
A ferrocene functionalized Schiff base containing Cu(ii) complex: synthesis, characterization and parts-per-million level catalysis for azide alkyne cycloaddition†‡
 a
Biswajit
Saha
*ab
a
Biswajit
Saha
*ab
Abstract
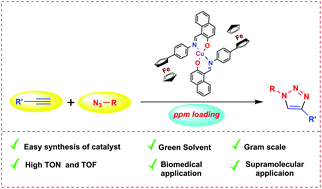
Atom economy is one of the major factors in developing catalysis chemistry. Using the minimum amount of catalyst to obtain the maximum product yield is of the utmost priority in catalysis, which drives us to use parts-per-million (ppm) levels of catalyst loadings in syntheses. In this context, a new ferrocene functionalized Schiff base and its copper(II) complex have been synthesized and characterized. This Cu(II) complex is employed as a catalyst for popular ‘click chemistry’, where 1,2,3-triazoles are the end product. As low as 5 ppm catalyst loading is enough to produce gram scale product, and highest turnover number (TON) and turnover frequency (TOF) values of 140 000 and 70 000 h−1 are achieved, respectively. Furthermore, this highly efficient protocol has been successfully applied to the preparation of diverse functionalized materials with pharmaceutical, labelling and supramolecular properties.
 Please wait while we load your content...
Please wait while we load your content...

