seen for Co1, Co2 and Co7, a very significant increase in the spin-splitting energy is computed for Co3 (−2.6 to +4.9 kcal mol−1).116 In contrast, significant reductions between 3.8 and 7.4 kcal mol−1 are seen for the remaining complexes.117
Overall, NEVPT2 shows varied outcomes for the Co(III) complexes studied, with generally good performance for Co1, Co2 and Co7, but much larger deviations for Co3, Co4, Co5 and Co6. Co5 consistently gave the most problems and further efforts to improve this situation, for example, further adapting the active space, proved unsuccessful. Therefore, a (10,12) active space was identified as optimal for these mononuclear pseudo-octahedral complexes. These outcomes for NEVPT2 find parallels in the literature. For instance, Pierloot et al. identified varied behaviour of NEVPT2 in over- and under-stabilizing the higher spin-states, relative to reference CCSD(T) energetics,16 while in Radoń's 2019 study NEVPT2 was also identified to yield variable spin-state energies with no systematic trend.20 In our case we also find NEVPT2 provides no systematic stabilization of one spin-state over the other, and this contrasts with the DFT approaches described below.
3.3 DFT functionals
Table 3 shows the deviations computed for the spin-splitting energies of the seven Co(III) complexes using nine density functionals, along with the associated MAD, MSD and Max. values. These outcomes are also summarized in Fig. 7. Of the hybrid functionals (entries 1 to 4) none provides good quantitative agreement with experiment due to an over-stabilization of the triplet spin-state that leads to underestimated 1Co → 3Co spin splitting energies. The poorest performer was M06 (entry 4, MAD = +10.4 kcal mol−1), with which all spin-splitting energies are significantly underestimated. Similar results have been seen in work on Fe(II) complexes by Cirera in 200924 and Pierloot in 2010,14 and in general, a correlation between the degree of Hartree–Fock exchange within hybrid DFT and over-stabilization of higher-spin-states has been established.118–120 Thus M06 (with a 27% admixture of HF exchange, the largest of the hybrid functionals studied here) is the worst offender in this regard and is closely followed by B3LYP (entry 4, MAD = +8.3 kcal mol−1). Pinter et al., have previously shown that reducing the %HF exchange to between 0–5% yields better performance than the 20% admixture in the original B3LYP functional used here.120 Of the four hybrid functionals TPSSh has the most balanced performance (entry 1, MAD = 4.3 kcal mol−1).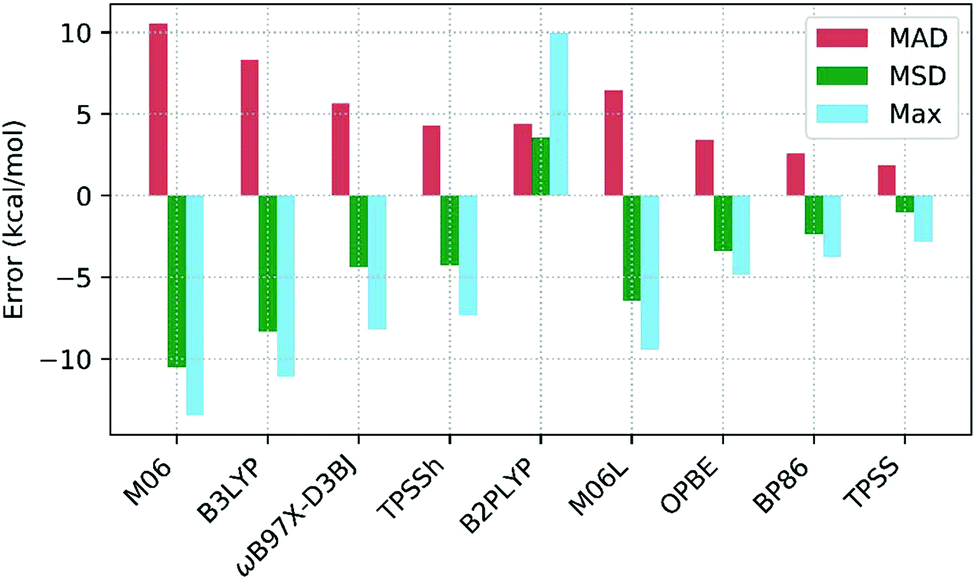 | ||
| Fig. 7 Mean absolute deviations (MAD, red), mean signed deviations (MSD, green) and Maximum deviations (Max, blue) for the 9 DFT functionals studied. | ||
|
The one double-hybrid functional in this study, B2PLYP (entry 5, MAD = +4.4 kcal mol−1), is something of an outlier as it tends to over-estimate the spin splitting energy and hence has a positive MSD of +3.6 kcal mol−1. B2PLYP has a large admixture of 53% HF exchange, but also an additional admixture of 27% MP2 correlation. The latter presumably more than offsets what would otherwise be a large over-stabilization of the triplet spin-state. While B2PLYP provides good performance for Co1, Co2 and Co3, the larger thioether complexes Co4–Co7 are more problematic. In 2018 Adamo evaluated the PBE0-DH double-hybrid functional and noted that the MP2 contribution gave a more pronounced low-spin stabilization effect on complexes featuring higher field-strength ligands.25 With the exception of one κ-O-sulfoxide ligand in Co3, the ligands in Co1–Co3 are amines while Co4–Co7 all feature at least one thioether ligand. Kraatz et al.121 have placed the field strength of thioethers between that of amines and phosphines, and so the effect noted in Adamo's study can also account for the larger errors obtained with B2PLYP for Co4–Co7. In particular, the largest errors are seen for Co4 (ΔE = +10.0 kcal mol−1) and Co6 (ΔE = +7.5 kcal mol−1), both of which have three thioether ligands.
In general, the best performance is obtained with the pure functionals (entries 6–9), although these still tend to underestimate the spin-splitting energy. The leading performer is TPSS (entry 6) with an MAD of 1.9 kcal mol−1. Radoń's 2019 study also identified good performances with pure GGA and mGGA functionals,20 where OPBE performs the second best (behind B2PLYP). In our case, OPBE (entry 8) is 3rd in terms of MAD, behind TPSS (entry 6) and BP86 (entry 7) with B2PLYP 4th (entry 5).122 However, comparing our results with Radoń's shows little consistency in the rank order of functionals: for example, TPSS performs very poorly in Radoń's study, yet, it is the strongest for Co(III) in this study.
4. Conclusions
This work has explored the performance of DLPNO-CCSD(T), NEVPT2 and DFT for the computation of the spin-state energetics of a range of pseudo-octahedral Co(III) complexes. Within the DLPNO-CCSD(T) framework, the use of “TightPNO” settings in combination with Kohn–Sham (BP86) reference orbitals achieved the most balanced and accurate performance, giving a MAD of 1.3 kcal mol−1 when a triple-ζ basis set is employed with a complete basis-set extrapolation scheme. In contrast, use of “NormalPNO” settings or HF reference orbitals gave poorer performance, especially for the larger Co complexes featuring thioether ligands. The precise DLPNO protocol therefore has an important effect on the computed spin-state energetics. In contrast, varying the basis sets used in this study had little impact on the computed outcome.With NEVPT2 the results obtained with the expanded (10,12) active space proved most promising, with a MAD of 2.5 kcal mol−1. However, the performance of NEVPT2 was in general less well balanced, with significant and counter-balancing variations in absolute values across the seven Co(III) complexes that were then masked in the MSD and MAD values.
Nine different density functionals were also tested, covering all rungs of Jacob's ladder. In general, pure functionals performed better than the hybrid and double hybrid functionals, with the mGGA functional TPSS giving the lowest MAD of 1.9 kcal mol−1. Hybrid functionals tended to over-stabilise the triplet state, with this effect increasing with the greater degree of HF exchange embedded in the functional. Comparison with other DFT studies suggests that identifying a functional that can provide a good and balanced performance across several 1st row metal centres remains problematic.
Within the context of the burgeoning field of organometallic catalysis based on Co(III) species, the DLPNO-CCSD(T) approach would appear to be a promising method, assuming an appropriate protocol is adopted (i.e. “TightPNO” settings and Kohn–Sham reference orbitals). Further work on the spin-state energetics and two-state reactivity in Co(III) complexes applied to C–H activation is currently underway and will be reported in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Heriot-Watt University for a James Watt Scholarship to SEN and ScotCHEM for a Postgraduate and Early Career Researcher Exchange (PECRE) award. This work used the Cirrus UK National Tier-2 HPC Service at EPCC (http://www.cirrus.ac.uk) funded by the University of Edinburgh and EPSRC (EP/P020267/1). DAP acknowledges support by the Max Planck Society. Open Access funding provided by the Max Planck Society.References
- R. Poli and J. N. Harvey, Chem. Soc. Rev., 2003, 32, 1–8 RSC
.
- J. N. Harvey, R. Poli and K. M. Smith, Coord. Chem. Rev., 2003, 238–239, 347–361 CrossRef CAS
- D. Schröder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef PubMed
- R. Poli, Acc. Chem. Res., 1997, 30, 494–501 CrossRef CAS
-
J.-F. Létard, P. Guionneau and L. Goux-Capes, in Spin Crossover in Transition Metal Compounds III, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 221–249 Search PubMed
- G. Molnár, L. Salmon, W. Nicolazzi, F. Terki and A. Bousseksou, J. Mater. Chem. C, 2014, 2, 1360–1366 RSC
- R. Tsuchida, Bull. Chem. Soc. Jpn., 1938, 13, 388–400 CrossRef CAS
- R. Tsuchida, Bull. Chem. Soc. Jpn., 1938, 13, 436–450 CrossRef CAS
- Y. Shimura and R. Tsuchida, Bull. Chem. Soc. Jpn., 1956, 29, 311–316 CrossRef CAS
- H. Elias, Ber. Bunsenges. Phys. Chem, 1995, 99, 901–901 CrossRef
- M. Swart, A. R. Groenhof, A. W. Ehlers and K. Lammertsma, J. Phys. Chem. A, 2004, 108, 5479–5483 CrossRef CAS
- L. Rousseau, E. Brémond and G. Lefèvre, New J. Chem., 2018, 42, 7612–7616 RSC
- R. K. Carlson, S. O. Odoh, S. J. Tereniak, C. C. Lu and L. Gagliardi, J. Chem. Theory Comput., 2015, 11, 4093–4101 CrossRef CAS PubMed
- S. Vancoillie, H. Zhao, M. Radoń and K. Pierloot, J. Chem. Theory Comput., 2010, 6, 576–582 CrossRef CAS PubMed
- L. M. Lawson Daku, F. Aquilante, T. W. Robinson and A. Hauser, J. Chem. Theory Comput., 2012, 8, 4216 CrossRef CAS PubMed
- K. Pierloot, Q. M. Phung and A. Domingo, J. Chem. Theory Comput., 2017, 13, 537–553 CrossRef CAS PubMed
- Q. M. Phung, M. Feldt, J. N. Harvey and K. Pierloot, J. Chem. Theory Comput., 2018, 14, 2446–2455 CrossRef CAS PubMed
- M. Radoń, J. Chem. Theory Comput., 2014, 10, 2306 CrossRef PubMed
- M. Radoń and G. Drabik, J. Chem. Theory Comput., 2018, 14, 4010–4027 CrossRef PubMed
- M. Radoń, Phys. Chem. Chem. Phys., 2019, 21, 4854–4870 RSC
- S. Ye and F. Neese, Inorg. Chem., 2010, 49, 772–774 CrossRef CAS PubMed
- J. Cirera, M. Via-Nadal and E. Ruiz, Inorg. Chem., 2018, 57, 14097–14105 CrossRef CAS PubMed
- M. Swart and M. Gruden, Acc. Chem. Res., 2016, 49, 2690 CrossRef CAS PubMed
- K. P. Jensen and J. Cirera, J. Phys. Chem. A, 2009, 113, 10033–10039 CrossRef CAS PubMed
- L. Wilbraham, C. Adamo and I. Ciofini, J. Chem. Phys., 2018, 148, 041103 CrossRef PubMed
- M. Roemelt and D. A. Pantazis, Adv. Theory Simul., 2019, 2, 1800201 CrossRef
-
Y. Garcia and P. Gütlich, in Spin Crossover in Transition Metal Compounds II, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 49–62 Search PubMed
- R. Hoppe, Recl. Trav. Chim. Pays-Bas, 1956, 75, 569–575 CrossRef CAS
- W. J. Hickinbottom, N. W. Rogers, H.
C. Clark, B. Cox, A. G. Sharpe, M. Z. Barakat, S. K. Shehab, M. M. El-Sadr, R. Pauncz, E. E. Aynsley, W. A. Campbell, J. Baddiley, J. G. Buchanan, B. Carss, D. Woodcock, D. R. Clifford, S. A. Faseeh, J. Harley-Mason, J. R. Merchant, R. M. Naik, A. J. Mountwalla, R. L. Williams, R. J. Pace, D. G. O'Sullivan, P. W. Sadler, W. Cocker, B. E. Cross, W. R. N. Williamson, R. J. McIlroy, D. A. Everest, C. W. Bird, J. K. Norymberski, G. F. Woods, D. F. Heath, B. J. Aylett, J. I. G. Cadogan and E. A. Johnson, J. Chem. Soc., 1957, 4131–4156 RSC
- W. Kläui, J. Chem. Soc., Chem. Commun., 1979, 700–700 RSC
- P. Guetlich, B. R. McGarvey and W. Kläui, Inorg. Chem., 1980, 19, 3704–3706 CrossRef CAS
- W. Eberspach, N. El Murr and W. Kläui, Angew. Chem., Int. Ed. Engl., 1982, 21, 1943–1949 CrossRef
- G. Navon and W. Kläui, Inorg. Chem., 1984, 23, 2722–2725 CrossRef CAS
- W. Kläui, W. Eberspach and P. Guetlich, Inorg. Chem., 1987, 26, 3977–3982 CrossRef
- D. T. Shay, G. P. A. Yap, L. N. Zakharov, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2005, 44, 1508–1510 CrossRef CAS PubMed
- D. T. Shay, G. P. A. Yap, L. N. Zakharov, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2006, 45, 7870–7870 CrossRef
- E. R. King, G. T. Sazama and T. A. Betley, J. Am. Chem. Soc., 2012, 134, 17858–17861 CrossRef CAS PubMed
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed
- P. G. Chirila and C. J. Whiteoak, Dalton Trans., 2017, 46, 9721–9739 RSC
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS
- O. Planas, C. J. Whiteoak, V. Martin-Diaconescu, I. Gamba, J. M. Luis, T. Parella, A. Company and X. Ribas, J. Am. Chem. Soc., 2016, 138, 14388–14397 CrossRef CAS PubMed
- J. Sanjosé-Orduna, D. Gallego, A. Garcia-Roca, E. Martin, J. Benet-Buchholz and M. H. Pérez-Temprano, Angew. Chem., Int. Ed., 2017, 56, 12137–12141 CrossRef PubMed
- J. Sanjosé-Orduna, J. M. S. Toro and M. H. Pérez-Temprano, Angew. Chem., Int. Ed., 2018, 57, 11369–11373 CrossRef PubMed
- J. Sanjosé-Orduna, J. Benet-Buchholz and M. H. Pérez-Temprano, Inorg. Chem., 2019, 58, 10569–10577 CrossRef PubMed
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed
- H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424–5431 CrossRef CAS PubMed
- X.-K. Guo, L.-B. Zhang, D. Wei and J.-L. Niu, Chem. Sci., 2015, 6, 7059–7071 RSC
- Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944–9947 CrossRef CAS PubMed
- R. Mei, H. Wang, S. Warratz, S. A. Macgregor and L. Ackermann, Chem. – Eur. J., 2016, 22, 6759–6763 CrossRef CAS PubMed
- M. Sen, B. Emayavaramban, N. Barsu, J. R. Premkumar and B. Sundararaju, ACS Catal., 2016, 6, 2792–2796 CrossRef CAS
- S. Qu and C. J. Cramer, J. Org. Chem., 2017, 82, 1195–1204 CrossRef CAS PubMed
- D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 10378–10382 CrossRef CAS PubMed
- X. Zhou, Y. Luo, L. Kong, Y. Xu, G. Zheng, Y. Lan and X. Li, ACS Catal., 2017, 7, 7296–7304 CrossRef CAS
- Q. Wang, F. Huang, L. Jiang, C. Zhang, C. Sun, J. Liu and D. Chen, Inorg. Chem., 2018, 57, 2804–2814 CrossRef CAS PubMed
- F. Neese, A. Hansen and D. G. Liakos, J. Chem. Phys., 2009, 131, 064103 CrossRef PubMed
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed
- M. Saitow, U. Becker, C. Riplinger, E. F. Valeev and F. Neese, J. Chem. Phys., 2017, 146, 164105 CrossRef PubMed
- M. Schütz, J. Chem. Phys., 2000, 113, 9986–10001 CrossRef
- M. Schütz and H.-J. Werner, J. Chem. Phys., 2000, 114, 661–681 CrossRef
- H.-J. Werner and M. Schütz, J. Chem. Phys., 2011, 135, 144116 CrossRef PubMed
-
Y. Liu, Ph Thesis, Inst. Für Theor. Chem. Univ. Stuttg, 2011
- M. Feldt, Q. M. Phung, K. Pierloot, R. A. Mata and J. N. Harvey, J. Chem. Theory Comput., 2019, 15, 922–937 CrossRef PubMed
- Q. M. Phung, C. Martín-Fernández, J. N. Harvey and M. Feldt, J. Chem. Theory Comput., 2019, 15, 4297–4304 CrossRef CAS PubMed
- P. Ma and H. Chen, ACS Catal., 2019, 9, 1962–1972 CrossRef CAS
- M. Espinal-Viguri, S. E. Neale, N. T. Coles, S. A. Macgregor and R. L. Webster, J. Am. Chem. Soc., 2019, 141, 572–582 CrossRef CAS PubMed
- T. F. Hughes and R. A. Friesner, J. Chem. Theory Comput., 2011, 7, 19–32 CrossRef CAS PubMed
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J. P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- F. Neese, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2006, 125, 074110 CrossRef PubMed
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS
- For species 3Co5 at the N-PNO(HF)/cc-QZ level, convergence issues emerged in the initial stages of the calculation relating to the calculation of the initial guess energy and construction of the open-shell PNO space. To resolve this, the TCutDO parameter was modified from 1.00 × 10−3 (default) to 9.00 × 10−2. Such a small change to one of the specific threshold settings was not expected to have a significant impact on the calculated spin-splitting energy.
- J. G. Hill and J. A. Platts, J. Chem. Phys., 2008, 128, 044104 CrossRef PubMed
- F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2002, 116, 3175–3183 CrossRef CAS
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed
- G. L. Stoychev, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2017, 13, 554–562 CrossRef CAS PubMed
- A. Karton and J. M. L. Martin, Theor. Chem. Acc., 2006, 115, 330–333 Search PubMed
- D. G. Truhlar, Chem. Phys. Lett., 1998, 294, 45–48 CrossRef CAS
- F. Neese and E. F. Valeev, J. Chem. Theory Comput., 2011, 7, 33–43 CrossRef CAS PubMed
- F. Neese, J. Am. Chem. Soc., 2006, 128, 10213–10222 CrossRef CAS PubMed
- V. Veryazov, P. Å. Malmqvist and B. O. Roos, Int. J. Quantum Chem., 2011, 111, 3329–3338 CrossRef CAS
- K. Pierloot, Mol. Phys., 2003, 101, 2083 CrossRef CAS
- J. P. Perdew and K. Schmidt, AIP Conf. Proc., 2001, 577, 1–20 CrossRef CAS
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- N. C. Handy and A. J. Cohen, Mol. Phys., 2001, 99, 403–412 CrossRef CAS
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- A. Najibi and L. Goerigk, J. Chem. Theory Comput., 2018, 14, 5725–5738 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC
- J. Andzelm, C. Kölmel and A. Klamt, J. Chem. Phys., 1995, 103, 9312–9320 CrossRef CAS
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed
- S. J. Paddison, D. W. Reagor and T. A. Zawodzinski Jr., J. Electroanal. Chem., 1998, 459, 91–97 CrossRef CAS
- We also explored the effect of the dielectric constant in the performance of the DLPNO-CCSD(T) splitting energies, where further single-point DFT calculations were carried out for Co4, Co5, Co6 and Co7 with three different dielectric constants at the CPCM approximation (ε = 2, ε = 20 and ε = 80.4). While a significant improvement to the spin splitting energies was obtained when going from ε = 2 to ε = 20, a much smaller difference was seen when going to H2O. Herein the energies reported were obtained with the CPCM continuum solvation model invoked where ε = 20 for Co4, Co5, Co6 and Co7.
- J. N. Harvey and D. P. Tew, Int. J. Mass Spectrom., 2013, 354–355, 263–270 CrossRef CAS
- W. Jiang, N. J. DeYonker and A. K. Wilson, J. Chem. Theory Comput., 2012, 8, 460–468 CrossRef CAS PubMed
- For this complex the energy and g value tolerance settings were lowered, along with the alteration of the convergence algorithms from the default, to achieve convergence of the CASSCF wavefunction for further calculation of the dynamic correlation at the NEVPT2 level. It was therefore anticipated that a significant part of this deterioration in accuracy in going to the (10,12) active space here could be as a result in this loosening of the convergence criteria for this complex.
- In some cases, notably Co3 at the (10,12) active space for both spin-states, and species 3Co5 at the (10,7) active space, the CASSCF calculations including the CPCM correction failed to converge fully to the default criteria. Full details of alternative approaches at the NEVPT2 level to overcome these issues can be found in the ESI.†.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed
- E. I. Ioannidis and H. J. Kulik, J. Chem. Phys., 2015, 143, 034104 CrossRef PubMed
- B. Pinter, A. Chankisjijev, P. Geerlings, J. N. Harvey and F. De Proft, Chem. – Eur. J., 2018, 24, 5281–5292 CrossRef CAS PubMed
- H. B. Kraatz, H. Jacobsen, T. Ziegler and P. M. Boorman, Organometallics, 1993, 12, 76–80 CrossRef CAS
- Radoń's study used OPBE without the D3BJ correction; recomputing with the OPBE functional gave a MAD of 4.2 kcal mol−1, slightly higher than the OPBE result in Table 3 that includes a D3BJ correction.
Footnotes |
| † Electronic supplementary information (ESI) available: An overview of the NEVPT2 procedure; additional test calculations on Co5; detailed results at the NEVPT2 level; T1 diagnostics; DLPNO-CCSD amplitudes; computed structures and energies for all species (PDF). See DOI: 10.1039/d0dt00993h |
| ‡ Present address: Center for Molecular Modeling, Ghent University, Technologiepark 46, 9052 Zwijnaarde, Belgium. |
| This journal is © The Royal Society of Chemistry 2020 |