
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral binary chalcogen–nitrogen and ternary S,N,P molecules: new structures, bonding insights and potential applications
Tristram
Chivers
 *a and
Risto S.
Laitinen
*b
*a and
Risto S.
Laitinen
*b
aDepartment of Chemistry, University of Calgary, AB, Canada T2N 1N4. E-mail: chivers@ucalgary.ca
bLaboratory of Inorganic Chemistry, Environmental and Chemical Engineering, University of Oulu, P. O. Box 3000, 90014, Finland. E-mail: risto.laitinen@oulu.fi
First published on 21st April 2020
Abstract
Early theoretical and experimental investigations of inorganic sulfur–nitrogen compounds were dominated by (a) assessments of the purported aromatic character of cyclic, binary S,N molecules and ions, (b) the unpredictable reactions of the fascinating cage compound S4N4, and (c) the unique structure and properties of the conducting polymer (SN)x. In the last few years, in addition to unexpected developments in the chemistry of well-known sulfur nitrides, the emphasis of these studies has changed to include nitrogen-rich species formed under high pressures, as well as the selenium analogues of well-known S,N compounds. Novel applications have been established or predicted for many binary S/Se,N molecules, including their use for fingerprint detection, in optoelectronic devices, as high energy-density compounds or as hydrogen-storage materials. The purpose of this perspective is to evaluate critically these new aspects of the chemistry of neutral, binary chalcogen–nitrogen molecules and to suggest experimental approaches to the synthesis of target compounds. Recently identified ternary S,N,P compounds will also be considered in light of their isoelectronic relationship with binary S,N cations.
 Tristram Chivers | Tristram Chivers, a native of Bath, England, received his BSc, PhD (with Prof. R. D. Chambers FRS) and DSc degrees all from the University of Durham (UK). He joined the University of Calgary in 1969 and served as Head of the Chemistry Department from 1977 to 1982. He is currently Professor Emeritus of Chemistry. His research interests are in the area of main group element chemistry with an emphasis on chalcogen compounds and inorganic ring systems. He is the author of two books: “A Guide to Chalcogen–Nitrogen Chemistry” and (with I. Manners) “Inorganic Rings and Polymers of the p-Block Elements: From Fundamentals to Applications”. |
 Risto S. Laitinen | Risto S. Laitinen received his Ph. D. (eng) in 1982 from Helsinki University of Technology (Finland). He joined University of Oulu in 1988 and is currently Professor Emeritus of inorganic chemistry. He has been the Head of Chemistry Department in 1993–2013 and the head of Research Unit of Environmental and Chemical Engineering in 2017–2019. His research activities comprise experimental and computational main group chemistry, in particular chemistry of chalcogens. He has edited two books: “Selenium and Tellurium Chemistry: From Small Molecules to Biomolecules and Materials” (with J. D. Woollins) and “Selenium and Tellurium Reagents in Chemistry and Materials Science” (with R. Oilunkaniemi). |
1. Introduction
In contrast to the oxides of nitrogen NO, NO2, and N2Ox (x = 1, 3, 5), which all have acyclic structures, sulfur and nitrogen form a variety of neutral binary compounds with cyclic, polycyclic or polymeric structures.1 These include the intriguing, orange cage molecule E4N4 (1, E = S) with D2d symmetry,2a the colourless, planar four-membered ring E2N2 (2, E = S),2b and the blue-black polymer (SN)x (3),2c which becomes superconducting at 0.26 K. Less well-known are the red, six-membered ring 1,3-S4N2 (4),2d which adopts a half-chair conformation, and the explosive S5N6 (5),2e an orange compound with a cradle-like structure (Scheme 1). A labile –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SN– bridging unit is a common feature of both 4 and 5. The extremely hazardous selenium nitride Se4N4 (1, E = Se)3a has a cage structure similar to that of the sulfur analogue as does the mixed chalcogen nitride 1,5-Se2S2N4.3b The binary selenium nitride Se2N2 (2, E = Se) has not been identified as the free ligand, but both main-group metal and transition–metal complexes have been structurally characterised.3c,d The six-membered ring 1,3-Se4N2 (4, E = Se) is unknown,4 consistent with ab initio molecular orbital calculations for the SxSe4−xN2 ring systems. The stability of these ternary chalcogen nitrides is estimated to decrease with increasing selenium content and those that contain an –NSeN– fragment were found to be less stable than those that incorporate an –NSN– moiety.5
SN– bridging unit is a common feature of both 4 and 5. The extremely hazardous selenium nitride Se4N4 (1, E = Se)3a has a cage structure similar to that of the sulfur analogue as does the mixed chalcogen nitride 1,5-Se2S2N4.3b The binary selenium nitride Se2N2 (2, E = Se) has not been identified as the free ligand, but both main-group metal and transition–metal complexes have been structurally characterised.3c,d The six-membered ring 1,3-Se4N2 (4, E = Se) is unknown,4 consistent with ab initio molecular orbital calculations for the SxSe4−xN2 ring systems. The stability of these ternary chalcogen nitrides is estimated to decrease with increasing selenium content and those that contain an –NSeN– fragment were found to be less stable than those that incorporate an –NSN– moiety.5
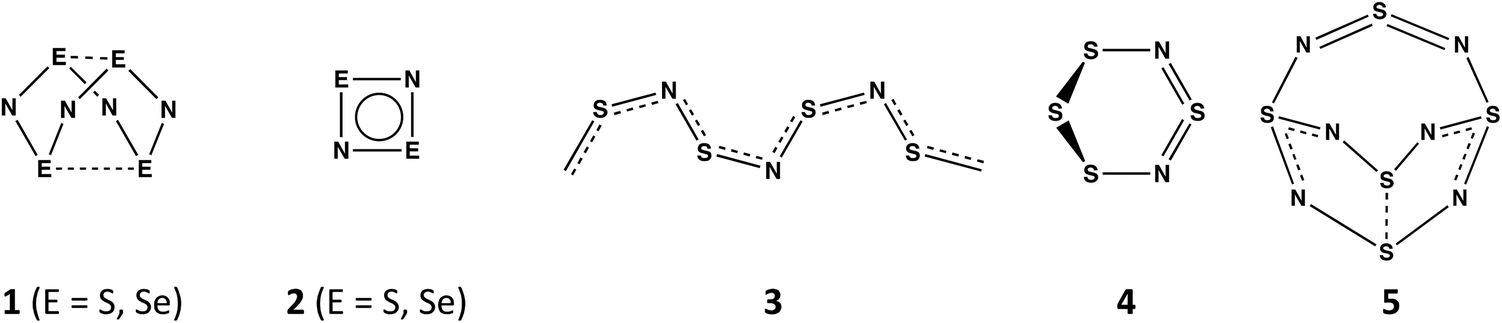 | ||
| Scheme 1 Structures of E4N4 (E = S, Se), E2N2 (E = S, Se), (SN)x, S4N2 and S5N6. | ||
Early studies of cyclic S,N species, including cations and anions as well as neutral molecules, focussed on the proposed aromatic character of planar ring systems as well as the properties and possible applications of the conducting polymer (SN)x. For example, the following species were considered to be aromatic on the basis of the Hückel 4n + 2π-electron rule: S2N2 (6π), S3N3− (10π), S4N3+ (10π) and S5N5+ (14π).1a A fuller understanding of (a) the nature of the transannular E⋯E interactions in E4N4 (1, E = S, Se) and related bicyclic sulfur–nitrogen molecules,6,7 (b) the electronic structures of the chalcogen nitrides E2N2 (2, E = S, Se), and (c) the mechanism of the topochemical polymerisation of S2N2 rings to generate the polymer (SN)x continues to occupy the attention of computational chemists.8 Furthermore, the serendipitous finding by Kelly and co-workers ca. 12 years ago that S2N2 vapour interacts with fingerprints on a glass vial to give a visible image of (SN)x has now reached the stage of potential commercial applications in forensic science.9 This development has generated impetus to the search for practical uses of other binary chalcogen–nitrogen species both known and unknown.
In the foregoing context nitrogen-rich chalcogen nitrides have been neglected, undoubtedly as a result of the difficulty in handling these energy-rich systems. However, recent experimental and computational studies of sulfur–nitrogen and selenium–nitrogen species under high pressure have revealed the formation or prediction of a number of novel binary chalcogen nitrides with unique structures and properties. In a similar vein optoelectronic or mechanical applications have been predicted for the as-yet-unknown sulfur-rich nitride S3N2.10 The focus of this perspective is an appraisal of recent experimental and computational studies of the structures and properties of both known and new binary chalcogen nitrides with an emphasis on the potential applications of these materials.11 The chemistry of the less well-studied selenium nitrides will be compared with that of sulfur nitrides throughout the discussion.
The article is organised by considering sulfur-rich nitrides SxN2 (x = 3, 4) initially, followed by new developments in the chemistry of molecules containing equal numbers of chalcogen and nitrogen atoms as exemplified by compounds 1–3. The treatise then focusses on the synthesis, unusual structures and predicted properties of nitrogen-rich chalcogen nitrides. Finally, the significance of the recent characterisation of both acyclic and cyclic S,N,P molecules12a,b will be evaluated in the context of the isoelectronic relationship of these ternary species with known binary S,N cations.1a,b
2. Sulfur-rich sulfur nitrides S3N2 and S4N2
The structures and properties of the sulfur–rich nitrides 1,3-S3N2 and 1,3-S4N2 (4) provide an interesting comparison. The putative five-membered ring cyclo-1,3-S3N2 is an antiaromatic (8π-electron) system, whereas the known six-membered ring cyclo-1,3-S4N2 is formally a 10π-electron molecule, which adopts a half-chair conformation in the solid state.2d The optimised geometries for 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 gave values of S–S and S–N bond lengths that are in close agreement with the experimental data.2d For example, the calculated S–S bond length is 2.090 Å,13cf. 2.061 Å from the X-ray structure.2d This congruence establishes confidence in the calculated S–S bond length of 2.302 Å for cyclo-1,3-S3N2.13 The elongated S–S bond is presumably a result of inherent strain in the five-membered ring, as suggested by the predicted value of the internal bond angle < NSS = 94.5°, cf. 105.2° in the six-membered ring 4.2d
13 gave values of S–S and S–N bond lengths that are in close agreement with the experimental data.2d For example, the calculated S–S bond length is 2.090 Å,13cf. 2.061 Å from the X-ray structure.2d This congruence establishes confidence in the calculated S–S bond length of 2.302 Å for cyclo-1,3-S3N2.13 The elongated S–S bond is presumably a result of inherent strain in the five-membered ring, as suggested by the predicted value of the internal bond angle < NSS = 94.5°, cf. 105.2° in the six-membered ring 4.2d
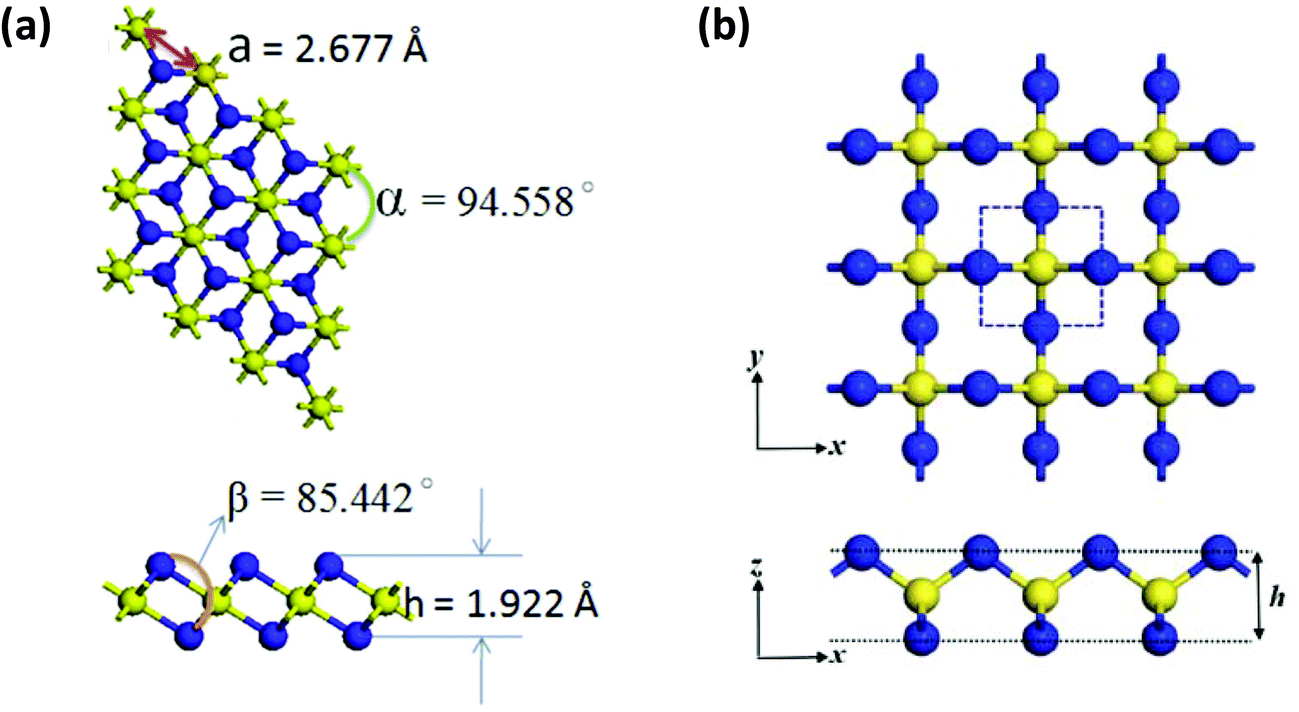
As an alternative to a cyclic structure, in 2018 Chen and co-workers have proposed a stable two-dimensional trisulfur dinitride with three polymorphs comprised solely of σ bonds.10a The crystal structure of the most stable form, α-S3N2 (space group Pmn21), consists of condensed 12-membered (S6N6) rings (Fig. 1). The S3N2 crystal is predicted to be dynamically, thermally and chemically stable on the basis of the computed phonon spectrum and ab initio molecular dynamics simulations. The predicted “chemical stability” is surprising in view of the known susceptibility of sulfur–nitrogen compounds to degradation by both nucleophiles and electrophiles.1a,b Calculations also indicate that α-S3N2 will be a wide, direct band gap (3.92 eV) semiconductor with possible optoelectronic applications such as blue or UV light-emitting diodes and photodetectors.
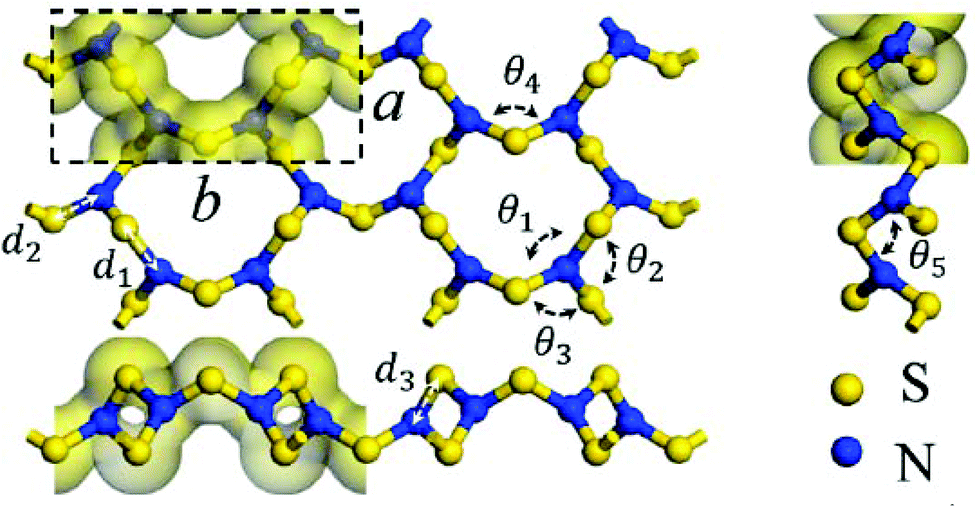 | ||
| Fig. 1 The PBE-predicted 2D crystal structure of α-S3N2: a = 4.24, b = 8.89 Å; d1 = 1.81, d2 = 1.72, d3 = 1.66 Å; θ1 = 116.8°, θ2 = 119.3°, θ3 = 119.2°, θ4 = 106.1°, θ5 = 103.7°.10a Bonding is depicted by an isosurface of the electron density.10a (Reproduced with permission from H. Xiao, X. Shi, X. Liao, Y. Zhang and X. Chen, Phys. Rev. Mater., 2018, 2, 024002. @ 2018 American Physical Society.) | ||
A subsequent study of 2D monolayer crystals of E3N2 (E = S, Se) employing DFT calculations predicted α-heart and β-heart structures, which exhibit different mechanical and electrical properties.10b The former structure resembles that depicted for α-S3N2 in Fig. 1. The E3N2 materials are predicted to have unusual mechanical properties as indicated by the values of Negative Poisson's Ratios indicating auxetic behaviour.10c
The recent computational predictions of the unique structure and mechanical properties of S3N2 provide a strong incentive for pursuing experimental approaches to this unknown binary sulfur nitride. A carefully controlled cyclocondensation reaction of ClSSCl with Me3SiNSNSiMe3 under high-dilution conditions is a plausible first attempt.14 An alternative route is the one-electron chemical or electrochemical reduction of the known radical cation [S3N2]˙+, which is prepared by oxidation of S4N4 with AsF5 in liquid SO2.1a The reaction of 1,3-S4N2 (4) with one equivalent of PPh3 is likely to result in attack of the nucleophile at the S(IV) centre rather than the required removal of a single sulfur atom from the –S–S–S– unit.
3. Molecules with equal numbers of chalcogen and nitrogen atoms
3.1. Tetrasulfur and tetraselenium tetranitride E4N4 (1, E = S, Se)
The S4N4 (1, E = S) molecule is regarded as the quintessential binary sulfur–nitrogen compound in view of its multifarious applications as a reagent in inorganic and organic chemistry, despite a tendency to detonate under the influence of heat or friction.1a Notwithstanding the availability of simple synthetic procedures, the selenium analogue 1 (E = Se) has undergone limited use for the preparation of selenium–nitrogen compounds owing to the explosive nature of this reagent.1a,b Some new aspects of the chemical reactivity of 1 (E = S), including the electrochemical reduction and photochemical behaviour, are discussed in a recent book chapter.1b This section will consider more recent developments in the chemistry of S4N4, especially those involving an understanding of the electronic structure and unusual reaction chemistry of this cage compound.A different aspect of the bonding interactions in 1 (E = S) was reported in 2016 by Jenkins and co-workers.17 By applying QTAIM (quantum theory of atoms in molecules) methodology to all 18 modes of vibration of the cage molecule, these authors found a considerable degree of metallicity in the S–S and S–N bonding interactions, on the basis of the delocalisation of the electron density away from the atoms. In particular, considerable metallic behaviour was apparent in the S–S bond critical points for all modes.17 The experimental implications of this finding are yet to be investigated.
A convenient synthesis of S4N4 involves the cyclocondensation reaction of [(Me3Si)2N]2S with an equimolar mixture of SCl2 and SO2Cl2.21 The complexity of the reaction pathway for this transformation is highlighted on the cover page of a recent textbook with a focus on “arrow-pushing” in inorganic chemistry.22 Ghosh and Berg propose a twelve-step process with the sequential participation of the reagents SCl2 followed by SO2Cl2. However, the combination of SCl2 and SO2Cl2 generates SCl4 (+SO2) spontaneously, so a cyclocondensation pathway involving this in situ reagent is more likely. The formation of an acyclic –SNSNS– intermediate and, subsequently, an eight-membered S4N4 ring via a series of thermodynamically favoured eliminations of Me3SiCl is suggested (Scheme 2).
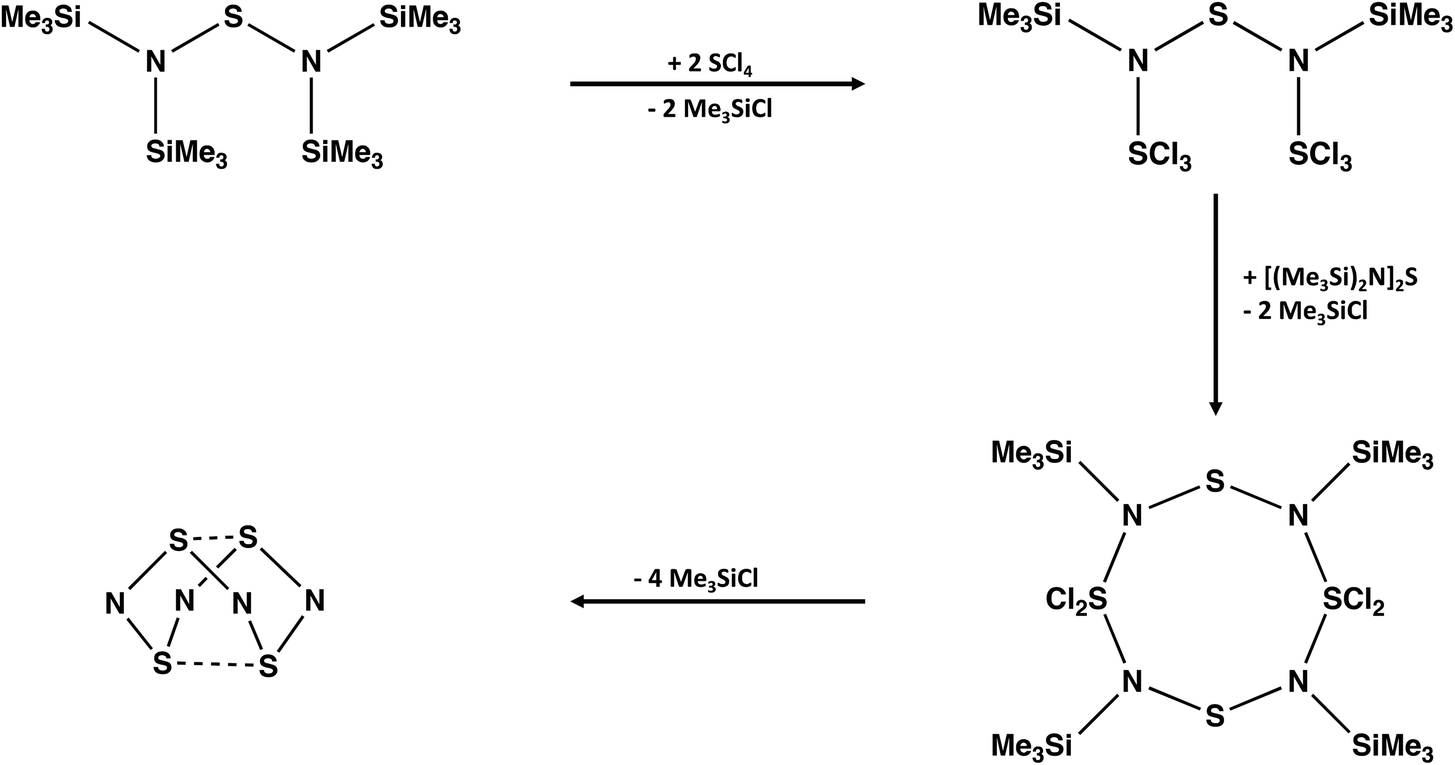 | ||
| Scheme 2 Formation of S4N4via cyclocondensation of [(Me3Si)2N]2S with SCl4. | ||
Kumar and Muralidharan have reported the hexamethyldisilazane (HMDS)-assisted synthesis of β-In2S3, CuS and Ag2S nanoparticles via reactions of sulfur with an appropriate metal salt in the presence of HMDS.23 In order to explain the role of HMDS in the synthesis of these metal sulfides, the formation of a novel polymeric intermediate [SN(SiMe3)]n, was invoked.23c,d The characterization of this purported polymer was based upon GPC (gel permeation chromatography) analysis of the distilled material, as well as 29Si NMR spectra and HRMS (high resolution mass spectrometry) data.23a The fact that this intermediate could not be isolated as a pure compound was attributed by the authors to the similarity of its boiling point with that of HMDS. Since the boiling point of HMDS is only 125 °C, this explanation is highly unlikely. Orange crystals obtained as a minor by-product in the synthesis of Ag2S were identified by XRD as S4N4 as a minor by-product;23b however, the formation of this binary sulfur nitride from the purported sulfur imide polymer is not readily explained.
3.2. Disulfur and diselenium dinitride E2N2 (2, E = S, Se)
In contrast to the numerous applications of S4N4 as a source of novel inorganic and organic sulfur–nitrogen compounds,1a,b the use of S2N2 in synthesis is rare owing to its highly labile nature. However, both electrochemical and chemical reduction produce the [S3N3]− anion, which can be isolated as ion-separated salts.1b,24 This section will consider recent developments in our understanding of the chemistry of S2N2 and its heavier chalcogen homologues, including electronic structures, spectroscopic characterisation, the isomers formed upon photolysis, topochemical polymerization to give (SN)x, and possible use as a hydrogen-storage material.In 2004 Head-Gordon et al. concluded that the diradical character of S2N2 is not significant on the basis of a large HOMO–LUMO gap (5.1 eV), a large singlet–triplet vertical transition energy (3.6 eV), and the lack of a spin-unrestricted solution at the level of DFT using the B3LYP functional.26 They asserted, however, that S2N2 is aromatic based on its negative NICS value (−26.2 ppm), albeit with a small aromatic stabilisation energy, since only two of the six π electrons contribute to the bonding.
Concurrently, Tuononen and co-workers investigated the electronic structures and molecular properties of S2N2,27a as well as the unknown heavier chalcogen analogues Se2N2, SSeN2 and Te2N2,27b by employing various computational methods. These authors agreed that all of these four-membered rings can be described as 2π-electron aromatics, but with minor singlet diradical character ranging from 6% in S2N2 to 10% Te2N2 located solely on the two nitrogen atoms.27b
In 2012 Braida et al. carried out an ab initio valence bond study of E2N2 which confirmed that the diradical sites are the two nitrogen atoms (E = Se, Te), however they estimated that the contribution from diradical structures is close to 50%.28 Their calculations of vertical resonance energies gave values of about 80% of the predicted value of benzene for S2N2 with somewhat lower values for Se2N2 and Te2N2, consistent with the notion of aromatic character for S2N2 based on magnetic criteria. An important conclusion from these results is that diradical character and aromaticity are not mutually exclusive.28 In a sequel to his earlier work on valence bond structures for S2N2, Harcourt29 postulated that the six Lewis structures assumed by Braida et al.28 are equivalent to resonance between two increased-valence structures with one-electron and fractional electron-pair π bonds.
In 2018 Karadakov et al. employed magnetic criteria to study the bonding and aromaticity in the ground, first singlet excited and lowest triplet electronic states of S2N2.25 Their results indicate that (a) the S0 electronic state is aromatic, but less so than the electronic ground state of benzene, (b) S1 is strongly antiaromatic, and (c) T1 is moderately antiaromatic to a similar extent to that observed in the electronic ground state of square cyclobutadiene. Thus, they assert that S2N2 is the first example of an inorganic ring for which theory predicts substantial changes in aromaticity upon vertical transition from the ground state to the first singlet or lowest triplet electronic states.25
The calculated IR-active vibrational frequencies27b are in reasonable agreement with experimental data from the IR and Raman spectra of S2N2 obtained as a solid condensate and in N2 or CH4 matrices at 15–35 K, which were reported by Downs et al.30 By using 30% 15N-enrichment, these authors confirmed that the isolated S2N2 molecule has essentially the same square-planar geometry as the crystalline solid; calculations of the S–N stretching force constant indicated a bond order only slightly greater than 1.30 In 2014 the high-resolution gas-phase FTIR spectrum of S2N2 together with a re-investigation of the IR spectrum in solid argon at 16 K were reported by Perrin, Beckers and co-workers.31 The main difference between the gas-phase and solid-state structures of S2N2 involves the bond angles; in the gas phase the angle at nitrogen <SNS is smaller than <NSN, whereas the opposite is true in the solid-state structures.31 These small disparities may be the consequence of the intermolecular interactions that occur between S2N2 rings in the plane of the chain of rings in the solid state. Cremer et al. have computed a pseudorotation barrier of <2 kJ mol−1 for S2N2 indicative of a floppy molecule, however these authors maintain that this characteristic does not alter the electronic structure of the four-membered ring.32
The predicted IR and Raman spectra of the selenium-containing systems Se2N2 and SSeN2 are depicted in Fig. 2.27b The band at ∼590 cm−1 in the IR spectrum of Se2N2 should be considerably less intense than the other two bands. In a similar vein, the intensity of the band at 822 cm−1 is estimated to be more than twice those of the other two bands.
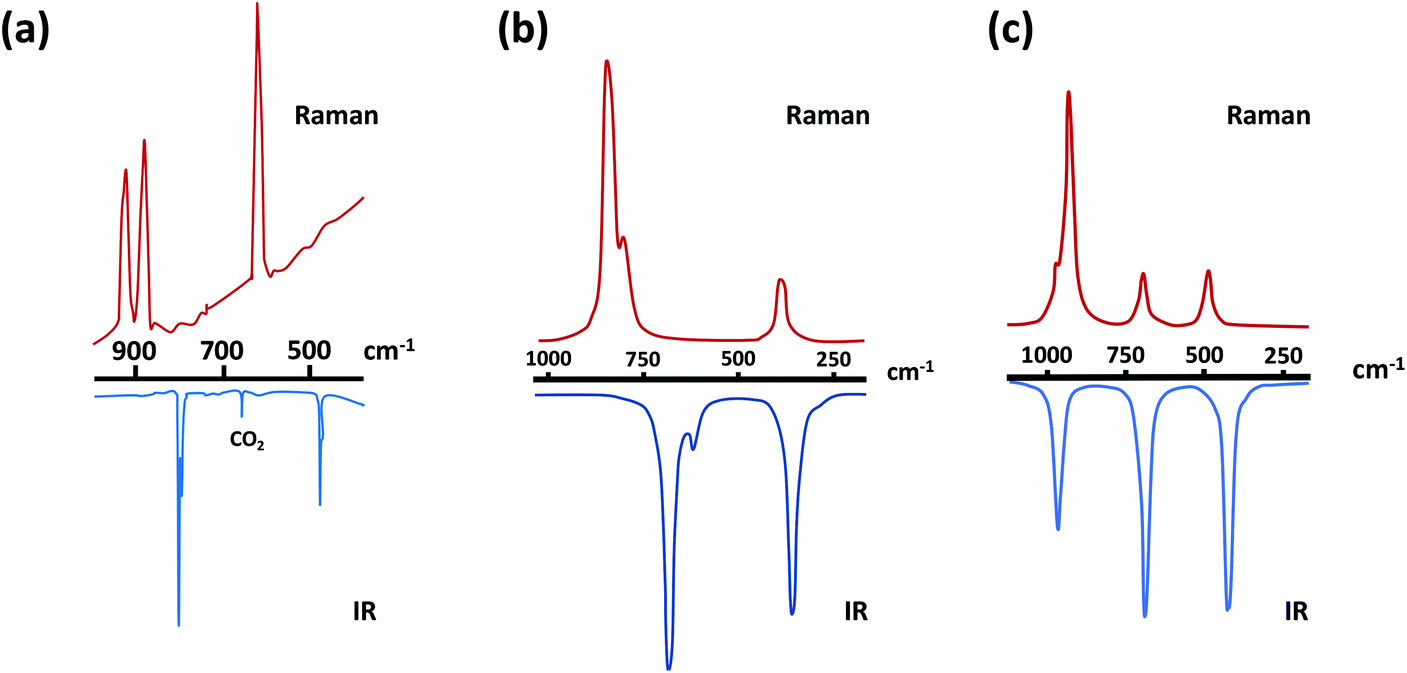 | ||
| Fig. 2 (a) IR/Raman spectra of S2N2.30 (Adapted with permission from R. Evans, A. J. Downs, R. Köppe and S. C. Peake, J. Phys. Chem. A, 2011, 115, 5127–5137. @ 2011 American Chemical Society), and the theoretically predicted IR/Raman spectra of (b) and Se2N2 and (c) SeSN227b (adapted with permission from H. M. Tuononen, R. Suontamo, J. Valkonen and R. S. Laitinen, J. Phys. Chem. A, 2005, 109, 6309–6317. @ 2005 American Chemical Society). | ||
Tuononen et al., have also calculated the 14N, 15N and 77Se NMR chemical shifts for the chalcogen nitrides S2N2, SeSN2, Se2N2, S4N4 and Se2S2N4.27b Surprisingly, no experimental NMR data are available for S2N2. However, the calculated 14N and 15N NMR chemical shifts for S4N4 and the known mixed selenium–sulfur nitride Se2S2N4 are in excellent agreement with the experimental data3c providing some confidence in the predicted values of 95 ± 20, 125 ± 20 and 185 ± 20 ppm for S2N2, SeSN2 and Se2N2, respectively. The calculated 77Se chemical shifts for 1,5-Se2S2N4 at different levels of theory are ca. 200 ppm lower than experimental values.3c,27b By contrast, the calculated PBEPE and CAS 77Se chemical shifts of 1906 and 1886 ppm for Se42+ are only slightly lower than the experimental value of 1936 ppm. Assuming a similar error for SeSN2 and Se2N2, 77Se chemical shifts of 1820 ± 20 and 1780 ± 20 ppm, respectively, are estimated for these structurally analogous four-membered rings.27b The ternary chalcogen nitride SeSN2 has been proposed as an intermediate in the formation of 1,5-Se2S2N4 from reactions of (Me3SiNSN)2E (E = S, Se) with E′Cl2 (E′ = S, Se),3c but this potential precursor of a mixed sulfur–selenium nitride polymer (SNSeN)x has eluded isolation.
The experimental UV spectrum of S2N2 consists of a broad band in the range 4.5–5.8 eV comprised of two overlapping absorptions centred at approximately 5.0 eV (∼250 nm) which is attributed to the π → π* electronic transition.33 The calculated band-gap energies decrease as the selenium content in the ring increases as illustrated by colourless S2N2 (3.32 eV, ∼375 nm) the predicted orange-yellow colour of SSeN2 (2.50 eV, ∼495 nm) and red colour of Se2N2 (2.20 eV, ∼560 nm).27b This information should be valuable for the identification of the selenium-containing four-membered rings.
 | ||
| Scheme 3 Structural arrangements of planar cyclic and acyclic isomers of S2N2.34 | ||
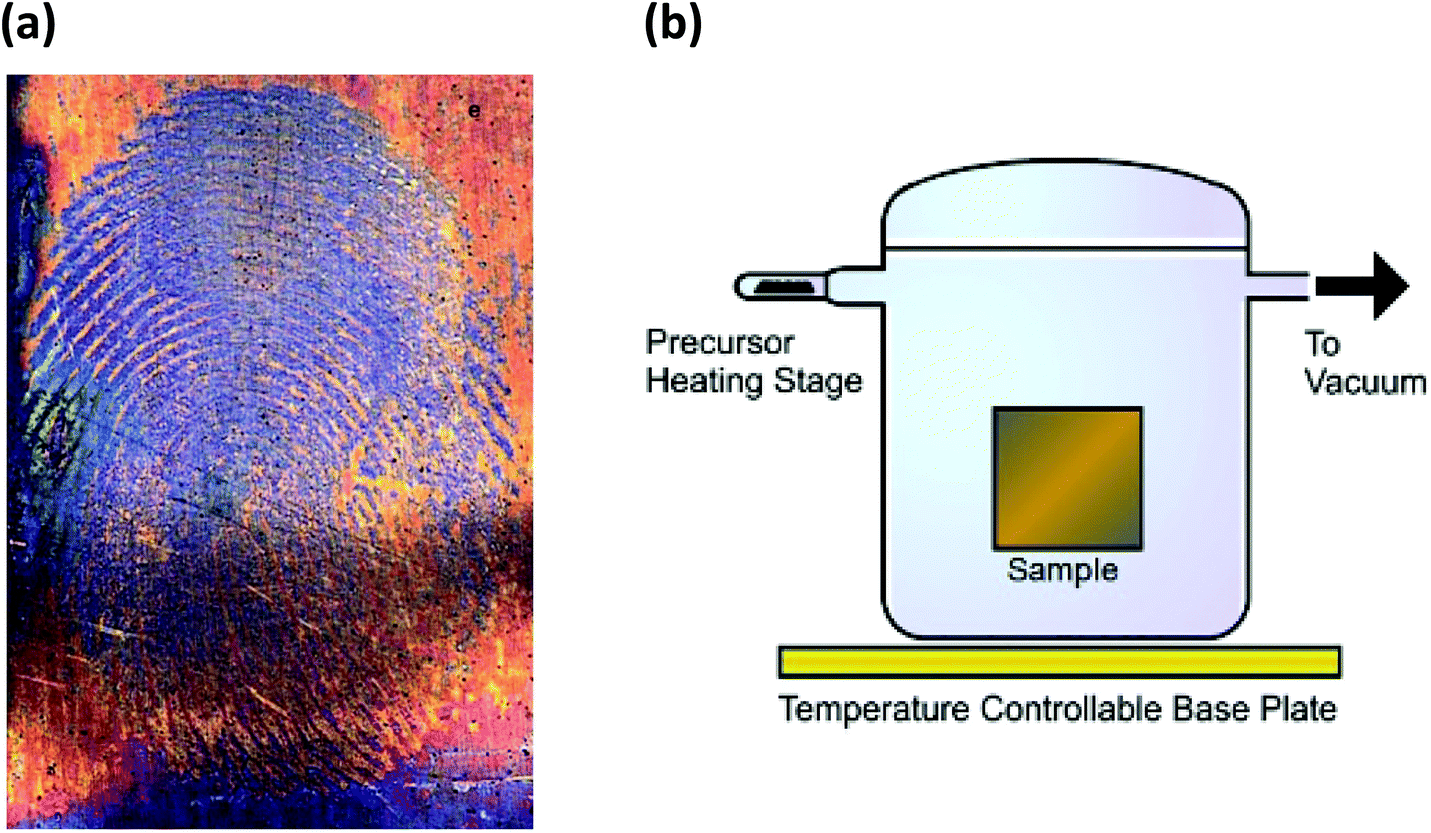 | ||
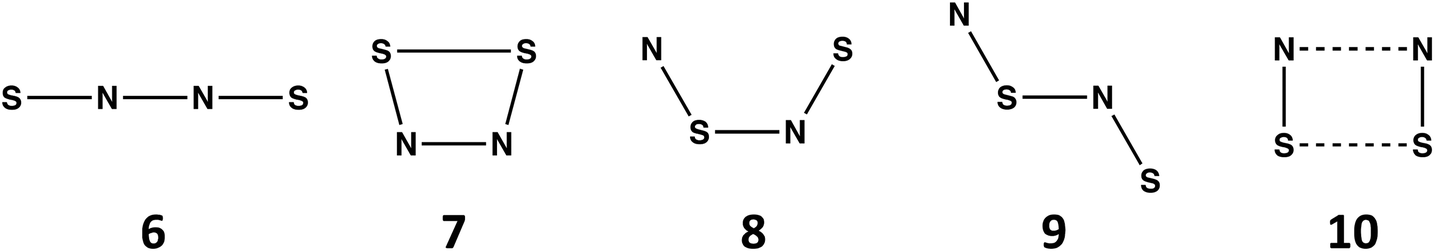
| Fig. 3 (a) Formation of (SN)x over latent fingermarks on metallic surfaces9c (reproduced with permission from S. M. Bleay, P. F. Kelly, R. S. B. King, J. Mater. Chem., 2010, 20, 10100–10102. @ 2010 The Royal Society of Chemistry.). (b) Diagram of S2N2 processing equipment9d (reprinted with permission from S. M. Bleay, P. F. Kelly, R. S. P. King and S. G. Thorngate, Sci. Justice, 2019, 59, 606–621. @ 2019 Elsevier B. V.). | ||
In 2013 Takaluoma et al. carried out molecular dynamics simulations of the topochemical polymerisation of S2N2 in the solid state using DFT methods and periodic functions.8 The calculations involved both high pressures and slightly elevated temperatures in order to increase the reaction rate. The formation of (SN)x is initiated by cleavage of one S–N bond in S2N2 followed by a very rapid attack of the resulting open-chain isomer, e.g.8 in Scheme 3, on a neighbouring ring (Fig. 4). Propagation occurs along the a axis throughout the lattice. The packing changes from the herringbone structure of the S2N2 lattice to the layered structure of (SN)x. Although the metrical data for the polymer chain are in good agreement with the experimental crystal structure, there is less long-range order between neighbouring chains, possibly due to the influence of pressure or the poor description of dispersion by density functional theory.8
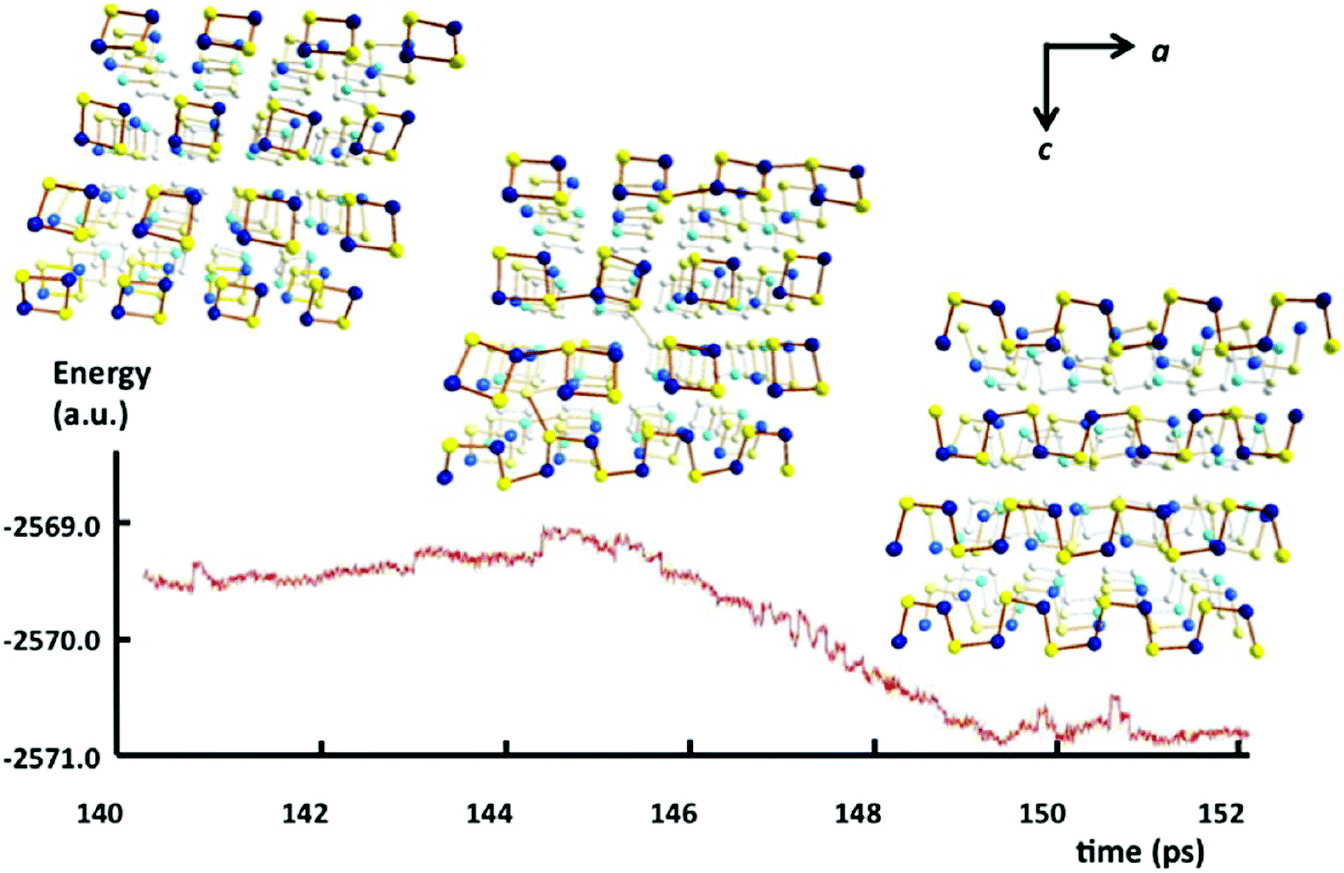 | ||
| Fig. 4 Simulated energy profile of the topochemical polymerisation of S2N2 rings to (SN)x at 50 GPa and 600 K. Sulfur atoms are indicated by yellow spheres and nitrogen atoms by blue spheres (b axis is directed away from the reader).8 (Reproduced with permission from T. T. Takaluoma, K. Laasonen and R. S. Laitinen, Inorg. Chem., 2013, 52, 4648–4657. @ 2013 American Chemical Society.) | ||
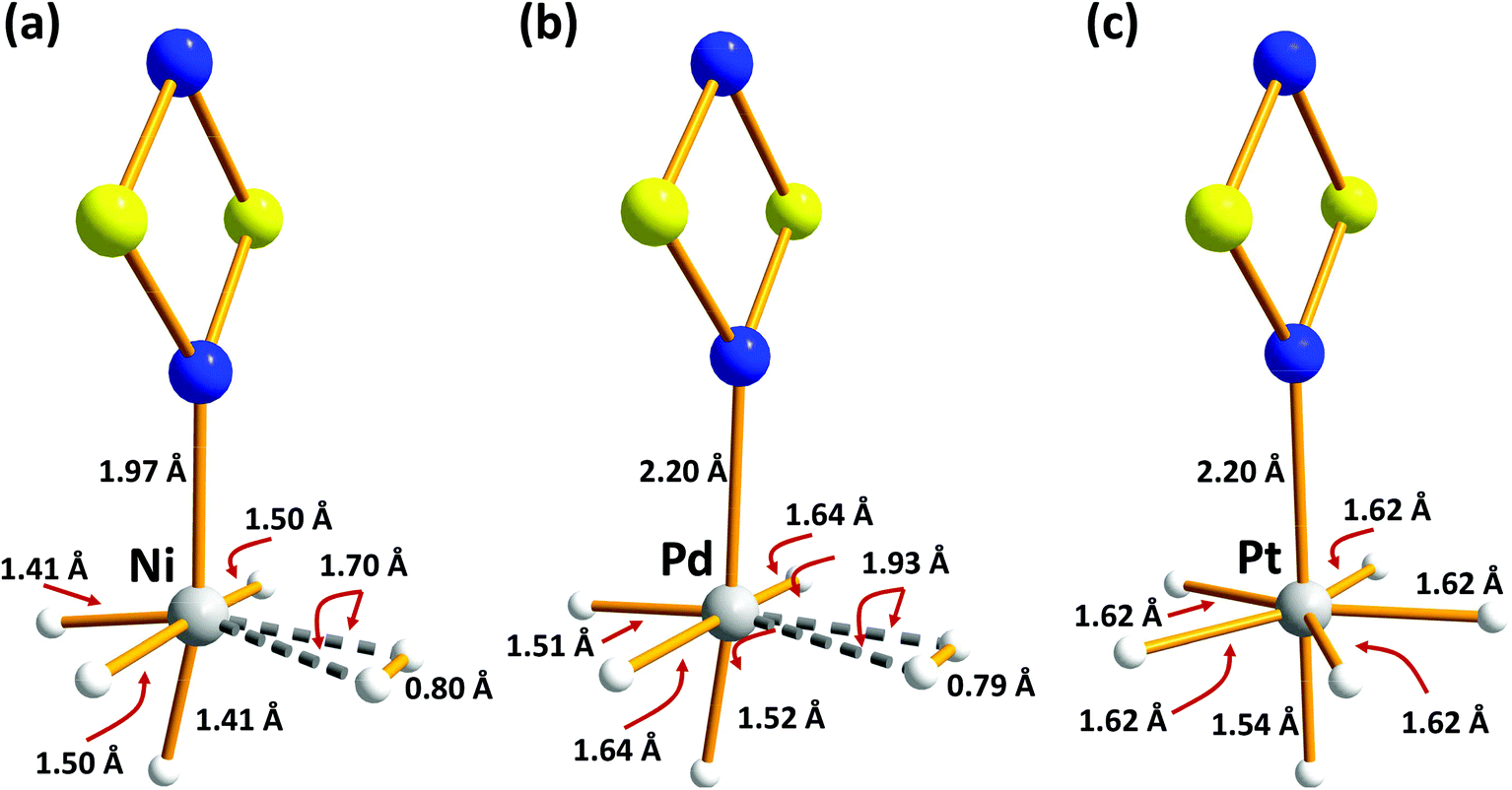 | ||
| Fig. 5 B3LYP/6-31G(d,p) optimised structures of H2-absorbed metal-doped S2N2 (a) M = Ni (b) M = Pd (c) M = Pt (redrawn from computational data in ref. 35). | ||
4. Nitrogen-rich chalcogen nitrides
Early work on nitrogen-rich sulfur nitrides such as SN2 and SN4 was mainly limited to the purported involvement of these species as fleeting intermediates. However, recent experimental and theoretical investigations of the formation and structures of this class of sulfur nitrides and their selenium analogues under high pressure have revealed fascinating properties with potential applications in optoelectronics and as high-energy-density materials. This section is organised according to increasing nitrogen content of these binary molecular species. Nitrogen-rich selenium nitrides are discussed in section 4.4.4.1. The isomers NNS and NSN
The lowest energy isomer NNS, a heavier analogue of the stable molecule N2O, decomposes above 160 K. It is generated by flash photolysis of 5-phenyl-1,2,3,4-thiatriazole and has been characterised by high-resolution mass spectrometry and IR spectroscopy.36 The symmetrical isomer NSN has been invoked as an elimination product during the transformations of polycyclic S–N compounds,1a but the synthesis and structure of this simple sulfur nitride remained elusive until very recently (vide infra).In 2016 Zhang and co-workers investigated the stability, electronic structure and optical properties of a P3m1 phase of 1T-EN2 (E = S, Se, Te) by means of DFT calculations (the prefix 1T refers to one layer per trigonal unit cell).37 The calculated crystal structure of the two-dimensional material 1T-SN2 is comprised of six-coordinated S atoms and three-coordinate N centres (Fig. 6a). These chalcogen nitrides 1T-EN2 (E = S, Se, Te) are endothermic materials. 1T-SN2 has dynamical stability based on phonon spectra and calculated cohesive energies, but the thermal and mechanical stabilities have yet to be determined. Band-structure calculations revealed indirect band gaps of 2.825, 2.351, and 2.336 eV for 1T-SN2, 1T-SeN2, and 1T-TeN2, respectively. Furthermore, the application of biaxial strain induced a transition from a wide-band-gap semiconductor to a metal. These findings suggest that the binary chalcogen nitrides 1T-EN2 are promising materials for applications in optoelectronic devices.37
 | ||
| Fig. 6 Optimised structures of (a) 1T-SN237 (reproduced with permission from J.-H. Lin, H. Zhang, X.-L. Cheng and Y. Miyamoto, Phys. Rev. B, 2016, 94, 195404. @ 2016 American Physical Society), and (b) S-SN238 (reproduced with permission from F. Li, X. Lv, J. Gu, K. Tu, J. Gong, P. Jin and Z. Chen, Nanoscale, 2020, 12, 85–92. @ 2020 The Royal Society of Chemistry). | ||
Earlier this year Chen and co-workers proposed a new phase of the SN2 monolayer, namely S-SN2, by using first-principles calculations combined with the particle-swarm optimisation method.38 The structure of S-SN2 belongs to the space group P4M2 and incorporates four-coordinate S atoms and two-coordinate nitrogen centres (Fig. 6b); the prefix S is used to indicate its square lattice and, hence, distinguish this new structure from 1T-SN2. The S-SN2 monolayer is a remarkably stable semi-conductor with an indirect band gap of 2.79 eV, cf. 2.825 eV for 1T-SN2.37 The co-existence of both out-of-plane and in-plane negative Poisson's ratios indicates 3D auxetic properties for this new material,10c which may have applications in mechanical or optoelectronics devices.
In 2017 Cui and co-workers performed particle-swarm optimisation calculations on the sulfur–nitrogen system up to 200 GPa.39 Three binary sulfur–nitrogen solids were determined to be thermodynamically stable: Pnma (SN)x, Pnnm SN2, and C2/c SN4 at pressures of 37, 43 and 48 GPa, respectively. Their crystal structures are depicted in Fig. 7. A polymeric 3D arrangement was predicted for SN2 (Fig. 8c). Partial density of states calculations indicated that SN2 is a semiconductor with a direct band gap of 0.66 eV at 60 GPa.
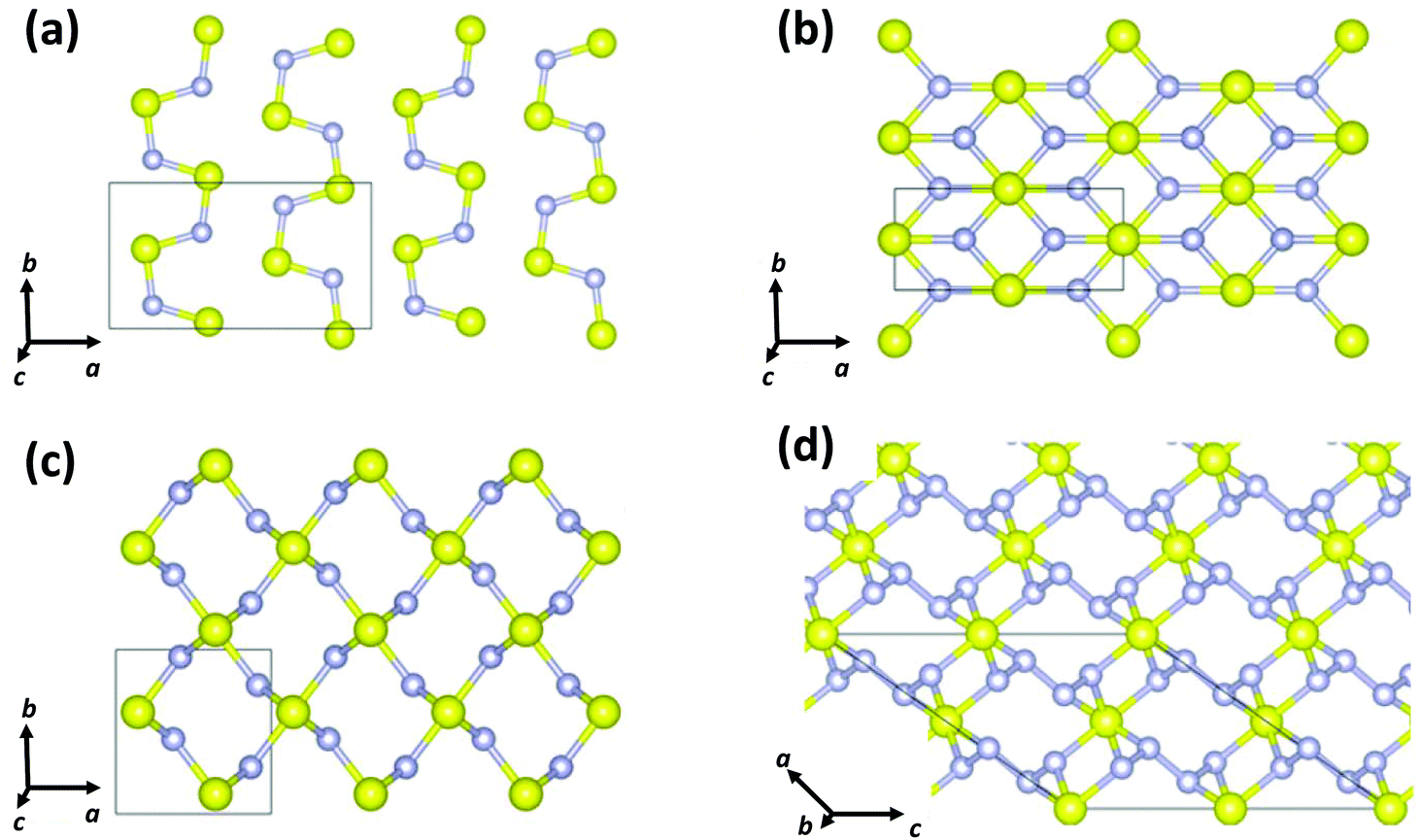 | ||
| Fig. 7 Crystal structures of (a) Pnma (SN)x, (b) Immm (SN)x, (c) Pnnm SN2, and (d) C2/c SN4.39 (Reproduced with permission from D. Li, F. Tian, Y. Z. Lv, S. Wei. D. Duan, B. Liu and T. Cui, J. Phys. Chem. C, 2017, 121, 1515–1520. @ 2017 The American Chemical Society.) | ||
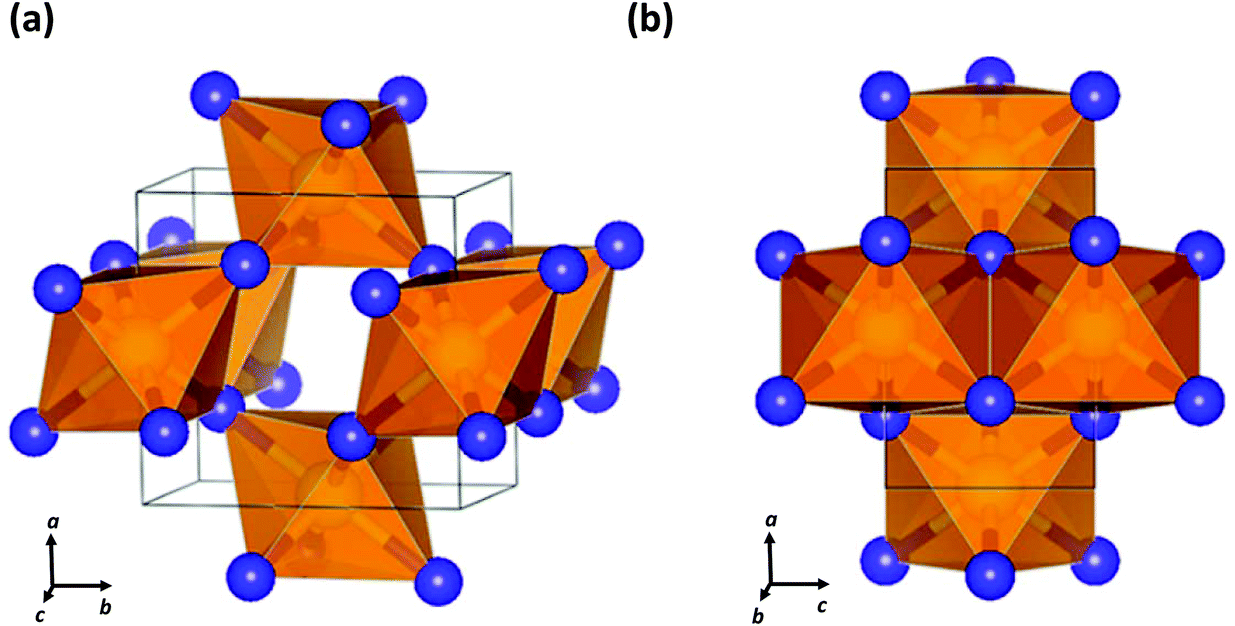 | ||
| Fig. 8 Crystal structure of SN2 formed at 81.6 GPa showing (a) the cross-linked octahedra and (b) the edge-sharing octahedra and apical S–N bonds.40 (Reproduced with permission from D. Laniel, M. Bykov, T. Fedotenko, A. V. Ponomareva, I. A. Abrikosov, K. Glazyrin, V. Svitlyk, L. Dubrovinsky and N. Dubrovinskaia, Inorg. Chem., 2019, 58, 9195–9204. @ 2019 The American Chemical Society.) | ||
Last year a seminal experimental investigation of the formation of sulfur nitrides in the S–N2 system at high pressures by Laniel and co-workers confirmed the predictions for SN2. These authors found that the reaction of elemental sulfur and N2 gas in a diamond anvil cell at pressures above 60 GPa with laser heating produces an orthorhombic (Pnnm) SN2 compound.40 The lattice parameters of this material obtained from powder diffraction data match closely the theoretically predicted crystalline structure of SN2.37 Single crystal X-ray diffraction analysis revealed a CaCl2-type structure composed of edge-sharing SN6 octahedra with N atoms that are triply coordinated by S atoms (Fig. 8).40 The SN2 solid was found to be metastable down to ca. 20 GPa, at which point it dissociates into its elements.40
4.2. Thiatetrazole SN4
The nitrogen-rich thiatetrazole SN4 has attracted the attention of both experimental and theoretical chemists for more than 20 years. Klapötke and Schulz attempted to prepare SN4 from the reaction of the cation [NS]+ in [NS][AsF6] with the azide ion [N3]− in cesium azide. The final product was a black precipitate identified as the polymer (SN)x; no experimental evidence (14N NMR) could be found for the expected SN4 intermediate.41 Furthermore, no spectroscopic evidence for the formation of SN4 was garnered from the recent study of the S–N2 system at high pressures, which led to the characterisation of metastable SN2 (section 4.1).40The main focus of early theoretical investigations of SN4 was an estimate of the aromaticity of the cyclic isomer, which is a 6π-electron five-membered ring formally related to thiophene by the replacement of four CH units by four nitrogen atoms. Schleyer and co-workers calculated NICS (nucleus independent chemical shift) and NICS(1) values of −18.40 and −17.48 for cyclo-SN4; the large negative values are indicative of substantial aromatic character in the ring.42a This conclusion is supported by the calculated value of 18.56 for the ASE (aromatic stabilisation energy).42b
Nitrogen-rich sulfur nitrides are of considerable interest as high energy-density materials. The energy content of cyclo-SN4 was first computed by Wang and co-workers using DFT methods at the B3LYP/6-311+G** level.43 It was found that cyclo-SN4 is ca. 100 kcal mol−1 higher in energy than the decomposition products 1/8S8 + N2. However, the low dissociation barrier of 7.0 kcal mol−1 for the decomposition of SN4 into N2 + N2S (Scheme 4) precludes the isolation of this sulfur nitride under ambient conditions.44
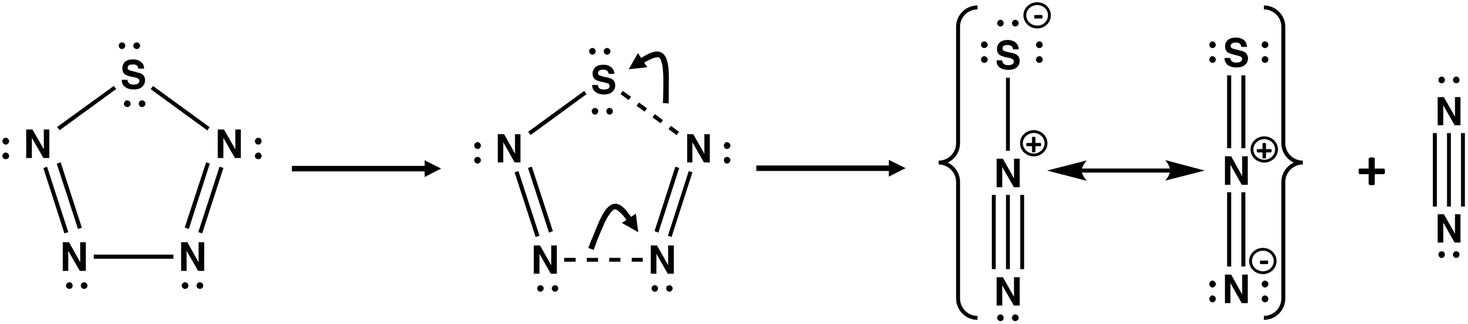 | ||
| Scheme 4 The most facile dissociation pathway for cyclo-SN4.44 | ||
4.3. Dithiatetrazine S2N4
A planar six-membered ring cyclo-S2N4 would be an 8π-electron (antiaromatic) system isoelectronic with dithiatriazines (RC)S2N3. The parent dithiatriazine (R = H) is not known, but aryl derivatives (R = Ph) exist as cofacial dimers in the solid state.1a Numerous acyclic and cyclic isomers are possible for the putative nitrogen-rich sulfur nitride S2N4. Schleyer and co-workers have carried out computational investigations of the thermochemical stability and kinetic persistence of various S2N4 isomers at the B3LYP/6-311+G(3df) DFT level.13 The results were compared with the corresponding data for the known sulfur nitrides S2N2, S4N2 and S4N4, as well as the nitrogen-rich species SN4 and S3N4. A boat conformation of 1,4-S2N4 (11), cf. 1,4-S2(CH)4, was predicted to be the most stable of the three possible six-membered rings. The other most thermodynamically stable isomers include two five-membered rings SN4(S) (12 and 13) and two acyclic species SNSNNN (14 and 15) (Scheme 5). From a consideration of the kinetic stability (dissociation barriers) of these five isomers it was concluded that the acyclic isomer 14 is the most viable candidate for synthesis. However, its dissociation barrier to give N2 and cyclo-S2N2 is only 21.6 kcal mol−1, cf. 51 kcal mol−1 for cyclo-S2N2 (2, E = S). Although this barrier is considerably higher than the value of 7 kcal mol−1 determined for the decomposition of cyclo-SN4 (section 4.2), the facile formation of S2N2 from decomposition of SNSNNN (14) may account for the rapid production of (SN)x observed in the reaction of [SNS][AsF6] with CsN3 in SO2(l) at −20 °C.45
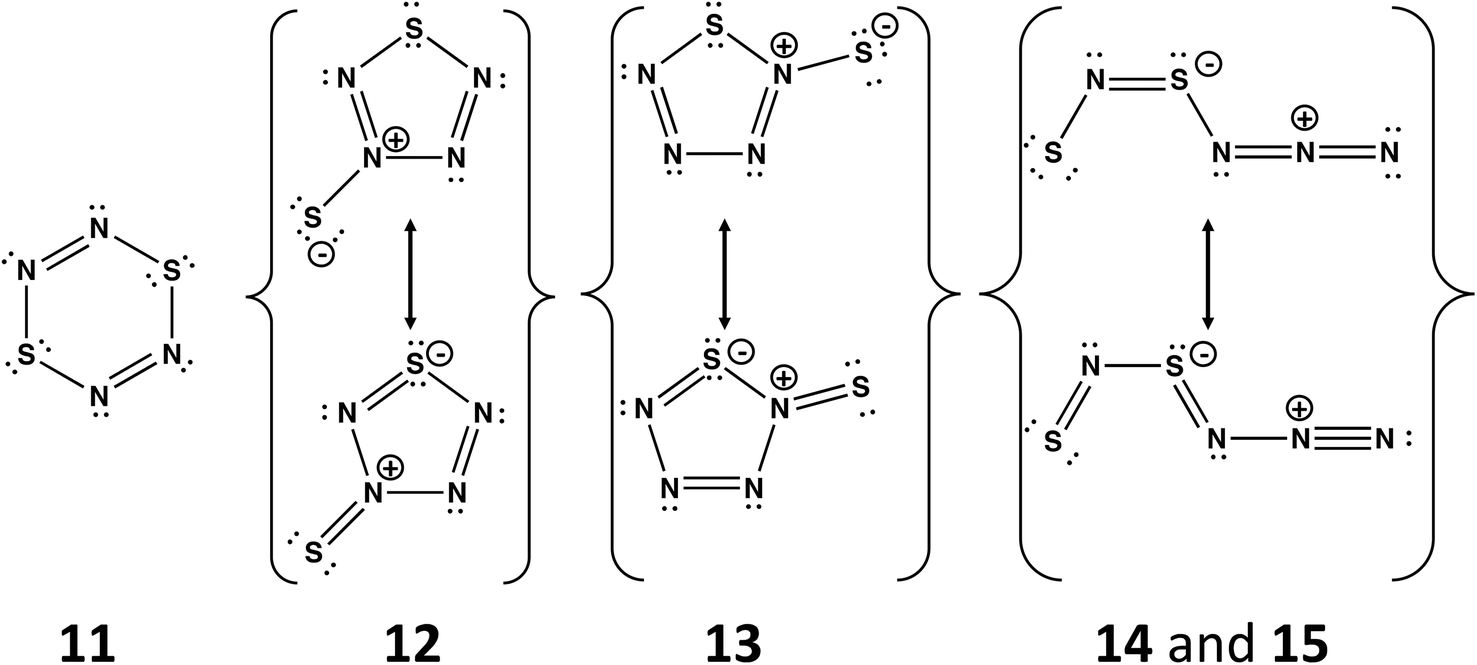 | ||
| Scheme 5 Structures of the five most stable isomers of S2N4. The acyclic isomers 14 and 15 differ only in the conformation; the –NNN fragment in 14 is oriented above the SNSN– plane, whereas 15 is planar.13 | ||
As a general conclusion, it is important to note that kinetic stability is more important than thermodynamic stability in determining which binary sulfur–nitrogen species can be isolated under ambient conditions. The isomers of the nitrogen-rich cation [S2N3]+ a 6π-electron system, provide a compelling example of this principle. Despite being less thermodynamically stable than the 1,3-isomer, the 1,2-isomer has been isolated and structurally characterised in the salt [S2N3]2[Hg2Cl6].46 Although it is the only known monocyclic, nitrogen-rich S,N cation, several salts of the tricyclic cation [S4N5]+ have been structurally characterised.47 The relative stability of 1,2-[S2N3]+ is attributed to the higher dissociation barriers for its fragmentation compared to the 1,3-isomer for which the dissociation pathway to form [SNS]+ + N2 has a barrier of only 2.6 kcal mol−1.44a
4.4. Trithiatetrazepine S3N4
Planar, cyclic S3N4 (16) would be a 10π-electron (aromatic) system (see Scheme 6)48 isoelectronic with the known trithiatriazepine (HC)S3N3.49 However, the isolation of 16 at room temperature is unlikely in view the low energy barrier (14.14 kcal mol−1) for dissociation into SNN and S2N2.13 The known compound S3N3–NPPh3 (17) formed from the reaction of S4N4 with PPh3via ring contraction can be considered as the triphenylphosphine adduct of S3N4.50 It is also noted that the related derivative S3N3–NS has been identified as a short-lived intermediate generated upon flash photolyis of S4N4.51
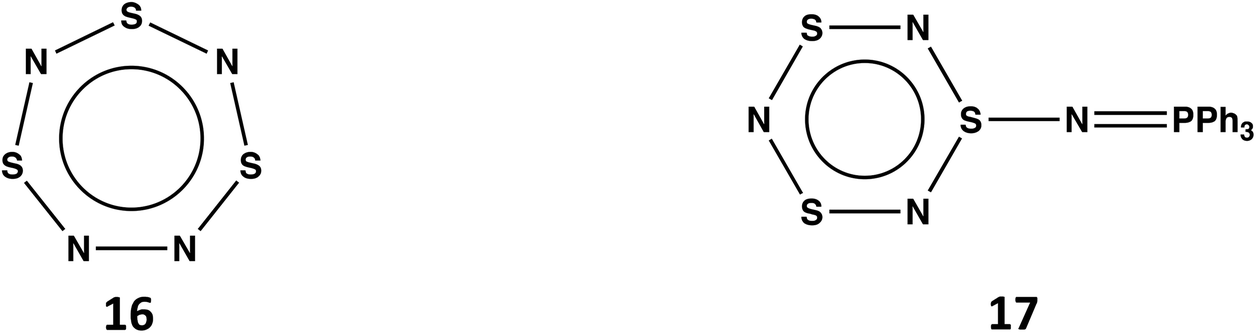 | ||
| Scheme 6 S3N4 (16) and S3N3–NPPh3 (17). | ||
5. Nitrogen-rich selenium nitrides
As mentioned in section 1, the only binary selenium nitride that has been isolated under ambient conditions and structurally characterised is Se4N4 (1, E = Se),3a although metal complexes of Se2N2 (2, E = Se) are also known.3c,d In that context investigations of the high-pressure phase diagrams of the binary selenium–nitrogen system by Cui and co-workers reported in 2019 are of considerable interest.52 These authors identified four stable compounds at high pressures: Cmc21-SeN2, P21/m-SeN3, P![[1 with combining macron]](https://www.rsc.org/images/entities/i_char_0031_0304.gif) -SeN4 and P-SeN5. As illustrated in Fig. 9, these novel nitrogen-rich materials are predicted to incorporate a variety of polynitrogen arrangements in the solid state.52
-SeN4 and P-SeN5. As illustrated in Fig. 9, these novel nitrogen-rich materials are predicted to incorporate a variety of polynitrogen arrangements in the solid state.52
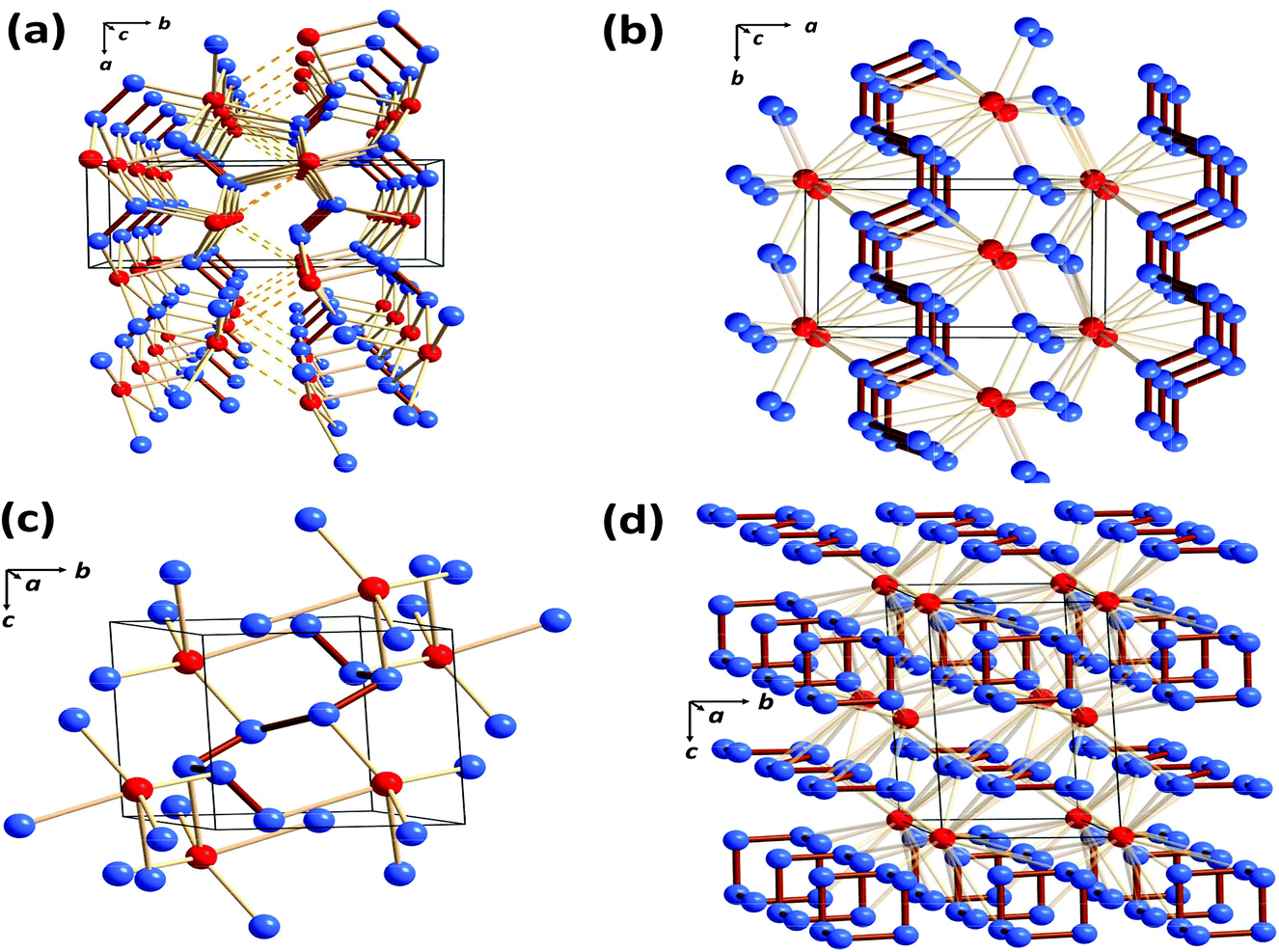 | ||
| Fig. 9 Crystal structures of the predicted selenium nitrides: (a) Cmc21-SeN2 at 90 GPa (b) P21/m-SeN3 at 80 GPa (c) P-SeN4 at 140 GPa and (d) P-SeN5 at 130 GPa. Selenium atoms are depiced in red and nitrogen atoms in blue (redrawn from the structural data in ref. 52). | ||
The binary selenium nitride Cmc21-SeN2 has a layer structure (Fig. 9a), while P21/m-SeN3, P-SeN4 and P-SeN5 incorporate N∞-chains (Fig. 9b), oligomeric N8-chains (Fig. 9c) or distorted [N6]3− anionic rings alternating with layers of N∞-chains (Fig. 9d). The high energy-content of the latter three phases is reflected in the values of their energy densities, which are 3.27, 3.26 and 4.08 kJ g−1, respectively.52
Information on the dissociation barriers for these binary selenium nitrides is not available.
6. Ternary S,N,P molecules
A phosphorus atom P has the same number of electrons as an S+ cation, consequently there can be an isoelectronic relationship between certain ternary S,N,P neutral molecules and well-known binary S,N cations. The latter species can be isolated as thermally stable salts, which have been structurally characterised, and the cations have been shown to exhibit a range of chemical reactions.1a,b The recent identification of the ternary systems SNP12a and SP2N212b provide cogent examples of this isoelectronic connection.
Francisco, Zeng et al., conducted an initial investigation of the pyrolysis and photolysis of the triazide SP(N3)3.12a The experimental work was presaged by DFT calculations of the three triatomic SNP isomers, viz. linear SNP (18), linear SPN (19) and the three-membered ring cyclo-PSN (thiazaphosphirine) (20) at the B3LYP/6-311+G(3df). As indicated in Fig. 10, the cyclic isomer 20 is separated from the linear isomers 18 and 19 by barriers of 138 and 95 kJ mol−1, respectively; the SNP arrangement 18 was predicted to be the global minimum. In one experimental approach the pyrolysis of SP(N3)3 at 1000 °C followed by trapping of the products in an argon matrix at 16 K generated SPN (19) [eqn (1)]. This species was identified with the help of 15N-labelling by comparison of calculated and experimental IR frequencies, which also revealed the formation of other nitrogen-containing species, PN, SN and SN2.12a
| SP(N3)3 → SPN + 4N2 | (1) |
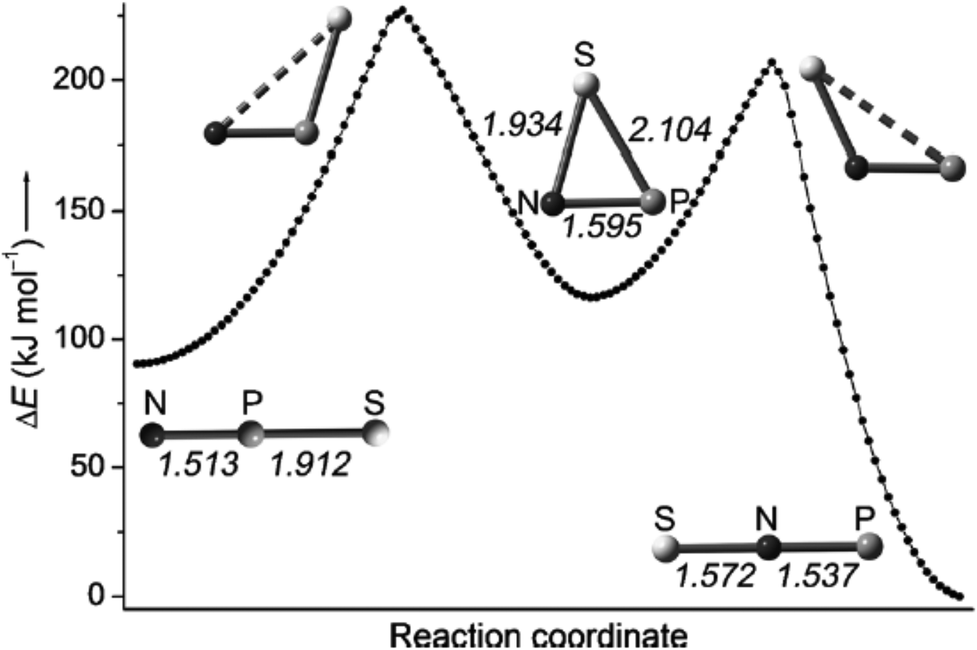 | ||
| Fig. 10 Calculated reaction coordinate of SPN isomers 18–20; bond lengths are given in Å.12a (Reproduced with permission from X. Zeng, H. Beckers, H. Willner and J. S. Francisco, Angew. Chem., Int. Ed., 2012, 51, 3334–3339. @ 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
In an alternative procedure the photolysis of SP(N3)3 in solid argon resulted in a stepwise dissociation to give the most stable SNP isomer 18 as the final product. In both the pyrolysis and photolysis experiments selective UV irradiation produced the linear SPN (19) and cyclo-PSN (20), which were identified by IR spectroscopy.12a
The structure of SNP (18) can be represented by the two Lewis structures shown in Fig. 11a. The calculated P–N bond length is 1.502 Å, slightly greater than the sum of the triple bond radii for P and N (1.48 Å), and the calculated P–N stretching frequency of 1314 cm−1 is close to that of diatomic PN (1327 cm−1).12a The SNP isomer (18) is isoelectronic with the linear sulfur–nitrogen cation [SNS]+, which has an extensive cycloaddition chemistry.53 The similarity of the frontier orbitals of SNP (18) (Fig. 11b) with those of [SNS]+ suggest that the ephemeral SNP species could be trapped via cycloaddition reactions, e.g. with alkynes or nitriles.
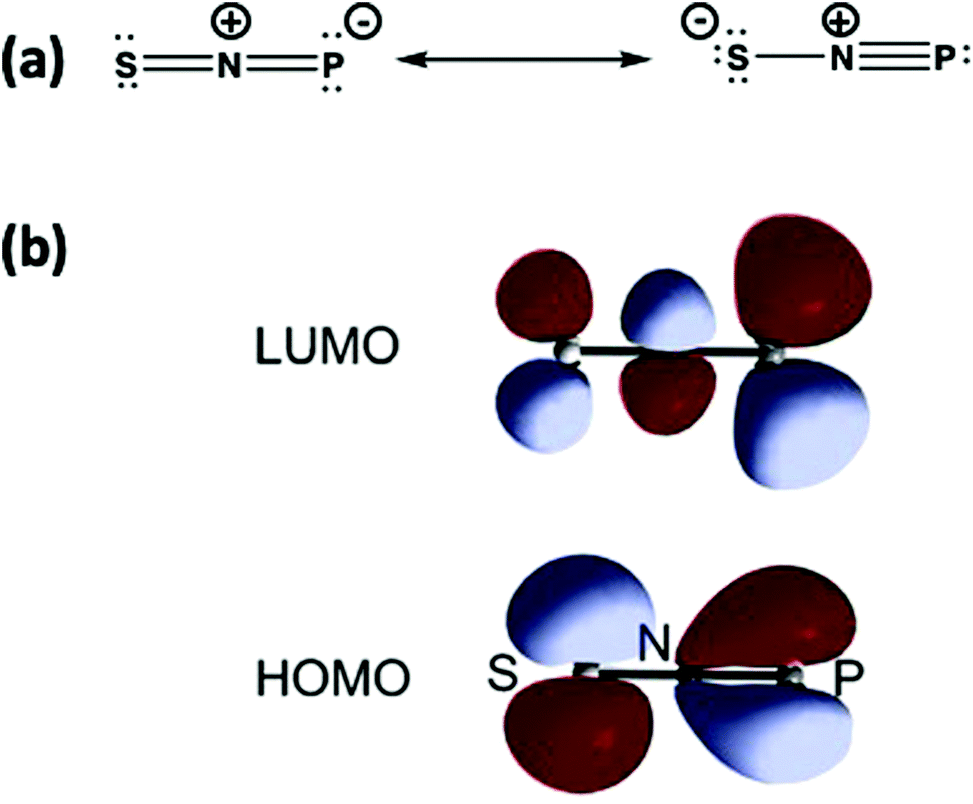 | ||
| Fig. 11 (a) Lewis resonance structures and (b) HOMO and LUMO of the isomer SNP.12 (Reproduced with permission from X. Zeng, H. Beckers, H. Willner and J. S. Francisco, Angew. Chem., Int. Ed., 2012, 51, 3334–3339. @ 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
A subsequent IR analysis of the products of the flash pyrolysis of SP(N3)3 under conditions used to generate SNP (18) (vide supra) revealed a new species with IR frequencies that increased in intensity with prolonged deposition time.12b The mid- and far-IR frequencies of this product appeared as quartets when 15N labelled SP(N3)3 was used as a precursor, indicating a molecule with two structurally non-equivalent N atoms.12b In order to elucidate the identity of this species, theoretical calculations of the dimerisation of SNP were carried out. As illustrated in Fig. 12, the activation energy for head to-tail association of SNP to give cyclo-SNPSNP is low. However, the structural equivalence of the two nitrogen atoms in this six-membered ring rules it out as the source of the new IR bands. Instead, the energetically favourable loss of a sulfur atom from cyclo-SNPSNP, which is formally an 8π-electron system, to give the five-membered ring cyclo-SNPNP (2,4-diphospha-3,5-diaza-thiole), a 6π-electron system, is invoked to explain the observed and calculated IR data (Fig. 12).12b
 | ||
| Fig. 12 Head-to-tail dimerisation of SNP and subsequent formation of cyclo-SNPNP; relative energies (kJ mol−1) and computed bond lengths (Å) at the B3LYP/6-311+G(3df) level.13 (Reproduced with permission from X. Zeng, H. Li, H. Sun, H. Beckers, H. Willner and H. F. Schaefer III, Angew. Chem., Int. Ed., 2015, 54, 1327–1330. @ 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
The aromatic nature of cyclo-SNPNP, which is isoelectronic with the known sulfur–nitrogen dication [S3N2]2+,1a may contribute to its stability. However, the 6π-electron cation cyclo-[SNSNP]+ is expected to exhibit higher stability owing to the positive charge and possible isolation as a salt. By analogy with the cycloaddition of [NS]+ with [SNS]+ to give [S3N2]2+,53 cycloaddition of SNP with an [NS]+ salt could generate the putative cation cyclo-[SNSNP]+.
A third example of the isoelectronic relationship between ternary S,N,P molecules and binary S,N cations involves the well-known planar, cyclic cation [S4N4]2+, a fully delocalised 10π-electron system.1a The replacement of two antipodal S atoms in the supposed planar molecule S4N4 by P atoms generates 1,5-P2N4S2, isoelectronic with [S4N4]2+. According to Gleiter, this ternary S,N,P molecule would incorporate an unpaired 3p electron at each P centre (21).7a The diradical species 21 would be stabilised by addition of two radicals R˙ at the phosphorus centres to give RP(NSN)2PR (22) with two 4π-electron –NSN– groups bridging the two RPIII centres (Scheme 7). Although the free ligand is not known, metal complexes of 22 (R = tBu, NiPr2) in which the P2N4S2 ring is almost planar have been structurally characterized 30 years ago.54
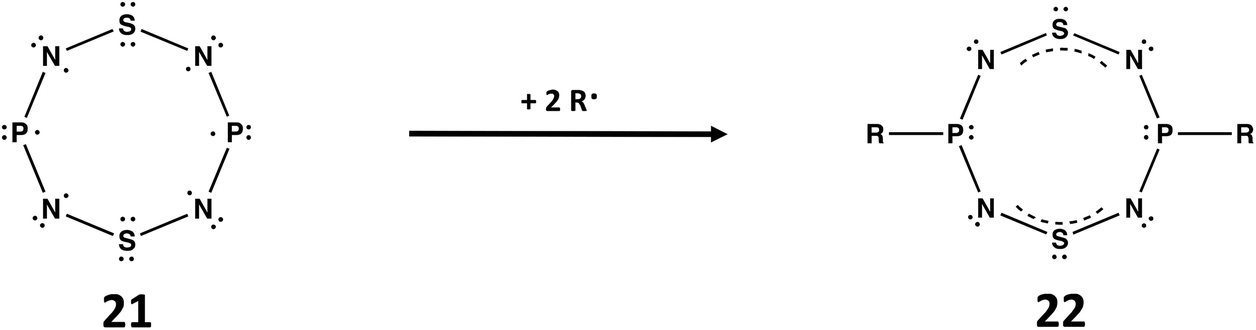 | ||
| Scheme 7 Stabilisation of P2N4S2 (21) by reaction with two R˙ radicals to give RP(NSN)2PR (22).7a | ||
The aromatic character of 1,5-P2N4S2 (21) has been compared with that of the isoelectronic 10π-electron heterocycle dithiatetrazocine (HC)2N4S2 using density functional methods.55,56 It was found that replacement of the two HC groups by the more electronegative P atoms decreases the aromatic character.55 However, the ternary ring system was suggested to be a reasonable synthetic target.
In contrast to the planar P(III)-containing heterocycle 22, the corresponding P(V) systems R2P(NSN)2PR2 (1,5-diphosphadithiatetrazocines) form bicyclic structures with a weak transannular S⋯S interaction similar to that discussed for S4N4 in section 3.1.1. A recent fragment-based energy analysis by Jacobsen estimates that the cross-ring connection in 1,5-diphosphadithiatetrazocines is about half as strong as a typical S–S bond.57 This class of S,N,P heterocycle is considered to be an unusual inorganic example of bishomoaromaticity.7a,58
7. Summary and outlook
Although the complexities of the electronic structures of the well-known binary sulfur nitrides S2N2 and S4N4 and their selenium analogues are still under consideration, the focus of recent investigations of this class of inorganic compounds has been on the potential applications of both known and unknown systems. The foremost example is the use of the facile transformation of S2N2 into the blue-black polymer (SN)x for the detection of fingerprints on metal surfaces, which is being considered as an alternative to existing processes for this application in forensic science (section 3.2.4). A more speculative function involves the possible use of S2N2 or other sulfur–nitrogen rings as hydrogen-storage materials (section 3.2.5). Although the feasibility of this application is questionable in the case of S2N2, sulfur–nitrogen systems with higher chemical and thermal stability, e.g. C,S,N heterocycles, may be worthy of computational and experimental interrogation for this purpose.In the area of sulfur-rich nitrides, the unknown molecule S3N2 is predicted to have quite different properties compared to those of the well-characterised six-membered ring 1,3-S4N2, which is a labile, low melting compound. By contrast, computational studies indicate that crystalline S3N2 will form a two-dimensional network involving only σ bonds rather than an 8π-electron five-membered ring. Furthermore, it has been proposed that S3N2 will exhibit chemical and thermal stability and behave as a wide direct band gap semiconductor with possible optoelectronic applications. As suggested in section 2, this neutral sulfur nitride should be experimentally accessible under ambient conditions so that these predictions can be tested.
Nitrogen-rich sulfur nitrides, e.g. thiatetrazole cyclo-SN4, have long been considered as potential high energy-density materials (section 4.2). Until recently, this purported 5-membered ring and the acyclic triatomic molecule NSN have only been invoked as fleeting intermediates in the decomposition of sulfur–nitrogen ring systems. In the last few years, however, seminal experimental and computational studies of the combination of a chalcogen (sulfur or selenium) and nitrogen gas (N2) at very high pressures have yielded unanticipated information about the structures and properties of this new class of chalcogen nitride. In general, the computational work indicates that at high pressures a variety of nitrogen-rich compounds with structures that are quite different from the metastable sulfur nitrides known to exist at ambient temperatures and pressures (Scheme 1) will be formed. Experimental verification of this prediction has been provided by the identification an SN2 solid at pressures above 64 GPa. This novel material has a CaCl2 structure comprised of SN6 octahedra with triply coordinated N atoms, as forecasted by earlier computational work (section 4.1). In the light of these experimental results, the predicted formation of stable binary selenium nitrides SeNx (x = 2, 3, 4 or 5) at high pressures should provide an incentive for an experimental study of the selenium–N2 system with the characterisation of the elusive (SeN)x polymer as one of the target molecules.
Finally, recently characterised acyclic and cyclic neutral S,N,P molecules are considered in the context of the known structures and reactions of binary sulfur–nitrogen cations, which are isolectronic with the ternary S,N,P systems. This comparison is intended to provide some guidance to the future development of the chemistry of these interesting ternary systems.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank an anonymous reviewer for the suggestion of computational and experimental investigations of the reaction S4N4 with molecular H2, which they had been considering. They are also grateful to Professor Dennis Salahub (University of Calgary) for helpful discussions of the electronic structure of S4N4.References
- (a) T. Chivers, A Guide to Chalcogen-Nitrogen Chemistry, World Scientific Publishing Co., Singapore, 2005 CrossRef; (b) T. Chivers and R. S. Laitinen, in Handbook of Chalcogen-Nitrogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova and W.-W. du Mont, 2nd edn, 2013, ch. 4, vol. 1 Search PubMed; (c) The chemistry of the ephemeral monomeric radical species NE (E = S, Se, Te) is covered comprehensively in a recent book chapter. R. T. Boeré and T. Roemmele, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, ch. 1.14, vol. 1, pp. 375–411 Search PubMed.
- (a) S4N4: M. L. Delucia and P. Coppens, Inorg. Chem., 1978, 17, 2336–2338 CrossRef CAS; (b) S2N2: C. M. Mikulski, P. J. Russo, M. S. Soran, A. G. MacDiarmid, A. F. Garito and A. J. Heeger, J. Am. Chem. Soc., 1975, 97, 6358–6363 CrossRef CAS; (c) (SN)x: M. J. Cohen, A. F. Garito, A. J. Heeger, A. G. MacDiarmid, C. M. Mikulski, M. S. Soran and J. Kleppinger, J. Am. Chem. Soc., 1976, 98, 3844–3848 CrossRef CAS; (d) S4N2: T. Chivers, P. W. Codding, W. G. Laidlaw, S. W. Liblong, R. T. Oakley and M. Trsic, J. Am. Chem. Soc., 1983, 105, 1186–1192 CrossRef CAS; (e) S5N6: T. Chivers and J. Proctor, J. Chem. Soc., Chem. Commun., 1978, 642–643 RSC.
- (a) Se4N4: H. Folkerts, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 1994, 620, 1011–1015 CrossRef, and references cited therein; (b) Se2S2N4: A. Maaninen, J. Siivari, R. S. Laitinen and T. Chivers, Inorg. Chem., 1999, 38, 3450–3454 CrossRef CAS PubMed; (c) Se2N2: P. F. Kelly and A. M. Z. Slawin, J. Chem. Soc., Dalton Trans., 1996, 4029–4030 RSC; (d) P. F. Kelly and A. M. Z. Slawin, Angew. Chem., Int. Ed. Engl., 1995, 34, 1758–1759 CrossRef CAS.
- The black powder obtained from the reaction of Se2Cl2 with trimethylsilyl azide in dichloromethane was originally claimed to be Se4N2, but it was subsequently identified as the selenium–nitrogen chloride Se3N2Cl2. (a) K. Dehnicke, F. Schmock, K. F. Kohler and G. Frenking, Angew. Chem., Int. Ed. Engl., 1991, 30, 577 CrossRef; (b) T. Chivers, J. Siivari and R. S. Laitinen, Angew. Chem., Int. Ed. Engl., 1992, 31, 1518–1519 CrossRef.
- A. Maaninen, J. Siivari, R. S. Laitinen and T. Chivers, Inorg. Chem., 1997, 36, 2170–2177 CrossRef CAS PubMed.
- For a summary of the literature prior to 2012, see: J. Moilanen, A. J. Karttunen, H. M. Tuononen and T. Chivers, J. Chem. Theory Comput., 2012, 8, 4249–4258 CrossRef CAS PubMed.
- (a) R. Gleiter, G. Haberhauer and S. Woitschetzki, Chem. – Eur. J., 2014, 20, 13801–13810 CrossRef CAS PubMed; (b) R. Gleiter and G. Haberhauer, Coord. Chem. Rev., 2017, 344, 263–298 CrossRef CAS.
- T. T. Takaluoma, K. Laasonen and R. S. Laitinen, Inorg. Chem., 2013, 52, 4648–4657 CrossRef CAS PubMed.
- (a) R. S. P. King, P. F. Kelly, S. E. Dean and R. J. Mortimer, Chem. Commun., 2007, 4812–4814 RSC; (b) P. F. Kelly, R. S. P. King and R. J. Mortimer, Chem. Commun., 2008, 6111–6113 RSC; (c) S. M. Bleay, P. F. Kelly and R. S. B. King, J. Mater. Chem., 2010, 20, 10100–10102 RSC; (d) S. M. Bleay, P. F. Kelly, R. S. P. King and S. G. Thorngate, Sci. Justice, 2019, 59, 606–621 CrossRef CAS PubMed . The identity of the S2N2 precursor is not revealed in this paper.
- (a) H. Xiao, X. Shi, X. Liao, Y. Zhang and X. Chen, Phys. Rev. Mater., 2018, 2, 024002 CrossRef CAS; (b) Y. Chen, X. Liao, X. Shi, H. Xiao, Y. Liu and X. Chen, Phys. Chem. Chem. Phys., 2019, 21, 5916–5924 RSC; (c) Auxetic materials become thicker when stretched and thinner in response to compression.
- For a recent review of the fundamental chemistry of binary S,N and ternary S,N,O anions, see: T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2017, 46, 5182–5192 RSC.
- (a) X. Zeng, H. Beckers, H. Willner and J. S. Francisco, Angew. Chem., Int. Ed., 2012, 51, 3334–3339 CrossRef CAS PubMed; (b) X. Zeng, H. Li, H. Sun, H. Beckers, H. Willner and H. F. Schaefer III, Angew. Chem., Int. Ed., 2015, 54, 1327–1330 CrossRef CAS PubMed.
- G.-H. Zhang, Y.-F. Zhao, J. I. Wu and P. V. R. Schleyer, Inorg. Chem., 2012, 51, 13321 CrossRef CAS PubMed.
- T. Chivers and R. S. Laitinen, Dalton Trans., 2017, 46, 1357–1367 RSC.
- (a) J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed; (b) the energies of strong hydrogen bonds falls within the range 15–40 kcal mol−1.
- R. Mews, J. Fluorine Chem., 1981, 18, 155–158 CrossRef CAS.
- Y. Xu, T. Xu, D. Jiajun, S. R. Kirk and S. Jenkins, Int. J. Quantum Chem., 2016, 116, 1025–1039 CrossRef CAS.
- J. Konu, T. Bajorek, R. S. Laitinen, T. Chivers, R. J. Suontamo and M. Ahlgrén, Eur. J. Inorg. Chem., 2006, 2951–2958 CrossRef CAS , and references cited therein.
- M. K. Si and B. Ganguly, Chem. Phys. Lett., 2018, 713, 160–165 CrossRef.
- D. V. Konarev, E. F. Valeev, Y. L. Slovokhotov and R. N. Lyobovskaya, J. Phys. Chem. Solids, 1997, 58, 1865–1867 CrossRef CAS.
- A. Maaninen, J. Siivari, R. S. Laitinen and T. Chivers, Inorg. Synth., 2002, 33, 196–199 CAS.
- A. Ghosh and S. Berg, Arrow Pushing in Inorganic Chemistry: A Logical Approach to the Chemistry of the Main-Group Elements, John Wiley & Sons, Inc., New Jersey, 2014, pp. 240–243 Search PubMed.
- (a) B. G. Kumar and K. Muralidharan, Eur. J. Inorg. Chem., 2013, 2102–2108 CrossRef CAS; (b) B. G. Kumar and K. Muralidharan, J. Mater. Chem., 2011, 21, 11271–11275 RSC; (c) B. G. Kumar and K. Muralidharan, RSC Adv., 2014, 4, 28219–28224 RSC; (d) Cyclic sulfur imides of the type (SNR)4 (R = Me, Et, Bz) have been characterised as colourless crystalline solids, but the corresponding polymers (SNR)n are unknown. H. G. Heal, The Inorganic Heterocyclic Chemistry of Sulfur, Nitrogen and Phosphorus, Academic Press Inc., London, U.K., 1980, pp. 16–40 Search PubMed.
- T. L. Roemmele, J. Konu, R. T. Boeré and T. Chivers, Inorg. Chem., 2009, 48, 9454–9462 CrossRef CAS PubMed.
- For an authoritative summary of earlier theoretical calculations for S2N2 see: P. B. Karadakov, M. A. H. Al-Yassiri and D. L. Cooper, Chem. – Eur. J., 2018, 24, 16791–16803 CrossRef CAS PubMed.
- Y. Jung, T. Heine, P. V. R. Schleyer and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 3132–3138 CrossRef CAS PubMed.
- (a) H. M. Tuononen, R. Suontamo, J. Valkonen and R. S. Laitinen, J. Phys. Chem. A, 2004, 108, 5670–5677 CrossRef CAS; (b) H. M. Tuononen, R. Suontamo, J. Valkonen and R. S. Laitinen, J. Phys. Chem. A, 2005, 109, 6309–6317 CrossRef CAS PubMed.
- B. Braida, A. Lo and P. C. Hiberty, ChemPhysChem, 2012, 13, 811–819 CrossRef CAS PubMed.
- R. D. Harcourt, ChemPhysChem, 2013, 14, 2859–2864 CrossRef CAS PubMed.
- R. Evans, A. J. Downs, R. Köppe and S. C. Peake, J. Phys. Chem. A, 2011, 115, 5127–5137 CrossRef CAS PubMed . The IR frequencies of S2N2 in the solid, vapour and matrix-isolated states obtained in previous studies are summarized in Table 3 of this paper.
- A. Perrin, A. F. Antognini, X. Zeng, H. Beckers, H. Willner and G. Rauhut, Chem. – Eur. J., 2014, 20, 10323–10331 CrossRef CAS PubMed.
- W. Zou, D. Izotov and D. Cremer, J. Phys. Chem. A, 2011, 115, 8731–8742 CrossRef CAS PubMed.
- A. J. Bridgeman and B. Cunningham, Spectrochim. Acta, Part A, 2004, 60, 471–480 CrossRef.
- X. Zeng, A. F. Antognini, H. Beckers and H. Willner, Angew. Chem., Int. Ed., 2015, 54, 2758–2761 CrossRef CAS PubMed.
- A. Datta, Phys. Chem. Chem. Phys., 2009, 11, 11054–11059 RSC.
- C. Wentrup and P. Kambouris, Chem. Rev., 1991, 91, 363–373 CrossRef CAS.
- J.-H. Lin, H. Zhang, X.-L. Cheng and Y. Miyamoto, Phys. Rev. B, 2016, 94, 195404 CrossRef.
- F. Li, X. Lv, J. Gu, K. Tu, J. Gong, P. Jin and Z. Chen, Nanoscale, 2020, 12, 85–92 RSC.
- D. Li, F. Tian, Y. Z. Lv, S. Wei, D. Duan, B. Liu and T. Cui, J. Phys. Chem. C, 2017, 121, 1515–1520 CrossRef CAS.
- D. Laniel, M. Bykov, T. Fedotenko, A. V. Ponomareva, I. A. Abrikosov, K. Glazyrin, V. Svitlyk, L. Dubrovinsky and N. Dubrovinskaia, Inorg. Chem., 2019, 58, 9195–9204 CrossRef CAS PubMed.
- T. Klapötke and A. Schulz, Polyhedron, 1996, 15, 4387–4390 CrossRef.
- (a) M. K. Cyrañski, T. M. Krygowski, A. R. Katrizky and P. V. R. Schleyer, J. Org. Chem., 2002, 67, 1333–1338 CrossRef PubMed; (b) M. K. Cyrañski, P. V. R. Schleyer, T. M. Krygowski, H. Jiao and G. Hohlneicher, Tetrahedron, 2003, 59, 1657–1665 CrossRef.
- (a) L. J. Wang, P. G. Mezey and M. Z. Zgierski, Chem. Phys. Lett., 2003, 369, 386–393 CrossRef CAS; (b) L. J. Wang and P. G. Mezey, Chem. Phys. Lett., 2004, 387, 233–237 CrossRef CAS.
- (a) G.-H. Zhang, Y.-F. Zhao, J. I. Wu and P. V. R. Schleyer, Inorg. Chem., 2009, 48, 6773–6780 CrossRef CAS PubMed; (b) The molecular structure, IR and UV spectra of SN4 have been calculated by a modification of DFT; a strong absorption at 212 nm is predicted. D. Glossman-Mitnik, Theor. Chem. Acc., 2007, 117, 57–68 Search PubMed.
- F. A. Kennett, G. K. MacLean, J. Passmore and M. N. S. Rao, J. Chem. Soc., Dalton Trans., 1982, 851–857 RSC.
- S. Herler, P. Mayer, H. Nöth, A. Schulz, M. Suter and M. Vogt, Angew. Chem., Int. Ed., 2001, 40, 3173–3175 CrossRef CAS PubMed.
- T. Chivers, L. Fielding, W. G. Laidlaw and M. Trsic, Inorg. Chem., 1979, 18, 3379–3388 CrossRef CAS.
- P. W. Fowler, C. W. Rees and A. Soncini, J. Am. Chem. Soc., 2004, 126, 11202–11212 CrossRef CAS PubMed.
- P. J. Dunn, J. L. Morris and C. W. Rees, J. Chem. Soc., Perkin Trans. 1, 1988, 1745–1748 RSC.
- J. Bojes, T. Chivers, A. W. Cordes, G. MacLean and R. T. Oakley, Inorg. Chem., 1981, 20, 16–21 CrossRef CAS , and references cited therein.
- E. A. Pritchina, D. S. Terpilovskaya, Y. P. Tsentalovich, H. S. Platz and N. P. Gritsan, Inorg. Chem., 2012, 51, 4747–4755 CrossRef CAS PubMed.
- W. Wang, H. Wang, Y. Liu, F. Tian, D. Duan, H. Yu and T. Cui, Inorg. Chem., 2019, 58, 2397–2402 CrossRef CAS PubMed.
- S. Parsons and J. Passmore, Acc. Chem. Res., 1994, 27, 101–108 CrossRef CAS , and references cited therein.
- T. Chivers, K. S. Dhathathreyan, C. Lensink, A. Meetsma, J. C. van de Grampel and J. L. de Boer, Inorg. Chem., 1989, 28, 4150–4154 CrossRef CAS.
- A. G. Papadopoulos, N. D. Charistos and A. Muñoz-Castro, New J. Chem., 2016, 40, 5090–5098 RSC.
- K. H. Moock, K. M. Wong and R. T. Boeré, Dalton Trans., 2011, 40, 11599–11604 RSC.
- H. Jacobsen, Inorg. Chem., 2013, 52, 11843–11849 CrossRef CAS PubMed.
- For a discussion of the nature of the transannular S⋯S interaction in 1,5-diphosphadithiatetrazocines, including the concept of bishomoaromaticity, see: (a) Q. Zhang, S. Yue, X. Lu, Z. Chen, R. Huang, L. Zheng and P. V. R. Schleyer, J. Am. Chem. Soc., 2009, 131, 9789–9799 CrossRef CAS PubMed; (b) H. S. Rzepa, Nat. Chem., 2009, 1, 510–512 CrossRef CAS PubMed; (c) T. Chivers, R. W. Hilts, P. Jin, Z. Chen and X. Lu, Inorg. Chem., 2010, 49, 3810–3816 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |