
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bulky 1,1′-bisphosphanoferrocenes and their coordination behaviour towards Cu(I)†‡
Subhayan
Dey
a,
Daniel
Buzsáki
 b,
Clemens
Bruhn
a,
Zsolt
Kelemen
*b and
Rudolf
Pietschnig
*a
b,
Clemens
Bruhn
a,
Zsolt
Kelemen
*b and
Rudolf
Pietschnig
*a
aInstitut für Chemie und CINSaT, University of Kassel, Heinrich Plett-Straße 40, 34132 Kassel, Germany. E-mail: pietschnig@uni-kassel.de
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, and MTA-BME Computation Driven Chemistry Research Group, Szent Gellért tér 4, 1111 Budapest, Hungary
First published on 8th April 2020
Abstract
Two bulky mesityl substituted dppf-analogues Fe(C5H4PMes2)2 (Mes = 2,4,6-Me3C6H2, 1) and Fe(C5H4PMes2)(C5H4PPh2) (Mes = 2,4,6-Me3C6H2, Ph = C6H5, 3) have been prepared and their properties as donor ligands have been explored using heteronuclear NMR spectroscopy and in particular via1JP–Se coupling, cyclic voltammetry and DFT calculations. Based on the results obtained, a series of mono- and dinuclear Cu(I) complexes have been prepared with these new diphosphane ligands using Br−, I−, and BF4− as counter anions. For the very bulky ligand 1 rare and unprecedented double bridging complexation modes have been observed containing two non-planar Cu2Br2 units, while for the other dinuclear complexes planar Cu2Br2 units have been found. The Cu(I) complexes of 1 and 3 were then used as catalysts for CO2-fixation reaction with terminal alkynes, and complexes with ligand 3 were found to be more efficient than those with 1. DFT calculations performed on compounds 1, 3 and their Cu(I) complexes were able to verify the trend of these catalytic reactions.
Introduction
Being an extremely useful and unique building block, ferrocene has remained a center of attraction for several decades. Apart from the fact that it is important for synthesizing organometallic polymers,1–5 redox-tunable substances,4,6–8 drugs,9,10 and other functional materials,11–14 ferrocene has played a vital role in homogeneous catalysis.15,16 Diphosphane ligand systems have emerged from their monophosphane counterparts, and ferrocene has provided them with an unprecedented backbone,17–19 which helped to stabilize a variety of metal centers by forming flexible geometries.15,20 This special arrangement has further been appreciated, when it emerged that the bite angles (βn, A, Fig. 1) of such ligands, which have an apparent positive effect on the efficiency of catalysts,21,22 can be manipulated by changing the substituents on phosphorus.23 Systematic investigations further revealed that the alteration of substituents on phosphorus can be achieved by simple and modular synthetic approaches.23,24 The aforementioned qualities made ferrocene-based diphosphane ligands remarkably successful for homogeneous catalysis.15,16 The quest for new ligands, with ideal steric demand and optimum donating ability, is still ongoing.25–30 | ||
| Fig. 1 Bite angle (βn, A),21,22 previously (B–E),23 and herein reported (F) achiral 1,1′-bisphosphanoferrocene ligands. Known symmetrically substituted dppf analogues (B): R = Me,17 Et,34iPr,31,45 Cy,19tBu,32o-tolyl,33,46o-MeO–C6H4,34p-MeO–C6H4,47p-PhO–C6H4,46p-CF3–C6H4,48 3,5-CF3–C6H3,33 2-furyl,33 5-Me-2-furyl,37o-iPr-C6H4,34 1-Nap,46 2-Nap,46 C6F5.34 Known 1,1′-unsymmetrically substituted dppf analogues (C): R = Ph, R′ = tBu;35,36 R = Ph, R′ = iPr;25 R = Ph, R′ = Cy;25 R = Ph, R′ = p-MeO–C6H4;38 R = Ph, R′ = 5-Me-2-furyl;37 R = Ph, R′ = Cl;39 R = Ph, R′ = OPh;40 R = Ph, R′ = OMen;39 R = Ph, R′ = p-Me–Ph;38 R = p-Me–Ph, R′ = p-CF3–Ph.38 | ||
Although many different 1,1′-symmetrically and unsymmetrically substituted bisphosphanoferrocenes have been reported in the past (B and C, Fig. 1),19,23,25,31–40 the catalytic discussions have been dominated by 1,1′-bis(diphenylphosphano)ferrocene (dppf, D, Fig. 1) for many decades.15,16,24 However, in 2007, it was reported that 1,1′-bis(di-tert-butylphosphano)ferrocene (dtbpf, E, Fig. 1) is superior to dppf for the Pd-catalyzed α-arylation of ketones and certain Pd-catalyzed Suzuki coupling reactions.41,42 Moreover, in the recent past, several reports have theoretically proved the fact that with the increase of the steric bulk on phosphorus, the donating ability and βn increase, and as a result the catalytic activity increases.43,44 In this context, two obvious questions arise: (1) what is the maximum attainable sterics before the complexation of 1,1′-bisphosphanoferrocene compromises? (2) How much steric bulk can be used without affecting the catalytic activity of the resulting complexes? Since the steric situation will differ from one metal to the other, we have explored a catalytic process and addressed the above questions using two novel sterically congested 1,1′-diphosphanoferrocene ligands, namely 1,1′-bis(dimesitylphosphano)ferrocene and 1-(dimesitylphosphano)-1′-(diphenylphosphano)ferrocene. The complete syntheses of these ligands and their electrochemical properties and donating abilities will be reported herein, and the first insight into their potential for catalysis will be provided.
Owing to current interest in using CO2 as sustainable feedstock in catalytic transformations, we set out to investigate the catalytic formation of propiolic acid derivatives from CO2 and terminal alkynes. The first report of such catalytic CO2-fixation reactions was published by the Inoue group in 1994, where CuI was used as a catalyst (Scheme 1a).49 Although the insertion of CO2 into alkynylcopper was successful, high temperature and a large excess of base were needed for the execution of this reaction.49,50 Recently, Rath et al. have reported similar carboxylation reactions with Cu-complexes of dtbpf, where ambient temperatures have been used for high yielding transformations (Scheme 1b).51 Considering the fact that copper is an affordable late transition metal and potentially active for the reaction of our interest,50,52,53 we first decided to synthesize the CuX (X = Br, I, (MeCN)nBF4) complexes of our ligands. The structural properties of these complexes and their catalytic behaviour have been further studied in this report. Finally, to obtain an insight into the energetic scenario of complexation and the related catalysis, we have used density functional theory (DFT), which further helped us to verify the experimentally obtained results.
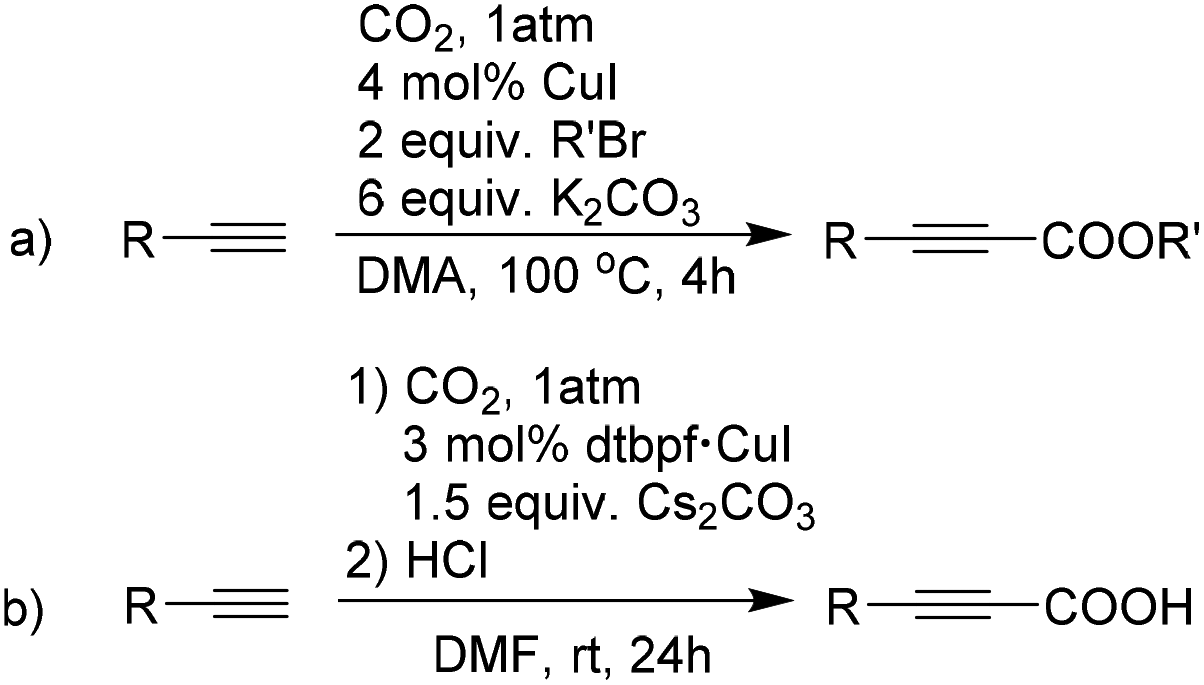 | ||
| Scheme 1 Cu-catalyzed carboxylation of terminal alkynes.50 | ||
Results and discussion
Syntheses and complexation
A few 1,1′-substituted ferrocenyl compounds with two dimesitylphosphanyl groups have been previously synthesized by reacting modified dilithioferrocene with Mes2PCl as reagents (G, H and I, Scheme 2).54,55 However, their properties as bidentate ligands in metal complexation and homogeneous catalysis have not been explored in detail, except for a few instances of silver-mediated nucleophilic fluorination.55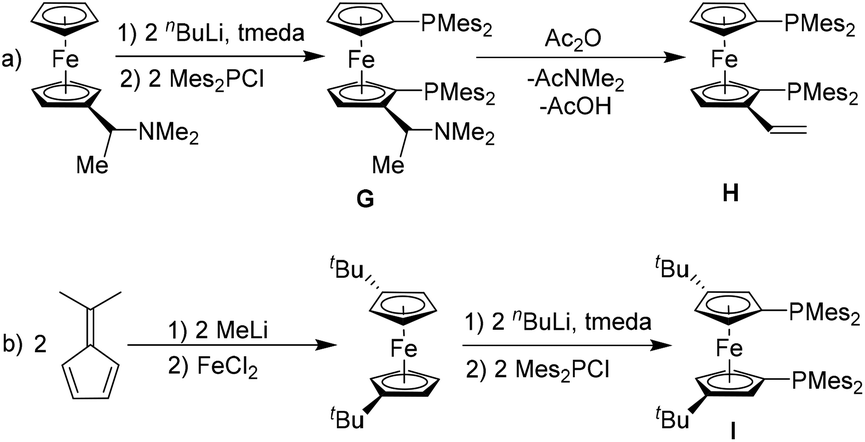 | ||
| Scheme 2 Syntheses of substituted 1,1′-bis(dimesitylphosphano)ferrocenes via routes (a) and (b).54,55 | ||
The synthesis of our mesityl-substituted bisphosphano ligand 1 was carried out by two complementary pathways. The first pathway involves PCl2-substituted ferrocene 2, which was synthesized by a known method56,57 and subsequently reacted with four equivalents of MesLi·LiBr·OEt2 (Scheme 3a). On the other hand, the second pathway (Scheme 3b) uses tmeda-stabilized dilithioferrocene, which was reacted with two equivalents of Mes2PX (X = Cl and Br). Although both the pathways can produce 1 in an acceptable purity, the first pathway (Scheme 3a) gives a slightly higher yield than the second (Scheme 3b).
 | ||
| Scheme 3 Syntheses of mesityl-substituted bisphosphano ligands 1 and 3. | ||
The unsymmetrically substituted bisphosphano ligand 3 was synthesized by following a simple and modular approach (Scheme 3c). At first, compound 4 was synthesized by selective monolithiation of 1,1′-dibromoferrocene58 and subsequent in situ reaction with Mes2PX. When compound 4 was further monolithiated and in situ reacted with Ph2PCl, compound 3 was obtained in an overall yield of 33%, starting from 1,1′-dibromoferrocene.
In order to explore the donating abilities of our ligands, the selenophosphorane derivatives of 1 and 3 were synthesized by adapting a reported procedure (Scheme 4), and their 1JP–Se values were compared with selenides of other phosphane ligands.59,60 Since compounds 5 and 6 are not soluble in common NMR solvents like CDCl3 and C6D6, our comparison became restricted to data available for toluene solutions. To this end we recorded the 31P and 77Se NMR spectra of Ph3P[Se] and dppf[Se]2 in toluene as the benchmark for this comparison as well, since the underlying phosphanes Ph3P and dppf are extremely popular and useful ligands in many synthetic applications.15,16,61–77 The 1JP–Se for compound 5 (723 Hz) is significantly lower than the corresponding values for Ph3P[Se] (732 Hz in CDCl3;78 758 Hz in toluene-d8, see Fig. S61 in ESI file‡) and dppf[Se]2 (737 Hz in CDCl3;59,79 761 Hz in toluene-d8, see Fig. S59 in ESI file‡), which indicates that the lone pairs of phosphorus centres in 1 have a lower s character, and therefore, a higher donating ability than Ph3P and dppf. A similar trend could also be noticed for the 77Se NMR chemical shifts, where the resonance of compound 5 at −82 ppm is deshielded by ca. δ 200 ppm and 220 ppm, compared to Ph3P[Se] and dppf[Se]2, respectively (see Fig. S21, S62 and S60 in the ESI file‡), which are in fact closer to values observed for the selenides of push pull substituted phosphanes.60
 | ||
| Scheme 4 Syntheses of diselenide derivatives of 5 and 6. | ||
In line with the lower symmetry in compound 6, two different values of 1JP–Se have been observed: 723 Hz for Mes2P[Se] and 763 Hz for Ph2P[Se], which are coherent with the corresponding values for 5 and dppf[Se]2, respectively. The 77Se NMR spectra of compound 6 show a set of two doublets, for the Se atoms at the two different phosphorus centres at −78 ppm (PSeMes2) and −299 ppm (PSePh2), which are consistent with the values observed for 5 and dppf[Se]2 (see Fig. S21, S25 and S60 in ESI file‡). It should be noted that although the 1JP–Se values for dtbpf[Se]2,80 dippf[Se]2 (dippf = 1,1′-bis(di-iso-propylphosphano)ferrocene),81 dchpf[Se]2 (dchpf = 1,1′-bis(dicyclohexylphosphano)ferrocene),82 dchpdppf[Se]2 [dchpdppf = 1-(dicyclohexylphosphano)-1′-(diphenylphosphano)ferrocene],25 dippdppf[Se]2 [dippdppf = 1-(diisopropylphosphano)-1′-(diphenylphosphano)ferrocene],25 and dppdtbpf[Se]2 [dppdtbpf = 1-(diphenylphosphano)-1′-(di-tert-butylphosphano)ferrocene]25 were reported in the literature, they were measured in CDCl3 and therefore could not be considered for this comparison.
Suitable single crystals for X-ray analyses were obtained for compounds 3–6. Fig. 2 and 3 show the molecular structures of 3 and 5 respectively, and their refinement data are listed in Table S1 (ESI file‡). The molecular structure of 3 in the solid state shows a sum of angles of 311.98(15)° at the phosphorus atom of the PMes2 unit which is larger than the respective value for the PPh2 unit in the same molecule (303.37(16)°), indicating increased steric interaction in the former.
 | ||
| Fig. 2 Ortep plots of the molecular structures of 3 in the solid state with ellipsoids drawn at the 50% probability level. Labels for some selected C atoms, solvent molecules and H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: C(1)–C(2) 1.428(5), C(1)–P(1) 1.816(3), C(11)–P(1) 1.850(3), C(11)–C(16) 1.418(5), C(15)–C(16) 1.391(5), C(14)–C(15) 1.382(5), C(16)–C(19) 1.504(5), C(6)–P(2) 1.823(3), C(29)–P(2) 1.835(4), C(29)–C(34) 1.394(5), C(33)–C(34) 1.384(5), C(32)–C(33) 1.372(6), C(5)–C(1)–P(1) 122.7(3), C(1)–P(1)–C(11) 108.70(15), P(1)–C(11)–C(16) 127.6(2), C(6)–P(2)–C(29) 101.70(15). | ||
 | ||
| Fig. 3 Ortep plots of the molecular structures of 5 in the solid state with ellipsoids drawn at the 50% probability level. Labels for some selected C atoms, solvent molecules and H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)–C(1) 1.798(2), P(1)–Se(1) 2.1216(6), P(1)–C(6) 1.847(2), C(15)–C(20) 1.411(3), C(20)–C(23) 1.507(3), C(1)–C(5) 1.440(3), C(1)–Fe(1) 2.051(2), C(1)–P(1)–Se(1) 109.11(7), C(1)–P(1)–C(6) 102.94(10), C(1)–P(1)–C(15) 112.25(10), C(1)–P(1)–C(15) 112.25(10), C(6)–P(1)–Se(1) 122.09(7), C(15)–P(1)–Se(1) 106.20(7). | ||
Similar trends are found for 5 and 6 which are the selenophosphorane derivatives of 1 and 3. The sum of the C–P–C angles in 5 carrying two PSeMes2 units is 319.5(1)° which is almost identical to the corresponding value of the PSeMes2 unit in mixed substituted 6 (321.8(3)°), while the PSePh2 unit shows only 315.2(2)°. Consistent with these findings the P–Se bond lengths are slightly longer in the sterically more demanding PSeMes2 units (5: 2.1216(6) Å, 6: 2.1246(14) Å) than in the PSePh2 unit (6: 2.0971(13) Å). These structural features indicate a more pronounced dative P–Se interaction for PPh2 than for the PMes2 unit in agreement with the NMR data outlined above, where the sterically less hindered phosphane unit entails larger 1JP–Se coupling values and stronger shielding of the 77Se resonance in the corresponding selenophosphorane. The solid-state structures of 4 and 6 are included in the ESI file (Fig. S65 and S66‡).
To explore the overall electronic effect of replacing phenyl with mesityl units in this molecular scaffold, the redox properties of the metallocene unit have been investigated using cyclic voltammetry (CV). The results, obtained by CV investigation, were further clarified by DFT calculations using the ω-B97XD/6-311+G** level of theory (more details in the ESI file‡) as the oxidation of P(III) substituted ferrocenes may involve iron or phosphorus centred redox events.83–89 The oxidation reactions of compounds 1 and 3 occur at 0.13 V and 0.16 V (see Fig. S69 and S70 in ESI file‡), respectively, which are slightly shifted to lower potentials in comparison to that of dppf (E° = 0.18 V). By investigating the Kohn–Sham molecular orbitals of 1, 3 and the parent dppf, it could be established that the lone pairs of the phosphorus atoms make a significant contribution to the HOMO (Fig. 4, Fig. S71 and S72 in ESI file‡). In agreement with the increased bond angles around phosphorus in the case of PMes2 units, the energy of this lone pair increases, thus contributing more to the HOMOs as well. This was in full agreement with the slightly shifted oxidation potentials of 1 and 3. The calculated spin density distribution of the corresponding cations of 1 and 3 is extensively localised at the iron center (Fig. S73 in ESI‡), and thus the iron centred redox process is reversible for compound 3 (Fig. S70 in ESI file‡) and quasi-reversible for compound 1 (Fig. S69 in ESI file‡). Moreover, for both complexes, several follow up oxidation processes can be found at higher anodic potentials, which are likely to involve PR2 moieties, in agreement with the significant contribution of the phosphorus lone pairs to the HOMO of the corresponding dicationic species. These follow up oxidations are irreversible and show small anodic shifts during reduction. To further characterize our ligands NBO calculations were performed, which revealed that upon changing the two phenyls to two mesityl groups, the p-character of the phosphorus lone pairs increased from 53% for dppf to 55–56% for 1 and 3 (Table S4 in ESI file‡). Recently it has been shown that the electronic effects of phosphorus containing bidentate ligands can be described by calculating the CO-stretching frequencies of the corresponding L2PdCO complexes (analogously to the experimental Tolman parameter).43 This method (more details in the ESI file‡) has further been used to show that the electron donating ability increases from dppf to 1 and 3 in the order of dppf < 3 < 1 (Table S4 in ESI file‡).
 | ||
| Fig. 4 The Kohn–Sham HOMOs of dppf, 1 and 3 (structures were optimized at the ω-B97XD/6-311+G** level of theory). Hydrogen atoms are omitted for clarity. | ||
The previous findings classify 1 and 3 as electron rich ligand systems with increased steric congestion along with the increased p-character of the phosphorus lone pair for the PMes2 unit. To explore their ligand properties towards d-block metals and possible effects on catalytic systems, Cu(I) complexes have been chosen, owing to their proclivity to adopt small coordination numbers for which steric effects should be less decisive. Using a common synthetic methodology, CuX-complexes 7–12 (X = Br, I, (MeCN)nBF4, Scheme 5) were synthesized from ligands 1 and 3. Among them, the solid state structures could only be obtained for complexes 10 and 12, which are depicted in Fig. 5 and 6, respectively. Although no suitable single crystals for X-ray analysis could be isolated for complexes 7–9, their formation was indicated by the upfield shifts of 31P signals (from δ −33.5 for 1 to δ −26.8 for 7, δ −25.0 for 8 and δ −27.8 for 9; see Fig. S8, S28, S31 and S34 in ESI file‡). Moreover, the broad lines in their 1H and 31P NMR spectra (see Fig. S26–S34 in ESI file‡) suggest a fast exchange of Cu+ ions in the solution. On the other hand, the formation of complex 11 could be confirmed by the shifts and multiplicities of its 31P signals [from δ −35.1 (s), −17.2 (s) for 3 to δ −30.1 (brm), −20.2 (d) for 11, see Fig. S17 and S45 in ESI file‡], which suggest a coupling between the two non-equivalent phosphorus atoms. It is needless to say that the evidence of a similar P–P coupling could also be noticed for complexes 10 and 12 (see Fig. S42 and S48 in ESI file‡).
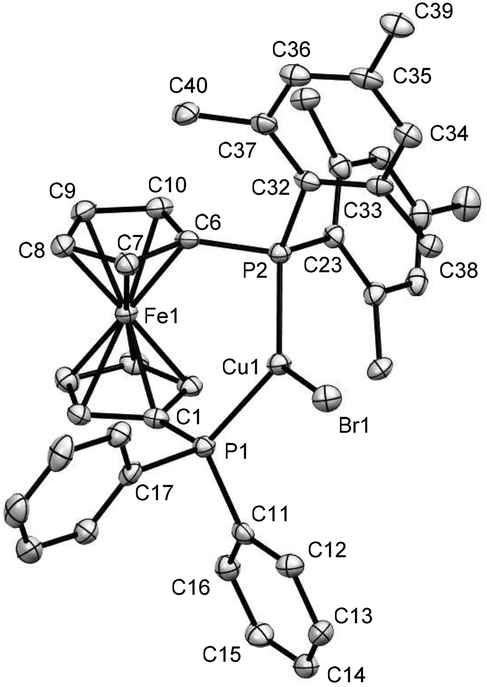 | ||
| Fig. 5 Ortep plots of the molecular structures of 10 in the solid state with ellipsoids drawn at the 50% probability level. Labels for some selected C atoms, solvent molecules and H atoms are omitted for clarity. The refinement data for this structure can be found in Table S2 (ESI file‡). Selected bond lengths [Å] and angles [°]: C(1)–P(1) 1.816(5), C(11)–P(1) 1.827(5), Cu(1)–P(1) 2.2540(15), Cu(1)–Br(1) 2.3428(9), Cu(1)–P(2) 2.2651(15), C(23)–P(2) 1.843(5), C(6)–P(2) 1.820(6), C(6)–P(2)–C(23) 103.7(2), C(23)–P(2)–C(32) 104.0(2), C(23)–P(2)–Cu(1) 120.25(17), C(32)–P(2)–Cu(1) 112.91(17), C(6)–P(2)–Cu(1) 104.52(17), C(6)–P(2)–C(32) 111.0(2), C(1)–P(1)–Cu(1) 115.88(17), C(1)–P(1)–C(11) 103.4(2), C(1)–P(1)–C(17) 102.4(2), C(11)–P(1)–Cu(1) 116.86(17), C(11)–P(1)–C(17) 102.4(2), P(1)–Cu(1)–Br(1) 113.81(5), P(2)–Cu(1)–Br(1) 128.24(5), P(1)–Cu(1)–P(2) 116.53(6). | ||
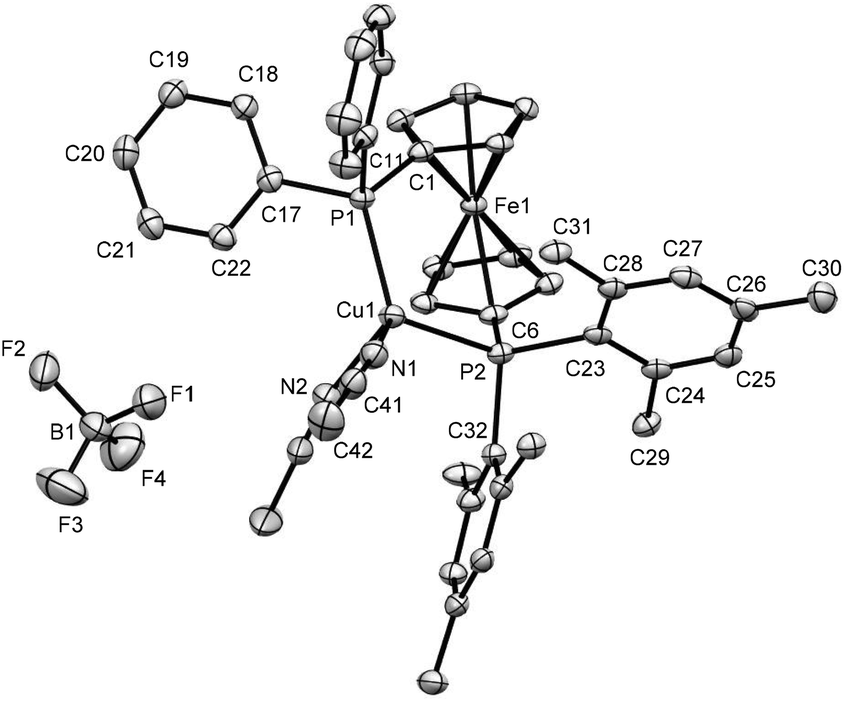 | ||
| Fig. 6 Ortep plots of the molecular structures of 12 in the solid state with ellipsoids drawn at the 50% probability level. One of the two coordinated acetonitrile molecules is disordered in the CCH3 part, for which only one position is shown without labels. Other solvent molecules and H atoms are omitted for clarity. The refinement data for this structure can be found in Table S2 (ESI file‡). Selected bond lengths [Å] and angles [°]: C(1)–P(1) 1.803(3), C(11)–P(1) 1.829(3), Cu(1)–P(1) 2.2437(8), Cu(1)–P(2) 2.3302(8), C(6)–P(2) 1.818(3), C(23)–P(2) 1.853(3), Cu(1)–N(1) 2.018(3), C(6)–P(2)–Cu(1) 103.33(10), C(6)–P(2)–C(23) 103.77(13), C(32)–P(2)–C(23) 104.36(13), C(6)–P(2)–C(32) 112.34(13), C(32)–P(2)–Cu(1) 103.22(9), C(1)–P(1)–Cu(1) 113.69(10), C(1)–P(1)–C(11) 102.41(13), C(1)–P(1)–C(17) 103.13(14), C(17)–P(1)–Cu(1) 117.22(10), P(1)–Cu(1)–P(2) 114.65(3), N(1)–Cu(1)–N(2) 91.98(12), P(1)–Cu(1)–N(1) 115.34(8), P(1)–Cu(1)–N(2) 118.70(9), F(1)–B(1)–F(2) 108.9(3), F(1)–B(1)–F(4) 112.5(4), F(2)–B(1)–F(3) 110.3(4). | ||
 | ||
| Scheme 5 CuX-complexes (X = Br, I, (MeCN)nBF4) of 1, 3 (eqn a) and 4 (eqn b). | ||
The X-ray crystal structures of 10 and 12 revealed that the copper atoms are bonded to two phosphorus units with bite angles 116.53(6)° and 114.65(3)°, respectively (see Fig. 5 and 6). As the mesityl group is bulkier than phenyl, the Cu–P bond in the P(Mes)2 unit [2.2651(15) Å for 10 and 2.3302(8) Å for 12] is slightly longer (1 pm for 10 and 9 pm for 12) than a similar bond in the PPh2 unit [2.2540(15) Å for 10 and 2.2437(8) Å for 12], which introduces a lower symmetry in complexes 10–12 for which a certain hemilability may be anticipated. To the best of our knowledge, there are only two complexes reported in the literature in which the ligands show a larger bite angle than 10 and both of them are based on dtbpf.20,51,90 For comparison, a few related complexes, ordered by increasing bite angles (in parentheses), are listed in the following: [Cu2(μ-SCN)2(κ2-P,P-dppf)2] (112.13(4)°),20,91 [Cu2(μ2-SCN)2(κ2-P,P-dppdtbpf)2] (112.82(3)°),90 [Cu2(μ-CN)2(κ2-P,P-dppf)2] (115.85(3)°),92 [Cu(κ2-P,P-dppdtbpf)(CH3CN)2]PF6 (116.36(8)°),90 [Cu2(μ-NO3-O)2(κ2-P,P-dppf)2] (117.8(1)°),93 and [CuI(dtbpf)] (120.070(19))°.51
Upon recrystallization of complex 7, crystals of complex 13 formed (see Scheme 5), along with micro-crystalline and powdery by-products. The X-ray crystal-structure of complex 13 shows a combination of ‘double-bridge’ and ‘quasi-closed bridge’ modes of coordination,15 where two molecules of ligand 1 are connected via two diamond shaped (Cu2Br2) units (Fig. 7). In order to avoid the formation of complex 13, several steps have been taken, such as changing the condition of crystallization and reaction of 1 with sub-stoichiometric amounts of CuBr (28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CuBr = 1:0.8, 1:0.7 and 1:0.5). However, all these experiments resulted in complex 13 as the only crystalline product and 13 can be prepared in a straightforward manner using an appropriate stoichiometry of metal to ligand.
:CuBr = 1:0.8, 1:0.7 and 1:0.5). However, all these experiments resulted in complex 13 as the only crystalline product and 13 can be prepared in a straightforward manner using an appropriate stoichiometry of metal to ligand.
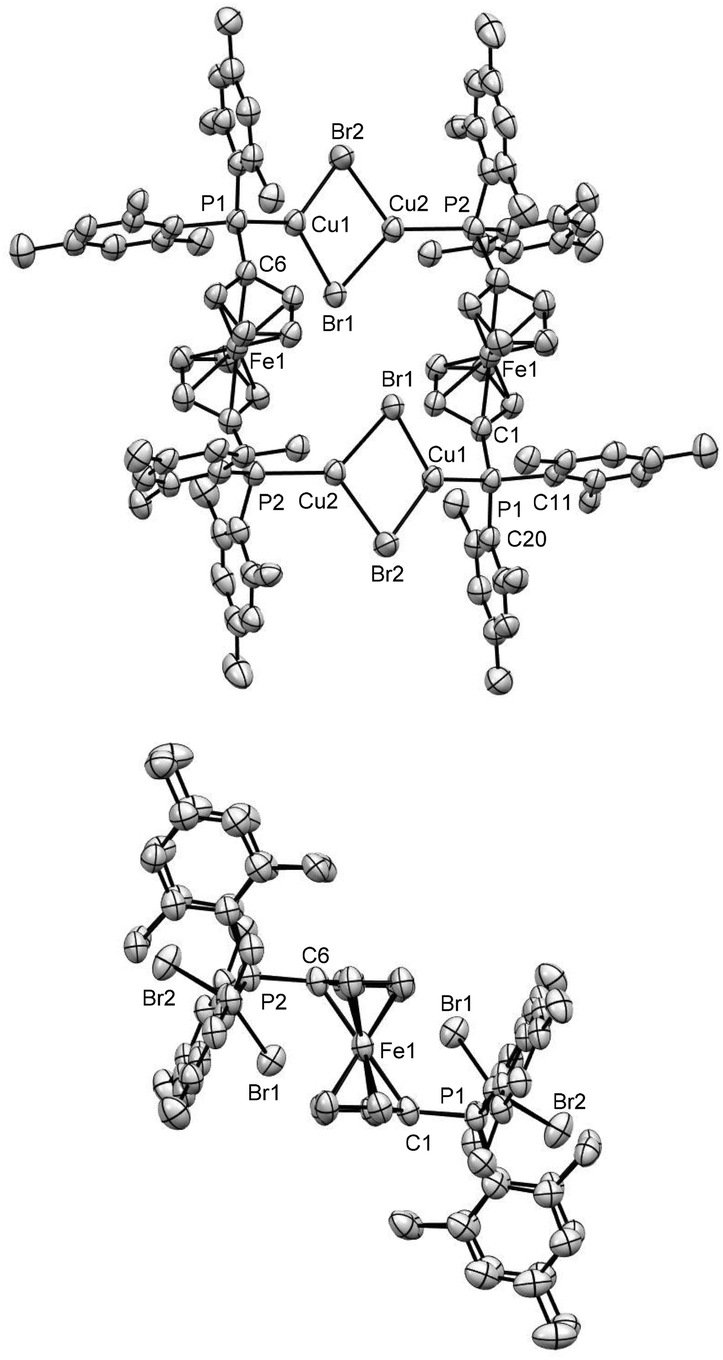 | ||
| Fig. 7 Ortep plot of the molecular structure of 13 in the solid state with ellipsoids drawn at the 50% probability level. Labels for some selected C atoms, solvent molecules and H atoms are omitted for clarity. The figure at the top shows the [Cu2Br2] bridges, and the figure at the bottom shows the side view of the molecule. The plane of symmetry, passing through the four Br atoms, made this molecule achiral. The refinement data for this structure can be found in Table S2, ESI file.‡ Selected bond lengths [Å] and angles [°]: C(1)–P(1) 1.804(6), C(11)–P(1) 1.831(6), Cu(1)–P(1) 2.2095(16), Cu(2)–P(2) 2.2059(15), Cu(1)–Br(1) 2.3949(10), Cu(1)–Cu(2) 2.9295(11), C(1)–P(1)–C(11) 102.7(3), C(1)–P(1)–C(20) 110.4(3), C(1)–P(1)–Cu(1) 110.21(19), Cu(1)–P(1)–C(11) 121.37(19), Cu(1)–P(1)–C(20) 103.6(2), C(20)–P(1)–C(11) 108.5(3), P(1)–Cu(1)–Br(1) 131.98(5), P(1)–Cu(1)–Br(2) 124.84(5), Br(1)–Cu(1)–Br(2) 102.34(4), Cu(1)–Br(1)–Cu(2) 74.46(3), Cu(2)–Cu(1)–Br(1) 53.57(3). | ||
The complexation via the formation of Cu2X2 (X = Cl, Br, I) bridges is common for dppf51,92–96 and some sterically encumbered dppf analogues.90,97–99 However, apart from this rare bonding motif, the classical chelating coordination mode of a single metal centre is also known for them.25,90,100 For the [Mes2P]-substituted ferrocene ligands, the formation of Cu2X2 bridges seems to be favoured as non-chelating ligand 4 similarly results in this motif for complex 14, which was synthesized by reacting 4 with one equivalent of CuBr. X-ray crystallographic analysis revealed that this complex shows a similar bonding motif to 13, where two molecules of 4 are bridged by one planar Cu2Br2 unit (Fig. 8). The distances between the Cu atoms in the Cu2Br2 moieties of complexes 13 and 14 are ca. 2.93 and 3.07 Å, respectively. These values are significantly lower, compared to the similar Cu–Cu distances of other halogen-bridged complexes reported in the literature.92,94–96
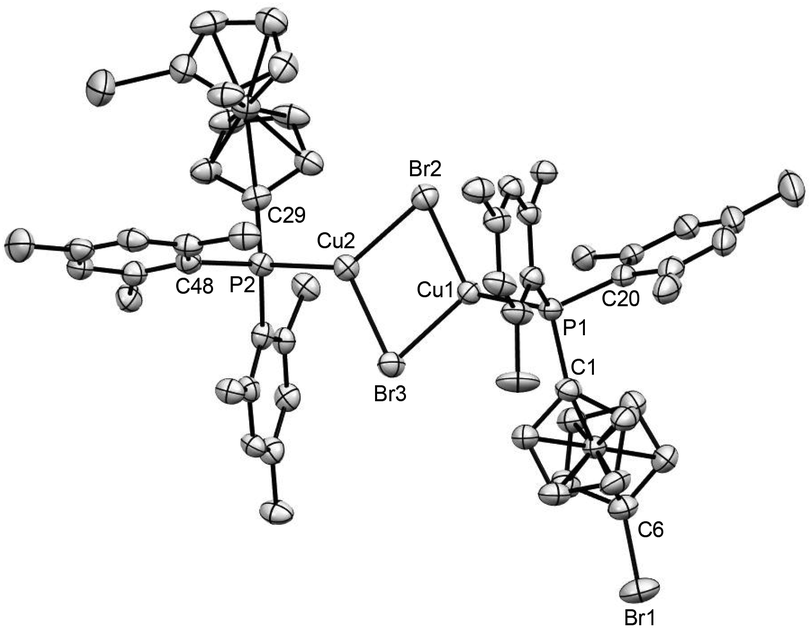 | ||
| Fig. 8 Ortep plot of the molecular structure of 14 in the solid state with ellipsoids drawn at the 50% probability level. Labels for some selected C atoms, solvent molecules and H atoms are omitted for clarity. The refinement data for this structure are listed in Table S2 (ESI file‡), and the side view of this molecule is shown in Fig. S67 (ESI file‡). The refinement data for this structure can be found in Table S2, ESI file.‡ Selected bond lengths [Å] and angles [°]: C(1)–P(1) 1.815(6), C(20)–P(1) 1.842(6), Cu(1)–P(1) 2.2044(15), Cu(1)–Br(2) 2.4592(9), Br(1)–C(6) 1.886(6), C(1)–P(1)–Cu(1) 111.33(18), C(1)–P(1)–C(20) 102.1(3), C(1)–P(1)–Cu(1) 111.33(18), Cu(1)–P(1)–C(20) 121.7(2), P(1)–Cu(1)–Br(2) 130.81(5), P(1)–Cu(1)–Br(3) 127.82(5), Cu(1)–Br(2)–Cu(2) 78.64(3). | ||
Catalysis
Complexes 7–12 were then used to explore their catalytic activity in the CO2-fixation of terminal alkynes as outlined in the introduction. In this investigation, the carboxylation of phenylacetylene was selected as a model reaction to study the influence of various ligands on catalysis compared with CuI, CuBr and Cu(MeCN)4BF4.As shown in Fig. 9, complexes 7–12 exhibited a higher catalytic activity compared to the free copper salts. The maximum increase of such catalytic activity can be seen for the Cu(MeCN)nBF4 analogues, where the yield increased from 29% for Cu(MeCN)4BF4 to 59% and 76% for complexes 9 and 12, respectively (entries 3, 6, and 9, Table 1 and Fig. 9). However, the catalytic activity of such complexes over the simple Cu halide decreases as the size of the halide increases [CuBr salts: 41% for CuBr to 62% for 7 and 88% for 10 (entries 2, 4 and 7, Table 1 and Fig. 9); CuI salts: 58% for CuI to 69% for 8 and 86% for 11 (entries 1, 5 and 8, Table 1 and Fig. 9)]. In order to make a fair comparison between dppf and our ligands (i.e.1 and 3), a similar catalytic reaction was further carried out with dppf·Cu(MeCN)2BF4 (entry 10, Table 1) as a catalyst, where 33% yield has been obtained, which was slightly higher than that of free Cu(MeCN)4BF4, but significantly lower than that of 9 and 12 (entries 6 and 9, Table 1). For the sake of completeness, it should be mentioned that CuCl and its respective complexes were not included in this comparison as they showed much lower catalytic conversion (ca. 5–10%) under otherwise identical conditions with no significant difference between complexes and free salts.
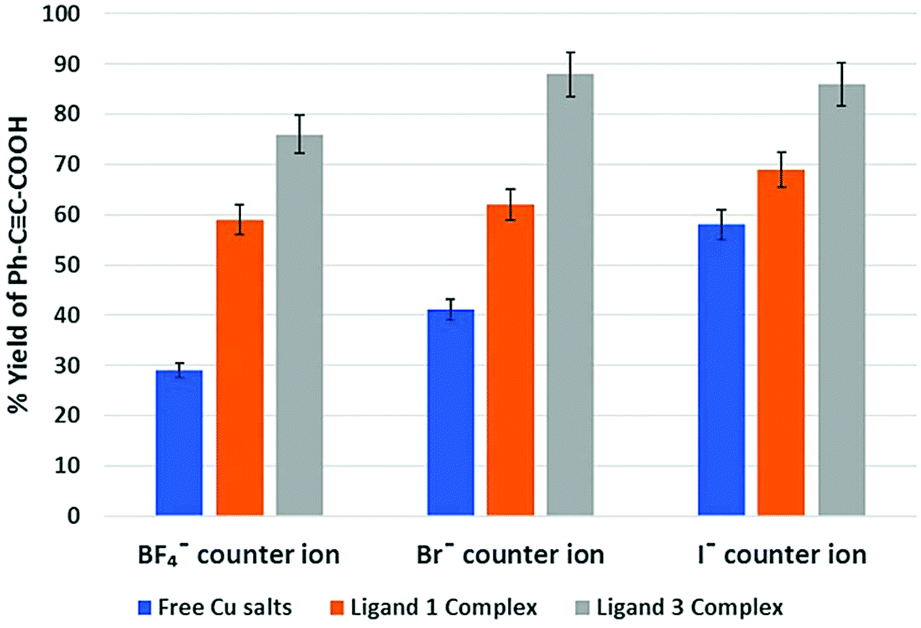 | ||
Fig. 9 Catalytic efficiency for the carboxylation reaction of Ph–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH to form Ph–CC–COOH. The blue, orange and grey bars show the yields resulting from free copper salts, and the corresponding Cu(I) complexes with ligands 1 and 3, respectively. As the yields listed here are the averages of two consecutive catalytic reactions, error bars of 5% are shown for all yields. CH to form Ph–CC–COOH. The blue, orange and grey bars show the yields resulting from free copper salts, and the corresponding Cu(I) complexes with ligands 1 and 3, respectively. As the yields listed here are the averages of two consecutive catalytic reactions, error bars of 5% are shown for all yields. | ||
| Entry | Catalyst | Product yield |
|---|---|---|
| 1 | CuI | 58 |
| 2 | CuBr | 41 |
| 3 | Cu(MeCN)4(BF4) | 29 |
| 4 | 7 | 62 |
| 5 | 8 | 69 |
| 6 | 9 | 59 |
| 7 | 10 | 88 |
| 8 | 11 | 86 |
| 9 | 12 | 76 |
| 10 | dppf·Cu(MeCN)2(BF4) | 33 |
In general, the catalytic activity of the complexes of phosphane ligands is raised by increasing the steric bulk of the substituents on phosphorus.21,51 An opposite trend is being noticed in our case, where complexes 10–12 produce higher yields than 7–9 (Table S3, ESI file‡) which may be attributed to the complexation mode observed for ferrocene based ligands where all phosphorus donor sites are mesityl substituted. Complexes 13 and 14 are the only examples for these ligands whose structural information is available and show this feature consistently. Since the conversion of 7 to 13 involves the transfer of CuBr units, it can be speculated that in complexes 7–9 the metal is more loosely bound, due to the high steric bulk of ligand 1. The lower complex stability of Cu(I) with 1 (in comparison to 3) was further corroborated by the 7 + 3 → 10 + 1 ligand-exchange reaction, which is slightly exothermic (−4.4 kcal mol−1 at the ω-B97X-D/6-311+G**//ω-B97X-D/6-31G* level of theory, Fig. S74 in ESI file‡). To test this hypothesis experimentally, the organic washings of the catalytic reactions were collected, dried under high vacuum, and analyzed by 31P NMR, which revealed that complexes 7–9 dissociate to give free ligand 1 during catalysis (Fig. S55 and S56 in ESI file‡), whereas for complexes 10–12 the Cu–P bonds remain intact (Fig. S57 and S58 in ESI file‡). It should be highlighted that we have further investigated computationally the CO2 insertion step in the catalytic system, which is usually the rate limiting step.101,102 The calculated reaction barriers are somewhat lower (by 1.2 kcal mol−1 at the ω-B97X-D/6-311+G**//ω-B97X-D/6-31G* level of theory) in the case of the complexes with 3 in comparison to those of 1, which can also be an explanation for the lower activity of its complexes (see Table S5 in the ESI file‡).
Conclusion
In summary, the tetra- and dimesityl analogues of dppf, 1 and 3 have been synthesized and their ligand properties were explored. The phosphorus lone pairs of tetramesityl substituted 1 show a lower s character, and therefore, a higher donating ability than those in Ph3P and dppf as indicated by spectroscopic, structural, electrochemical and computational means. In line with these experimental findings, the lone pair at phosphorus makes a significant contribution to the HOMO in 1 and 3, which explains the cathodic shift of the oxidation potentials with increasing mesityl substitution. For a series of Cu(I) complexes of these ligands, a variety of structural motifs have been found, ranging from the rare double bridging mode over dimeric bridging to isolated Cu-centers. With unsymmetrical ligand 3 stable complexes were formed with isolated Cu(I) centers, while the sterically more challenged tetramesityl substituted 1 is prone to dimeric bridging with increased separation of the phosphorus atoms and its adjacent mesityl substituents. As a consequence of its hemilabile nature, the Cu(I) complexes of 3 showed improved catalytic activity in the addition of CO2 to terminal alkynes as compared with the respective complexes of 1 which in turn showed superior performance compared to the respective dppf complexes. The potential of these ligands for complexation with other metals and the catalytic activity of these complexes will be explored in the near future.Experimental section
All manipulations were performed under an argon atmosphere unless mentioned otherwise. Prior to use, glassware was dried in a drying oven at 120 °C. Solvents were distilled over drying agents, prescribed in the CRC Handbook of Chemistry and subsequently stored under an argon atmosphere over 4 Å molecular sieves. Solvents for column chromatography and aqueous workups were used from bottles (analytical grade supplied by VWR and Alfa-Aesar) without further purification. NMR solvents (purchased from Deutero) were degassed via a few cycles of freeze, pump and thaw and finally stored over 3 Å molecular sieves under an argon atmosphere.Reagents and chemicals were purchased from commercial suppliers (Sigma-Aldrich, ABCR, Alfa-Aesar) and used as received. Fc′(PCl2)2, MesLi·LiBr·OEt2, and dppf·Cu(MeCN)2BF4 were synthesized by following the procedure reported in the literature.56,57,103,104 Mes2PX (X = Cl/Br, 48%/52%; Fig. S1 and S2 in ESI file‡) was synthesized by following the procedure reported for Xyl2PX (X = Cl, Br).105 It should be noted that all the chemical manipulations, involving Mes2PX as a reagent, require careful calculations of its formula weights. This is because the 1H NMR spectra of Mes2PX, obtained from different preparation attempts have shown different proportional ratios of Mes2PCl and Mes2PBr (e.g. 48:52 and 53:47). Due to minor side reactions, such as unwanted dilithiation and subsequent in situ hydrolysis, compounds 3 and 4 contain an impurity, wherein only the dimesitylphosphano group is present on ferrocene. We could not remove this compound from the targeted species and, therefore, used species 3 and 4 contaminated with ca. 1–4% of dimesitylphosphanoferrocene for the subsequent chemical transformations (see Fig. S9, S12 and S13 in ESI file‡).
NMR spectra were recorded with Varian 500VNMRS and Varian MR-400 spectrometers at 22 °C. Chemical shifts (δ in ppm) were expressed with respect to the following standards, set as 0 ppm: SiMe4 (for 1H and 13C), aqueous H3PO4 (for 31P), BF3·OEt2 (in CDCl3 for 11B) and CFCl3 (for 19F). The signals, resulting from the residual non-deuterated NMR solvents, were locked as indicated in the literature.106 In addition to the standard notation of signal multiplicity (s = singlet, d = doublet, m = multiplet, dd = doublet of doublet etc.), pst, brs, brd and brm were used to abbreviate pseudo-triplet, broad singlet, broad doublet and broad multiplet, respectively. The amount of residual solvents (if present) was verified by NMR analysis and the expected values for elemental analyses were calculated accordingly. The NMR spectra of compounds 1 and 3–6 were recorded in toluene-d8 and thf-d8, and those for the corresponding complexes (7–12 and 14) were only recorded either in thf-d8 or CD3CN. This is because, the complexes could not be dissolved in toluene-d8 by any means. Due to the reason of lower solubility even in the donating solvents (like thf-d8 or CD3CN), the 13C NMR spectra of some complexes are relatively poor and as a result, signals for the ipso-carbons could not be seen after a substantially high number of scans. On the other hand, the diphosphano ferrocene ligands were insoluble in CD3CN and, therefore, the NMR spectra in the corresponding solvent could not be recorded.
Infrared spectra recorded for the neat substances of 9 and 12 were obtained using a Bruker Alpha Platinum ATR spectrometer, and Opus 6.5 (from Bruker Optics) was used for analysing the data. Strong, medium strong and weak peaks for these species were denoted as s, m and w, respectively. For the sake of comparison, the infrared spectra of Cu(MeCN)4BF4 and dppf·Cu(MeCN)2BF4 were also recorded under identical conditions for comparison and are depicted in the ESI file (Fig. S75‡). Electrospray ionisation (ESI) and atmospheric pressure chemical ionization (APCI) mass spectra were recorded with a Finnigan LCQ Deca (ThermoQuest, San Jose, USA) instrument using samples dissolved in HPLC-quality thf, and MALDI was recorded with an UltraFlex ToF/ToF (Bruker Daltonics, Bremen, D) instrument, where an N2 laser with a 337 nm wavelength and 3 ns pulse duration was used. The matrix used for MALDI measurements was DCTB (2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene] malononitrile). Elemental analyses were performed without the presence of any external oxidizer (like V2O5) in an EA 3000 Elemental Analyzer (EuroVector). X-ray diffraction experiments were performed using either a STOE IPDS II [using Mo-Kα source (λ = 0.71073 Å)] or a STOE StadiVari [using either an Mo-GENIX source (λ = 0.71073 Å), or a Cu-GENIX source (λ = 1.54186 Å)] diffractometer. Structures were solved using the dual space method (SHELXT) and were refined with SHELXL-2018.107 All non-hydrogen atoms were refined anisotropically, whereas hydrogen atoms were placed on adjacent atoms using a riding model. Further programs used in the structure analyses were Mercury and Platon.108–110
All cyclic voltammetry measurements were carried out in an MBraun acrylic glove box GB2202-C-VAC under an argon atmosphere. Samples were measured as a solution (0.1 M) in dry and deoxygenated CH2Cl2, where anhydrous [Bu4N][PF6] was used as a conducting salt at a concentration of 0.1 M. The three-electrode cell consisted of a platinum working electrode, a silver counter electrode and a silver pseudoreference electrode. The potential was driven on a WaveDriver 20 Bipotentiostat from the Pine Research Instrument and the electrochemical data were recorded via AfterMath (Ver. 1.5.9807, Pine Instrument). All redox processes were referenced using half wave potentials of (C5Me5)2Fe as the standard, which was added to the analyzed solution. Its corresponding value was then subtracted from the recorded potentials to convert them to the Fc/Fc+ scale following established procedures111 and then finally evaluated with AfterMath and Excel.
Fc′(PMes2)2 (1)
A solution of Fc′(PCl2)2 (0.388 g, 1.00 mmol) in thf (10 mL) was added dropwise to a cold (−84 °C) and stirred suspension of MesLi·LiBr·OEt2 (1.145 g, 3.99 mmol) in hexanes (100 mL). The reaction mixture was slowly warmed to rt over 3–4 h and stirred for 18 h at rt. After all volatiles were removed under vacuum (10−3 mbar), the resulting compound was extracted with toluene (60 mL) and tested by 7Li NMR. A small peak at δ 7.33 ppm in 7Li NMR indicated the presence of soluble LiCl in the crude, which was further evacuated to dryness and extracted with toluene (30 mL). The volume of the thus obtained toluene solution was reduced to ca. 5 mL and finally precipitated on a cold (−20 °C) and vigorously stirred mixture of Et2O and pentane (40 mL, 1:1). The supernatant solution was carefully decanted and the precipitate was dried under high vacuum (10−3 mbar), resulting in compound 1 as a pale yellow amorphous solid (70%). Note: this compound has also been synthesized (with a yield of 66%) by the reaction of FcLi2·(tmeda)2/3 (1 mmol) and Mes2PX (X = Cl/Br, 48%/52%, FW = 327.914 g mol−1, 0.669 g, 2.04 mmol) in thf at −84 °C, with a subsequent workup as mentioned above. 1H NMR (toluene-d8): δ 2.08 (s, 12H, p-CH3 of Mes), 2.31 (s, 24H, o-CH3 of Mes), 4.20 (m, 4H, β-H of Cp), 4.25 (m, 4H, α-H of Cp), 6.66 (brs, 8H, m-H of Mes). 1H NMR (thf-d8): δ 2.17 (s, 24H, o-CH3 of Mes), 2.18 (s, 12H, p-CH3 of Mes), 4.19 (m, 4H, β-H of Cp), 4.22 (m, 4H, α-H of Cp), 6.74 (brs, 8H, m-H of Mes). 13C{1H} NMR (toluene-d8): δ 21.28 (s, p-CH3 of Mes), 23.47 (d, o-CH3 of Mes, J = 15 Hz), 72.50 (d, β-C of Cp, J = 4 Hz), 76.00 (d, α-C of Cp, J = 19 Hz), 80.14 (d, ipso-C of Cp, J = 13 Hz), 127.34 (s, p-Aryl C of Mes), 130.48 (d, m-Aryl C of Mes, J = 3 Hz), 132.65 (d, ipso-Aryl C of Mes, J = 21 Hz), 142.53 (d, o-Aryl C of Mes, J = 15 Hz). 13C{1H} NMR (thf-d8): δ 20.72 (s, p-CH3 of Mes), 23.29 (d, o-CH3 of Mes, J = 15 Hz), 72.68 (d, β-C of Cp, J = 4 Hz), 76.24 (d, α-C of Cp, J = 18 Hz), 80.27 (d, ipso-C of Cp, J = 13 Hz), 130.52 (d, p-Aryl C of Mes, J = 4 Hz), 132.82 (d, ipso-Aryl C of Mes, J = 20 Hz), 137.95 (s, m-Aryl C of Mes), 142.75 (d, o-Aryl C of Mes, J = 15 Hz). 31P{1H} NMR (toluene-d8): δ −35.1. 31P{1H} NMR (thf-d8): δ −33.5. MS (APCI-DIP): m/z (%) 722 (100) [M]+. HRMS (APCI-DIP; m/z): [M]+ calc. for C46H52FeP2, 722.28937; found 722.28882. Anal. calcd for C46H52FeP2: C, 76.45; H, 7.25. Found: C 75.23; H, 7.16. Probably due to the presence of a little amount of LiCl, the CHN values for this compound differ significantly than expected. As these values could not be improved after several attempts, the purity of the species was further clarified by mass spectrometry (see Fig. S68 in ESI file‡).
Fc′(PMes2)Br (4)
n BuLi (2.5 M in hexanes, 0.84 mL, 2.10 mmol) was added dropwise to a cold (−84 °C) and stirred thf (20 mL) solution of dibromoferrocene (0.687 g, 2.00 mmol). After the gradual color change from pale yellow to bright orange, the solution was stirred at −84 °C for another 30 min. Another solution of Mes2PX (X = Cl/Br, 48%/52%, FW = 327.914 g mol−1, 0.679 g, 2.07 mmol) in thf (20 mL) was slowly added to the previous cold solution over 5 min. After warming up to the ambient temperature, the reaction mixture was stirred overnight. All volatiles were removed under high vacuum (10−3 mbar) and the product was extracted with hexanes (50 mL). The volume of the filtrate was reduced to ca. 10 mL and the almost pure compound was obtained as yellow crystals upon refrigeration at −78 °C. This product had an impurity of dimesitylphosphanoferrocene (ca. 5%) which could be reduced to ca. 2–3% upon further crystallization in hexanes at −10 °C and the final product was obtained as bright orange crystals (53%). 1H NMR (C6D6): δ 2.08 (s, 6H, p-CH3 of Mes), 2.37 (s, 12H, o-CH3 of Mes), 3.85 (pst, 2H, β-H of CpBr), 4.19 (pst, 2H, α-H of CpBr), 4.23 (pst, 2H, β-H of CpPMes2), 4.32 (m, 2H, α-H of CpPMes2), 6.71 (d, 4H, m-H of Mes, J = 3 Hz). 13C{1H} NMR (C6D6): δ 20.89 (s, p-CH3 of Mes), 23.51 (d, o-CH3 of Mes, J = 15 Hz), 69.04 (s, β-C of CpBr), 71.59 (s, α-C of CpBr), 74.20 (d, β-C of CpPMes2, J = 4 Hz), 77.23 (d, α-C of CpPMes2, J = 18 Hz), 78.09 (s, ipso-C of CpBr), 81.55 (d, ipso-C of CpPMes2, J = 13 Hz), 130.57 (d, m-Aryl C of Mes, J = 3 Hz), 132.47 (d, o-Aryl C of Mes, J = 21 Hz), 137.81(s, p-Aryl C of Mes), 142.57 (d, ipso-Aryl C of Mes, J = 14 Hz). 31P{1H} NMR (C6D6): δ −35.5. MS (APCI-DIP): m/z (%) 533 (100) [M]+. HRMS (APCI-DIP; m/z): [M]+ calc. for C28H30BrFeP, 533.06179; found 533.069069. Anal. calcd for C28H30BrFeP: C, 63.07; H, 5.67. Found: C, 63.32; H, 5.75.Fc′(PMes2)(PPh2) (3)
n BuLi (2.5 M in hexanes, 0.15 mL, 0.38 mmol) was added dropwise to a cold (0 °C) thf (20 mL) solution of Fc′(PMes2)Br (0.200 g, 0.38 mmol). After the gradual color change from orange to bright red, the solution was stirred at 0 °C for 30 min. Another solution of Ph2PCl (72 μL, 0.086 g, 0.40 mmol) in hexanes (10 mL) was slowly added to the above cold solution over 5 min. After warming up to the ambient temperature, the reaction mixture was stirred overnight. All volatiles were removed under high vacuum (10−3 mbar) and the product was extracted with hexanes (20 mL). The volume of the filtrate was reduced to ca. 5 mL and the almost pure compound was obtained as yellow crystals (62%) upon refrigeration at −78 °C. This product had an impurity of dimesitylphosphanoferrocene (ca. 3–4%) which could not be reduced upon further crystallization. 1H NMR (toluene-d8): δ 2.08 (s, 6H, p-CH3 of Mes), 2.30 (s, 12H, o-CH3 of Mes), 4.04 (m, 2H, β-H of CpPPh2), 4.10 (pst, 2H, α-H of CpPPh2), 4.17 (m, 2H, β-H of CpMes), 4.21 (pst, 2H, α-H of CpMes), 6.66 (brd, 4H, m-H of Mes, J = 3 Hz), 7.01–7.02 (m, 6H, m and p-H of Ph), 7.38 (m, 4H, o-H of Ph). 1H NMR (thf-d8): δ 2.16 (s, 12H, o-CH3 of Mes), 2.19 (s, 6H, p-CH3 of Mes), 4.01 (m, 2H, β-H of CpPPh2), 4.14 (m, 2H, α-H of CpPPh2), 4.18 (m, 2H, β-H of CpMes), 4.26 (pst, 2H, α-H of CpMes), 6.74 (brd, 4H, m-H of Mes, J = 3 Hz), 7.23–7.29 (m, 10H, o, m and p-H of Ph). 13C{1H} NMR (toluene-d8): δ 20.87 (s, p-CH3 of Mes), 23.47 (d, o-CH3 of Mes, J = 15 Hz), 72.27 (d, β-C of CpPPh2, J = 3 Hz), 72.80 (d, β-C of CpPMes2, J = 4 Hz), 74.28 (d, α-C of CpPPh2, J = 15 Hz), 75.86 (d, α-C of CpPMes2, J = 18 Hz), 77.34 (d, ipso-C of CpPPh2, J = 10 Hz), 80.57 (d, ipso-C of CpPMes2, J = 13 Hz), 128.34 (d, m-Aryl C of Ph, J = 7 Hz), 128.57 (s, p-Aryl C of Ph), 130.49 (d, m-Aryl C of Mes, J = 3 Hz), 132.61 (d, o-Aryl C of Mes, J = 21 Hz), 133.92 (d, o-Aryl C of Ph, J = 21 Hz), 140.05 (d, ipso-Aryl C of Ph, J = 15 Hz), 142.54 (d, ipso-Aryl C of Mes, J = 15 Hz). 13C{1H} NMR (thf-d8): δ 20.90 (s, p-CH3 of Mes), 23.48 (d, o-CH3 of Mes, J = 15 Hz), 72.68 (dd, β-C of CpPPh2, J = 4, 1 Hz), 73.11 (d, β-C of CpPMes2, J = 4 Hz), 74.63 (d, α-C of CpPPh2, J = 15 Hz), 76.28 (d, α-C of CpPMes2, J = 18 Hz), 77.71 (d, ipso-C of CpPPh2, J = 10 Hz), 80.81 (d, ipso-C of CpPMes2, J = 13 Hz), 128.78 (d, m-Aryl C of Ph, J = 7 Hz), 129.09 (s, p-Aryl C of Ph), 130.70 (d, m-Aryl C of Mes, J = 3 Hz), 132.95 (d, o-Aryl C of Mes, J = 21 Hz), 134.27 (d, o-Aryl C of Ph, J = 20 Hz), 138.11 (s, p-Aryl C of Mes), 140.43 (d, ipso-Aryl C of Ph, J = 11 Hz), 142.91 (d, ipso-Aryl C of Mes, J = 15 Hz). 31P{1H} NMR (toluene-d8): δ −34.8 (PMes2), −17.0 (PPh2). 31P{1H} NMR (thf-d8): δ −35.1 (PMes2), −17.2 (PPh2). MS (APCI-DIP): m/z (%) 639 [M + 1]+. HRMS (APCI-DIP; m/z): [M + 1]+ calc. for C40H40FeP2, 639.19547; found 639.20281. Anal. calcd for C40H40FeP2: C, 75.24; H, 6.31. Found: C, 74.92; H, 6.45.Diselenide derivatives of 1 and 3
A suspension of red Se (0.120 g, 1.52 mmol) and 1 (0.170 g, 0.24 mmol) or 3 (0.153 g, 0.24 mmol) in thf (20 mL) was stirred for 1 h at r.t. All volatiles were removed under high vacuum (10−3 mbar) and the product was extracted with hot toluene. The analytically pure compound was crystallized from the hot toluene solution by slow cooling up to ambient temperature. Note: if all residual Se is not removed by single filtration, the procedure of filtration must be repeated multiple times before crystallization.Fc′(PSeMes2)2 (5)
Yield: 67%. 1H NMR (toluene-d8): δ 1.99 (s, 12H, p-CH3 of Mes), 2.38 (brs, 24H, o-CH3 of Mes), 4.79 (m, 4H, Cp), 4.90 (brs, 4H, Cp), 6.52 (m, 8H, m-H of Mes). 13C{1H} NMR (toluene-d8): δ 24.43 (d, o-CH3 of Mes, J = 6 Hz), 75.72 (d, β-C of Cp, J = 9 Hz), 78.25 (brs, α-C of Cp), 82.45 (d, ipso-C of Cp, J = 78 Hz), 125.63 (s, p-Aryl C of Mes), 132.15 (d, o-Aryl C of Mes, J = 11 Hz), 139.72 (d, m-Aryl C of Mes, J = 2 Hz), 140.49 (brm, ipso-Aryl C of Mes). 31P{1H} NMR (toluene-d8): δ 14.7 (1JP,Se = 723 Hz). 77Se{1H} NMR (toluene-d8): δ −82.6 (PSeMes2, 1JP,Se = 723 Hz). MS (MALDI): m/z (%) 880 (100) [M]+. Anal. calcd for C46H52FeP2Se2: C, 62.74; H, 5.95. Found: C, 62.89; H, 5.90.Fc′(PSeMes2)(PSePh2) (6)
Yield: 65%. 1H NMR (toluene-d8): δ 1.99 (s, 6H, p-CH3 of Mes), 2.35 (brs, 12H, o-CH3 of Mes), 4.42 (brs, 2H, β-H of CpPPh2), 4.69 (brs, 2H, α-H of CpPPh2), 4.73 (brs, 2H, α-H of CpMes), 4.76 (brs, 2H, β-H of CpMes), 6.49 (brd, 4H, m-H of Mes, J = 4 Hz), 6.92–6.94 (m, 6H, m and p-H of Ph), 7.70 (m, 4H, o-H of Ph). 13C{1H} NMR (toluene-d8): δ 20.69 (d, p-CH3 of Mes, J = 2 Hz), 24.32 (d, o-CH3 of Mes, J = 6 Hz), 74.94 (d, β-C of CpPPh2, J = 12 Hz), 75.38 (d, α-C of CpPPh2, J = 9 Hz), 75.99 (d, ipso-C of CpPPh2, J = 86 Hz), 76.12 (d, β-C of CpPMes2, J = 10 Hz), 78.05 (brd, α-C of CpPMes2, J = 13 Hz), 82.82 (d, ipso-C of CpPMes2, J = 78 Hz), 128.27 (d, p-Aryl C of Ph, J = 3 Hz), 131.10 (d, m-Aryl C of Ph, J = 3 Hz), 132.08 (d, o-Aryl C of Ph, J = 11 Hz), 132.38 (d, o-Aryl C of Mes, J = 11 Hz), 134.34 (d, ipso-Aryl C of Ph, J = 78 Hz), 139.62 (d, m-Aryl C of Mes, J = 3 Hz), 140.39 (brd, ipso-Aryl C of Mes). 31P{1H} NMR (toluene-d8): δ 14.8 (PMes2, 1JP,Se = 723 Hz), 30.8 (PPh2, 1JP,Se = 763 Hz). 77Se{1H} NMR (toluene-d8): δ −298.8 (PSePh2, 1JP,Se = 763 Hz), −77.5 (PSeMes2, 1JP,Se = 723 Hz). MS (ESI): m/z (%) 797 (100) [M + 1]+. HRMS (ESI; m/z): [M + 1]+ calc. for C40H40FeP2Se2, 799.02851; found 799.03579. Anal. calcd for C40H40FeP2Se2: C, 60.32; H, 5.06. Found: C, 60.44; H, 5.17.Copper complexes of 1
A suspension of 1 (0.144 g, 0.20 mmol), CuX (0.20 mmol), toluene (10 mL), thf (10 mL) and CH3CN (2 mL) was refluxed for 24 h. After removal of all insoluble materials by filtration, the volume of the filtrate was reduced to ca. 15 mL and analytically pure crystalline or semi-crystalline materials were obtained by slow introduction of dry pentane and kept in a double arm H-tube at ambient temperature. For all compounds, mentioned under this section, the mass spectrometric measurements gave similar data, where the corresponding peak for Fc′(PMes2)2Cu+ was identified as a molecular ion peak. One example is shown in the following: MS (ESI): m/z (%) 787 [M + 1]+. HRMS (ESI; m/z): [M]+ calc. for C46H52CuFeP2, 785.21897; found 785.21842.Fc′(PMes2)2·CuBr (7)
Yield: 63%. 1H NMR (thf-d8): δ 2.00–2.50 (brs, 36H, o- and p-CH3 of Mes), 4.24 (brs overlapped with another brs, 8H, α- and β-H of Cp), 6.79 (brs, 8H, m-H of Mes). 13C{1H} NMR (thf-d8): δ 20.88 (brs, p-CH3 of Mes), 73.12 (brs, α- and β-H of Cp), 78.46 (brs, ipso-C of Cp), 126.01 (s, p-Aryl C of Mes), 128.88 (s, m-Aryl C of Mes), 129.64 (s, o-Aryl C of Mes), 131.52 (brs, ipso-Aryl C of Mes). 31P{1H} NMR (thf-d8): δ −26.8. Anal. calcd for C46H52BrCuFeP2: C, 63.79; H, 6.05. Found: C, 63.72; H, 5.88.Fc′(PMes2)2·CuI (8)
Yield: 58%. 1H NMR (thf-d8): δ 2.00–2.50 (brs, 36H, o- and p-CH3 of Mes), 4.27 (brs overlapped with another brs, 8H, α- and β-H of Cp), 6.83 (brs, 8H, m-H of Mes). 13C{1H} NMR (thf-d8): δ 20.87 (brs, p-CH3 of Mes), 72.72 (brs, α- and β-H of Cp), 126.01 (s, p-Aryl C of Mes), 128.88 (s, m-Aryl C of Mes), 129.64 (s, o-Aryl C of Mes), 131.52 (brs, ipso-Aryl C of Mes). 31P{1H} NMR (thf-d8): δ −25.0. Anal. calcd for C46H52CuFeIP2: C, 60.50; H, 5.74. Found: C, 60.72; H, 5.35.Fc′(PMes2)2·Cu(BF4) (9)
Yield: 69%. 1H NMR (CD3CN): δ 2.00–2.50 (brs, 36H, o- and p-CH3 of Mes), 4.24 (brs, 4H, β-H of Cp), 4.45 (brs, 4H, α-H of Cp), 6.91 (brs, 8H, m-H of Mes). 13C{1H} NMR (CD3CN): δ 20.82 (s, p-CH3 of Mes), 24.98 (brs, o-CH3 of Mes), 73.47 (brs, α- and β-H of Cp), 78.70 (t, ipso-H of Cp, J = 17 Hz), 126.21 (s, p-Aryl C of Mes), 129.18 (s, m-Aryl C of Mes), 129.87 (s, o-Aryl C of Mes), 131.89 (brs, ipso-Aryl C of Mes). 31P{1H} NMR (CD3CN): δ −27.8. 11B{1H} NMR (CD3CN): δ −1.2. 19F{1H} NMR (CD3CN): δ −151.9. IR (ATR) ν: 1024 (m), 1159 (m), 1444 (m), 1466 (m), 1602 (w), 2918 (w). Anal. calcd for C46H52BCuF4FeP2: C, 63.28; H, 6.00. Found: C, 63.25; H, 6.01.Copper complexes of 3
A suspension of 3 (0.128 g, 0.20 mmol), CuX (0.2 mmol), toluene (10 mL), thf (10 mL) and CH3CN (2 mL) was refluxed for 24 h. After removal of all insoluble materials by filtration, the volume of the filtrate was reduced to ca. 15 mL and analytically pure crystalline or semi-crystalline materials were obtained by slow evaporation under an inert atmosphere at ambient temperature. For all compounds, mentioned under this section, the mass spectrometric measurements gave similar data, where the corresponding peak for Fc′(PMes2)(PPh2)Cu+ was identified as a molecular ion peak. One example is shown in the following: MS (ESI): m/z (%) 701 [M]+. HRMS (ESI; m/z): [M]+ calc. for C40H40CuFeP2, 701.12507; found 701.12452.Fc′(PMes2)(PPh2)·CuBr (10)
Yield: 71%. 1H NMR (thf-d8): δ 2.22 (s, 6H, p-CH3 of Mes), 2.32 (s, 12H, o-CH3 of Mes), 4.14 (s, 2H, β-H of CpPPh2), 4.27 (pst, 2H, β-H of CpPMes2), 4.42 (s, 4H, α-H of CpPPh2), 4.49 (brs, 4H, α-H of CpPMes2), 6.82 (brm, 4H, m-H of Mes), 7.38 (m, 6H, m and p-H of Ph), 7.92 (m, 4H, o-H of Ph). 13C{1H} NMR (thf-d8): δ 20.67 (s, p-CH3 of Mes), 30.46 (s, o-CH3 of Mes), 72.69 (s, β-C of CpPPh2,PMes2), 74.48 (s, α-C of CpPPh2), 77.41 (s, α-C of CpPMes2), 129.12 (d, p-Aryl C of Ph, J = 10 Hz), 130.67 (s, m-Aryl C of Ph), 131.63 (s, m-Aryl C of Mes), 134.95 (d, p-Aryl C of Mes, J = 15 Hz), 139.66 (s, o-Aryl C of Ph), 142.92 (s, o-Aryl C of Mes). 31P{1H} NMR (thf-d8): δ −31.5 (d, PMes2, J = 135 Hz), −19.3 (d, PPh2, J = 135 Hz). Anal. calcd for C40H40BrCuFeP2: C, 61.44; H, 5.16. Found: C, 61.04; H, 5.15.Fc′(PMes2)(PPh2)·CuI (11)
Yield: 74%. 1H NMR (thf-d8): δ 2.22 (s, 6H, p-CH3 of Mes), 2.29 (s, 12H, o-CH3 of Mes), 4.12 (pst, 2H, β-H of CpPPh2), 4.26 (pst, 2H, β-H of CpPMes2), 4.42 (s, 4H, α-H of CpPPh2), 4.52 (brs, 4H, α-H of CpPMes2), 6.82 (d, 4H, m-H of Mes, J = 3 Hz), 7.39 (m, 6H, m and p-H of Ph), 7.92 (m, 4H, o-H of Ph). 13C{1H} NMR (thf-d8): δ 20.85 (s, p-CH3 of Mes), 24.75 (dd, o-CH3 of Mes, J = 9 and 1 Hz), 72.91 (m, β-C of CpPPh2,PMes2), 74.82 (d, α-C of CpPPh2, J = 10 Hz), 77.59 (m, α-C of CpPMes2), 129.28 (d, p-Aryl C of Ph, J = 10 Hz), 130.88 (m, m-Aryl C of Ph), 131.83 (d, m-Aryl C of Mes, J = 6 Hz), 135.20 (d, p-Aryl C of Mes, J = 15 Hz), 139.86 (s, o-Aryl C of Ph), 142.94 (d, o-Aryl C of Mes, J = 11 Hz). 31P{1H} NMR (thf-d8): δ −30.1 (brm, PMes2), −20.2 (d, PPh2, J = 98 Hz). Anal. calcd for C40H40CuFeIP2: C, 57.95; H, 4.86. Found: C, 57.66; H, 4.90.Fc′(PMes2)(PPh2)·Cu(MeCN)2(BF4) (12)
Yield: 77%. 1H NMR (thf-d8): δ 2.08 (s, 6H, CH3 of MeCN) 2.23 (s, 6H, p-CH3 of Mes), 2.30 (s, 12H, o-CH3 of Mes), 4.11 (m, 2H, β-H of CpPPh2), 4.32 (pst, 2H, β-H of CpPMes2), 4.54 (pst, 4H, α-H of CpPPh2), 4.63 (pst, 4H, α-H of CpPMes2), 6.90 (, 4H, m-H of Mes), 7.48 (m, 6H, m and p-H of Ph), 7.68 (m, 4H, o-H of Ph). 13C{1H} NMR (thf-d8): δ 1.16 (s, CH3CN), 20.84 (s, p-CH3 of Mes), 24.45 (dd, o-CH3 of Mes, J = 11 and 1 Hz), 73.27 (d, β-C of CpPPh2, J = 5 Hz), 73.61 (d, β-C of CpPMes2, J = 6 Hz), 74.62 (d, α-C of CpPPh2, J = 9 Hz), 75.25 (d, ipso-C of CpPPh2, J = 47 Hz), 77.71 (d, α-C of CpPMes2, J = 14 Hz), 78.94 (d, ipso-C of CpPMes2, J = 39 Hz), 119.76 (s, CH3CN), 126.06 (dd, ipso-C of Ph, J = 28 and 3 Hz), 130.08 (d, p-Aryl C of Ph, J = 10 Hz), 131.57 (d, m-Aryl C of Ph, J = 2 Hz), 132.02 (d, p-Aryl C of Mes, J = 7 Hz), 133.34 (d, ipso-C of Mes, J = 31 Hz), 134.70 (d, o-Aryl C of Ph, J = 16 Hz), 140.61 (d, m-Aryl C of Mes, J 2 = Hz), 142.94 (d, o-Aryl C of Mes, J = 11 Hz). 31P{1H} NMR (thf-d8): δ −33.5 (d, PMes2, J = 119 Hz), −11.8 (d, PPh2, J = 119 Hz). 11B{1H} NMR (thf-d8): δ −0.9. 19F{1H} NMR (thf-d8): δ −153.2. IR (ATR) ν: 1025 (s), 1034 (s), 1053 (s), 1093 (m), 1436 (m), 1600 (w), 2228 (w), 2922 (w). Anal. calcd for C44H46BCuF4FeN2P2: C, 60.67; H, 5.32; N, 3.22. Found: C, 60.45; H, 5.34; N, 2.96.[Fc′(PMes2)2·(CuBr)2]2 (13)
A suspension of 1 (0.144 g, 0.20 mmol), CuBr (0.057 g, 0.40 mmol), toluene (10 mL), thf (15 mL) and CH3CN (5 mL) was refluxed for 48 h. After the removal of all insoluble materials by filtration, the volume of the filtrate was reduced to ca. 20 mL and the crystalline substances along with the semi-crystalline materials were obtained by slow introduction of dry pentane and kept in a double arm H-tube at ambient temperature. After washing several times with a mixture of dry toluene and pentane (1:1), followed by the removal of all volatiles under high vacuum (10−3 mbar), compound 13 was obtained in a yield of 56%. 1H NMR (CD3CN): δ 2.00–2.50 (brs overlapped with another brs, 72H, o- and p-CH3 of Mes), 4.25 (brs, 8H, α-H of Cp), 4.60 (brs, 8H, β-H of Cp), 6.88 (brs, 16H, m-H of Mes). 31P{1H} NMR (thf-d8): δ −26.9. 31P{1H} NMR (CD3CN): δ −27.5. MS (MALDI): m/z (%) 850 [M]+ for [Fc′(PMes2)2·Cu2]2+, 786 [M]+ for [Fc′(PMes2)2·Cu]+. Anal. calcd for C46H52BrCuFeP2: C, 54.72; H, 5.19. Found: C, 54.59; H, 5.06. Note: compound 13 is highly insoluble in commonly available organic solvents (including thf-d8 and CD3CN). After a prolonged NMR experiment only 1H (with a low S/N ratio and unresolved broad signals near the baseline) and 31P{1H} NMR spectra (with a satisfactory S/N ratio) could be obtained, which revealed the absence of any starting ligand (1) in the resulting mixture.
[Fc′(PMes2)Br·CuBr]2 (14)
A suspension of 4 (0.107 g, 0.20 mmol), CuBr (0.029 g, 0.20 mmol), toluene (10 mL), thf (10 mL) and CH3CN (2 mL) was refluxed for 24 h. After the removal of all insoluble materials by filtration, the volume of the filtrate was reduced to ca. 15 mL and analytically pure crystalline materials (72%) were obtained by slow evaporation under an inert atmosphere. 1H NMR (thf-d8): δ 2.22 (brs, 12H, p-CH3 of Mes), 2.29 (brs, 12H, o-CH3 of Mes), 4.19 (pst, 2H, β-H of CpPMes2), 4.32 (pst, 2H, β-H of CpBr), 4.51 (s, 2H, α-H of CpBr), 4.77 (s, 2H, α-H of CpPMes2), 6.83 (d, 8H, m-H of Mes, J = 5 Hz). 13C {1H} NMR (thf-d8): 20.87(s, p-CH3 of Mes), 24.77 (s, o-CH3 of Mes), 70.76 (s, β-C of CpBr), 72.48 (s, α-C of CpBr), 76.10 (d, β-C of CpPMes2, J = 7 Hz), 77.96 (d, ipso-C of CpPMes2, J = 41 Hz), 78.41 (s, ipso-C of CpBr), 79.09 (brd, α-C of CpPMes2), 128.79 (d, ipso-Aryl C of Mes, J = 36 Hz), 131.74 (d, p-Aryl C of Mes, J = 7 Hz), 139.97 (s, m-Aryl C of Mes), 142.11 (d, o-Aryl C of Mes, J = 11 Hz). 31P{1H} NMR (thf-d8): δ −26.5. MS (MALDI and APCI): m/z (%) 532 (100) [M]+ for the starting ligand Fc′(PMes2)Br and no peak for its corresponding CuBr complex 14 could be found. Anal. calcd for C56H60Br4Cu2Fe2P2: C, 49.70; H, 4.47. Found: C, 49.99; H, 4.43.Catalytic reaction
A mixture of Ph–CC–H (0.204 g, 0.22 mL, 2 mmol, 1 equiv.), the respective catalyst (mole% with respect to Cu, mentioned in Table S3, ESI file‡), dry DMF (10 mL), and Cs2CO3 (0.978 g, 3 mmol, 1.5 equiv.) was degassed by three consecutive cycles of freeze (at −98 °C, MeOH and liquid N2), pump and thaw. A balloon, filled with dry CO2, was placed on it and the resulting reaction mixture was stirred for 36 h at rt, followed by quenching with water (20 mL). The organic layer was then separated by washing with DCM (3 × 20 mL) and the combined DCM phases were stored for further investigation. The aqueous layer was acidified with conc. HCl (up to pH 1) and extracted with EtOAc (3 × 20 mL). The resulting EtOAc phases were combined and the residual DMF was removed by washing with water (2 × 30 mL) and brine (30 mL), followed by drying upon anhydrous Na2SO4. Volatiles were removed using a rotatory evaporator and the resulting colourless oil was subjected to controlled vacuum (5 × 10−2 mbar), until colourless crystals formed (see Fig. S54 in ESI file‡). The previously collected DCM phase was then washed with water (2 × 30 mL) and brine (30 mL), dried under a rotatory evaporator and finally subjected to high vacuum (10−3 mbar) overnight at 40 °C. The thus-obtained yellow solids were then characterized by 31P NMR, which revealed them as the remnants of catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the funding programs LOEWE and MASH for financial support. Zsolt Kelemen is grateful for the general support of Hungarian Academy of Science under the Premium Postdoctoral Research Program 2019.References
- D. E. Herbert, U. F. Mayer and I. Manners, Angew. Chem., Int. Ed., 2007, 46, 5060 CrossRef CAS PubMed.
- V. Bellas and M. Rehahn, Angew. Chem., Int. Ed., 2007, 46, 5082 CrossRef CAS PubMed.
- A. Alkan, L. Thomi, T. Gleede and F. R. Wurm, Polym. Chem., 2015, 6, 3617 RSC.
- R. Pietschnig, Chem. Soc. Rev., 2016, 45, 5216 RSC.
- L. Cao, I. Manners and M. A. Winnik, Macromolecules, 2002, 35, 8258 CrossRef CAS.
- M. Saleem, H. Yu, L. Wang, A. Zain ul, H. Khalid, M. Akram, N. M. Abbasi and J. Huang, Anal. Chim. Acta, 2015, 876, 9 CrossRef CAS PubMed.
- X. Sui, X. Feng, M. A. Hempenius and G. J. Vancso, J. Mater. Chem. B, 2013, 1, 1658 RSC.
- J. Elbert, J. Mersini, N. Vilbrandt, C. Lederle, M. Kraska, M. Gallei, B. Stühn, H. Plenio and M. Rehahn, Macromolecules, 2013, 46, 4255 CrossRef CAS.
- C. Ornelas, New J. Chem., 2011, 35, 1973 RSC.
- G. Jaouen, A. Vessieres and S. Top, Chem. Soc. Rev., 2015, 44, 8802 RSC.
- R. L. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358 RSC.
- S. Takahashi and J. I. Anzai, Materials, 2013, 6, 5742 CrossRef CAS PubMed.
- P. Debroy and S. Roy, Coord. Chem. Rev., 2007, 251, 203 CrossRef CAS.
- D. P. Puzzo, A. C. Arsenault, I. Manners and G. A. Ozin, Angew. Chem., Int. Ed., 2009, 48, 943 CrossRef CAS PubMed.
- K.-S. Gan and T. S. A. Hor, in Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, John Wiley & Sons, 2007, pp. 3–104 Search PubMed.
- S. W. Chien and T. S. Andy Hor, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons, 2008, pp. 33–116 Search PubMed.
- J. J. Bishop, A. Davison, M. L. Katcher, D. W. Lichtenberg, R. E. Merrill and J. C. Smart, J. Organomet. Chem., 1971, 27, 241 CrossRef CAS.
- G. M. Whitesides, J. F. Gaasch and E. R. Stedronsky, J. Am. Chem. Soc., 1972, 94, 5258 CrossRef CAS.
- L. E. Hagopian, A. N. Campbell, J. A. Golen, A. L. Rheingold and C. Nataro, J. Organomet. Chem., 2006, 691, 4890 CrossRef CAS.
- G. Bandoli and A. Dolmella, Coord. Chem. Rev., 2000, 209, 161 CrossRef CAS.
- M. N. Birkholz, Z. Freixa and P. W. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099 RSC.
- P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519 RSC.
- S. P. Thomas and J. Colacot, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons, 2008, pp. 117–140 Search PubMed.
- A. Fihri, P. Meunier and J.-C. Hierso, Coord. Chem. Rev., 2007, 251, 2017 CrossRef CAS.
- P. Vosáhlo, I. Císařová and P. Štěpnička, J. Organomet. Chem., 2018, 860, 14 CrossRef.
- K. Skoch, I. Cisarova, J. Schulz, U. Siemeling and P. Stepnicka, Dalton Trans., 2017, 46, 10339 RSC.
- P. Štěpnička, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons, 2008, pp. 177–204 Search PubMed.
- H.-U. Blaser, W. Chen, F. Camponovo and A. Togni, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons, 2008, pp. 205–235 Search PubMed.
- P. Štěpnička and M. Lamač, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons, 2008, pp. 237–277 Search PubMed.
- U. Siemeling, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons, 2008, pp. 141–176 Search PubMed.
- A. L. Boyes, I. R. Butler and S. C. Quayle, Tetrahedron Lett., 1998, 39, 7763 CrossRef CAS.
- W. R. Cullen, T. J. Kim, F. W. B. Einstein and T. Jones, Organometallics, 1983, 2, 714 CrossRef CAS.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 3694 CrossRef CAS.
- O. V. Gusev, T. y. A. Peganova, A. M. Kalsin, N. V. Vologdin, P. V. Petrovskii, K. A. Lyssenko, A. V. Tsvetkov and I. P. Beletskaya, Organometallics, 2006, 25, 2750 CrossRef CAS.
- I. R. Butler, W. R. Cullen, T. J. Kim, S. J. Rettig and J. Trotter, Organometallics, 1985, 4, 972 CrossRef CAS.
- T. Y. Dong and C. K. Chang, J. Chin. Chem. Soc., 1998, 45, 577 CrossRef CAS.
- A. Fihri, J. C. Hierso, A. Vion, D. H. Nguyen, M. Urrutigoïty, P. Kalck, R. Amardeil and P. Meunier, Adv. Synth. Catal., 2005, 347, 1198 CrossRef CAS.
- M. Yamashita, J. V. Cuevas-Vicario and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 16347 CrossRef CAS PubMed.
- J. C. Hierso, F. Lacassin, R. Broussier, R. Amardeil and P. Meunier, J. Organomet. Chem., 2004, 689, 766 CrossRef CAS.
- M. Laly, R. Broussier and B. Gautheron, Tetrahedron Lett., 2000, 41, 1183 CrossRef CAS.
- T. J. Colacot and H. A. Shea, Org. Lett., 2004, 6, 3731 CrossRef CAS PubMed.
- G. A. Grasa and T. J. Colacot, Org. Lett., 2007, 9, 5489 CrossRef CAS PubMed.
- T. R. Kegl, N. Palinkas, L. Kollar and T. Kegl, Molecules, 2018, 23, 3176 CrossRef PubMed.
- G. Cavinato and L. Toniolo, Molecules, 2014, 19, 15116 CrossRef PubMed.
- I. R. Butler, W. R. Cullens and T.-J. Kim, Synth. React. Inorg. Met.-Org. Chem., 2006, 15, 109 CrossRef.
- L. Qin, X. Ren, Y. Lu, Y. Li and J. Zhou, Angew. Chem., Int. Ed., 2012, 51, 5915 CrossRef CAS PubMed.
- M. Ogasawara, K.-i. Takizawa and T. Hayashi, Organometallics, 2002, 21, 4853 CrossRef CAS.
- L. A. Evans, N. Fey, J. N. Harvey, D. Hose, G. C. Lloyd-Jones, P. Murray, A. G. Orpen, R. Osborne, G. J. J. Owen-Smith and M. Purdie, J. Am. Chem. Soc., 2008, 130, 14471 CrossRef CAS PubMed.
- Y. Fukue, S. Oi and Y. Inoue, J. Chem. Soc., Chem. Commun., 1994, 2091 RSC.
- S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666 RSC.
- M. Trivedi, G. Singh, A. Kumar and N. P. Rath, Dalton Trans., 2015, 44, 20874 RSC.
- L. J. Gooßen, N. Rodríguez, F. Manjolinho and P. P. Lange, Adv. Synth. Catal., 2010, 352, 2913 CrossRef.
- W. H. Wang, L. Jia, X. Feng, D. Fang, H. Guo and M. Bao, Asian J. Org. Chem., 2019, 8, 1501 CrossRef CAS.
- R. Sihler, U. Werz and H.-A. Brune, J. Organomet. Chem., 1989, 368, 213 CrossRef CAS.
- J. Roger, S. Royer, H. Cattey, A. Savateev, R. V. Smaliy, A. N. Kostyuk and J. C. Hierso, Eur. J. Inorg. Chem., 2017, 330 CrossRef CAS.
- I. E. Nifant'ev, A. A. Boricenko, L. F. Manzhukova and E. E. Nifant'ev, Phosphorus, Sulfur Silicon Relat. Elem., 1992, 68, 99 CrossRef.
- C. Moser, A. Orthaber, M. Nieger, F. Belaj and R. Pietschnig, Dalton Trans., 2006, 3879 RSC.
- S. Dey, J. W. Quail and J. Müller, Organometallics, 2015, 34, 3039 CrossRef CAS.
- M. N. Chevykalova, Russ. Chem. Bull., Int. Ed., 2003, 52, 78 CrossRef CAS.
- A. Orthaber, M. Fuchs, F. Belaj, G. N. Rechberger, C. O. Kappe and R. Pietschnig, Eur. J. Inorg. Chem., 2011, 2588 CrossRef CAS.
- H. Zahra and R. Ali, Curr. Org. Chem., 2018, 22, 1589 CrossRef.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2013, 52, 176 CrossRef CAS PubMed.
- G. J. Grant, Dalton Trans., 2012, 41, 8745 RSC.
- J. Rodrigues, M. G. Jardim, J. Figueira, M. Gouveia, H. Tomás and K. Rissanen, New J. Chem., 2011, 35, 1938 RSC.
- R. H. Crabtree, in Iridium Catalysis, ed. P. G. Andersson, Springer Berlin Heidelberg, 2011, pp. 1–10 Search PubMed.
- Z. Qiu, S. Ren and Z. Xie, Acc. Chem. Res., 2011, 44, 299 CrossRef CAS PubMed.
- A. Sinha, T. Ghatak and J. K. Bera, Dalton Trans., 2010, 39, 11301 RSC.
- M. Tanabe and K. Osakada, Organometallics, 2010, 29, 4702 CrossRef CAS.
- E. Farnetti and S. Filipuzzi, Inorg. Chim. Acta, 2010, 363, 467 CrossRef CAS.
- F. Blank and C. Janiak, Coord. Chem. Rev., 2009, 253, 827 CrossRef CAS.
- P. Mastrorilli, Eur. J. Inorg. Chem., 2008, 4835 CrossRef CAS.
- G. Schmid, Chem. Soc. Rev., 2008, 37, 1909 RSC.
- L. Pedrosa, Synlett, 2008, 1581 CrossRef CAS.
- G. Schmid, Angew. Chem., Int. Ed., 2008, 47, 3496 CrossRef CAS PubMed.
- A. B. Chaplin, Chimia, 2008, 62, 217 CrossRef CAS.
- C. Lau, S. Ng, G. Jia and Z. Lin, Coord. Chem. Rev., 2007, 251, 2223 CrossRef CAS.
- S. Krompiec, N. Kuźnik, M. Krompiec, R. Penczek, J. Mrzigod and A. Tórz, J. Mol. Catal. A: Chem., 2006, 253, 132 CrossRef CAS.
- D. W. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51 RSC.
- P. Arsenyan, A. Petrenko, K. Oberte and S. Belyakov, Chem. Heterocycl. Compd., 2012, 48, 1263 CrossRef CAS.
- F. N. Blanco, L. E. Hagopian, W. R. McNamara, J. A. Golen, A. L. Rheingold and C. Nataro, Organometallics, 2006, 25, 4292 CrossRef CAS.
- M. Necas, M. Beran, J. D. Woollins and J. Novosad, Polyhedron, 2001, 20, 741 CrossRef CAS.
- K. D. Reichl, C. L. Mandell, O. D. Henn, W. G. Dougherty, W. S. Kassel and C. Nataro, J. Organomet. Chem., 2011, 696, 3882 CrossRef CAS.
- S. Isenberg, S. Weller, D. Kargin, S. Valic, B. Schwederski, Z. Kelemen, C. Bruhn, K. Krekic, M. Maurer, C. M. Feil, M. Nieger, D. Gudat, L. Nyulaszi and R. Pietschnig, ChemistryOpen, 2019, 8, 1235 CrossRef CAS PubMed.
- A. Lik, D. Kargin, S. Isenberg, Z. Kelemen, R. Pietschnig and H. Helten, Chem. Commun., 2018, 54, 2471 RSC.
- A. Jakob, P. Ecorchard, M. Linseis, R. F. Winter and H. Lang, J. Organomet. Chem., 2009, 694, 655 CrossRef CAS.
- F. Barrière, R. U. Kirss and W. E. Geiger, Organometallics, 2005, 24, 48 CrossRef.
- P. Zanello, G. Opromolla, G. Giorgi, G. Sasso and A. Togni, J. Organomet. Chem., 1996, 506, 61 CrossRef CAS.
- C. L. Mandell, S. S. Kleinbach, W. G. Dougherty, W. S. Kassel and C. Nataro, Inorg. Chem., 2010, 49, 9718 CrossRef CAS PubMed.
- A. Orthaber, R. H. Herber and R. Pietschnig, J. Organomet. Chem., 2012, 719, 36 CrossRef CAS PubMed.
- M. Trivedi, S. K. Ujjain, G. Singh, A. Kumar, S. K. Dubey and N. P. Rath, J. Organomet. Chem., 2014, 772–773, 202–209 CrossRef CAS.
- D. Li, Y.-F. Luo, T. Wu and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m927 CrossRef CAS.
- M. Trivedi, R. Nagarajan, A. Kumar, N. P. Rath and P. Valerga, Inorg. Chim. Acta, 2011, 376, 549 CrossRef CAS.
- S. P. Neo, Z.-Y. Zhou, T. C. W. Mak and T. S. A. Hor, J. Chem. Soc., Dalton Trans., 1994, 3451 RSC.
- P. Pinto, M. J. Calhorda, V. Félix, T. Avilés and M. G. B. Drew, Monatsh. Chem., 2000, 131, 1253 CrossRef CAS.
- C. Di Nicola, Effendy, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2005, 358, 695 CrossRef CAS.
- J. Díez, M. P. Gamasa, J. Gimeno, M. Lanfranchi and A. Tiripicchio, J. Organomet. Chem., 2001, 637–639, 677 CrossRef.
- M. Trivedi, G. Singh, A. Kumar and N. P. Rath, Dalton Trans., 2014, 43, 13620 RSC.
- P. S. Prasad, M. K. Pandey and M. S. Balakrishna, Polyhedron, 2019, 158, 173 CrossRef.
- S. K. Gibbons, R. P. Hughes, D. S. Glueck, A. T. Royappa, A. L. Rheingold, R. B. Arthur, A. D. Nicholas and H. H. Patterson, Inorg. Chem., 2017, 56, 12809 CrossRef CAS PubMed.
- K. M. Gramigna, J. V. Oria, C. L. Mandell, M. A. Tiedemann, W. G. Dougherty, N. A. Piro, W. S. Kassel, B. C. Chan, P. L. Diaconescu and C. Nataro, Organometallics, 2013, 32, 5966 CrossRef CAS.
- H. Tran-Vu and O. Daugulis, ACS Catal., 2013, 3, 2417 CrossRef CAS PubMed.
- L. Dang, Z. Lin and T. B. Marder, Organometallics, 2010, 29, 917 CrossRef CAS.
- S. S. Zigler, L. M. Johnson and R. West, J. Organomet. Chem., 1988, 341, 187 CrossRef CAS.
- X. Wu, W. Zhang, X. Zhang, N. Ding and T. S. A. Hor, Eur. J. Inorg. Chem., 2015, 2015, 876 CrossRef CAS.
- J. Campos, M. F. Espada, J. Lopez-Serrano and E. Carmona, Inorg. Chem., 2013, 52, 6694 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem. B, 1999, 103, 6713 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Manfred Scheer on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: NMR spectra, crystal structures of 4 and 6, crystal refinement data, CV and details of DFT calculations. CCDC 1980151–1980158 contains the supplementary crystallographic data for compounds 3–6, 10, and 12–14. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/D0DT00941E |
| This journal is © The Royal Society of Chemistry 2020 |