
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dinuclear rhenium complex in the electrochemically driven homogeneous and heterogeneous H+/CO2-reduction†
Lucas A.
Paul
a,
Sheida
Rajabi
a,
Christian
Jooss
 bc,
Franc
Meyer
ac,
Fatemeh
Ebrahimi
b and
Inke
Siewert
*ac
bc,
Franc
Meyer
ac,
Fatemeh
Ebrahimi
b and
Inke
Siewert
*ac
aUniversität Göttingen, Institut für Anorganische Chemie, Tammannstr. 4, 37077 Göttingen, Germany. E-mail: inke.siewert@chemie.uni-goettingen.de
bUniversität Göttingen, Institut für Materialphysik, Friedrich-Hund-Platz 1, 37077 Göttingen, Germany
cUniversität Göttingen, International Center for Advanced Studies of Energy Conversion (ICASEC), Tammannstr. 6, 37077 Göttingen, Germany
First published on 3rd June 2020
Abstract
A dinculear Re(CO)3 complex with a proton responsive phenol unit and a pyrene anchor in the ligand backbone was investigated in the electrochemical CO2/H+ conversion in solution and adsorbed on multi walled carbon nanotubes (MWCNT) on an GC electrode surface. The pyrene group unit is introduced at the end of the ligand synthesis via a coupling reaction, which allows for a versatile ligand modification in order to tune the electronic properties or to introduce various anchor groups for heterogenisation at a late stage. The redox chemistry of the pyrene-α-diimine-Re(CO)3 complex, 1, was investigated in N,N-dimethylformamide (dmf), including IR-spectroelectrochemical (IR-SEC) characterisation of the short lived, reduced species. Subsequently, the electrochemical H+/CO2-reduction catalysis in dmf/water was investigated. The complex catalyses syngas formation yielding CO and H2 with similar rates, namely in Faraday yields of 45% and 35%, respectively. Since the similar complex without the pyrene anchor in the backbone, I, prefers CO2 over H+ reduction, the formation of syngas was rationalised by the small differences in the redox properties and pKa values of the phenol-pyrene unit in regard to phenol unit as in I. Subsequently, the complex was adsorbed on multi walled carbon nanotubes (MWCNT) on a GC electrode surface. Scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS) confirmed coating of the electrode. The immobilised complex was utilised in the electrochemical CO2/H+ reduction in dmf/water, however, the complex quickly desorbed under reductive conditions, likely due to the good solubility of the reduced species. Water as a solvent prevents desorption as confirmed by XPS, however, then a preference for H2 formation over syngas formation was observed under electrocatalytic conditions. Thus, these experiments show, that the results obtained in aqueous organic solution are not easily transferable to the heterogeneous systems operating in water due to changes in the reaction rates for competing pathways.
Introduction
Electrochemical CO2 reduction as an alternative access to CO has been investigated intensively in the last few decades. Currently, CO is formed mainly via burning of fossil fuels and subsequent water gas shift reaction. The electrochemical reduction of CO2 represents an appealing alternative, green approach to synthetic fuels.1 The 2e−/2H+ reduction of CO2 exhibits a potential of −0.73 V vs. Fc+/0 in dmf, however, it competes with the thermodynamically favoured proton reduction (−0.66 V).2 Concomitant electrochemical CO and H2 formation, e.g. similar reaction rates for both reaction pathways, leads to the formation of syngas, which represents a crucial intermediate in chemical industry for the synthesis of bulk chemicals. Currently syngas is mainly produced by natural gas, coal, and steam reforming, which is very redox inefficient as reduced carbonic material is oxidised and subsequently reduced again to yield, e.g. methanol. Thus, alternative pathways are highly desirable.A well-known catalysts family mediating the electrochemical CO2-to-CO transformation are the α-diimine-Re(CO)3 complexes, which were introduced already in 1984 by Lehn and co-workers and were shown to catalyse selectively the electrochemical CO2-to-CO reduction.3 Investigation of the reaction led to a basic understanding of the mechanism.4 Catalysis can proceed via the slow one electron route, in which the singly reduced species reacts after chloride loss with CO2. Subsequently, the reduced Re–CO2 adduct reacts with a second equivalent of CO2 and a second equivalent of the reduced Re species forming a dimer. The disproportionation of the dimer leads to the re-formation of the catalyst and formation of the products CO and carbonate. In the faster two-electrons pathway, the doubly reduced species reacts after chloride ligand loss with CO2. Protonation of the Re–CO2 intermediate initiates C–O bond cleavage. Reduction of the tetracarbonyl species leads to CO release and regeneration of the active species. The protonation induced CO bond cleavage step in the two-electrons pathway prompted others and us to investigate the impact of a proton relay in such Re(CO)3 complexes on the reactivity (Fig. 1).5
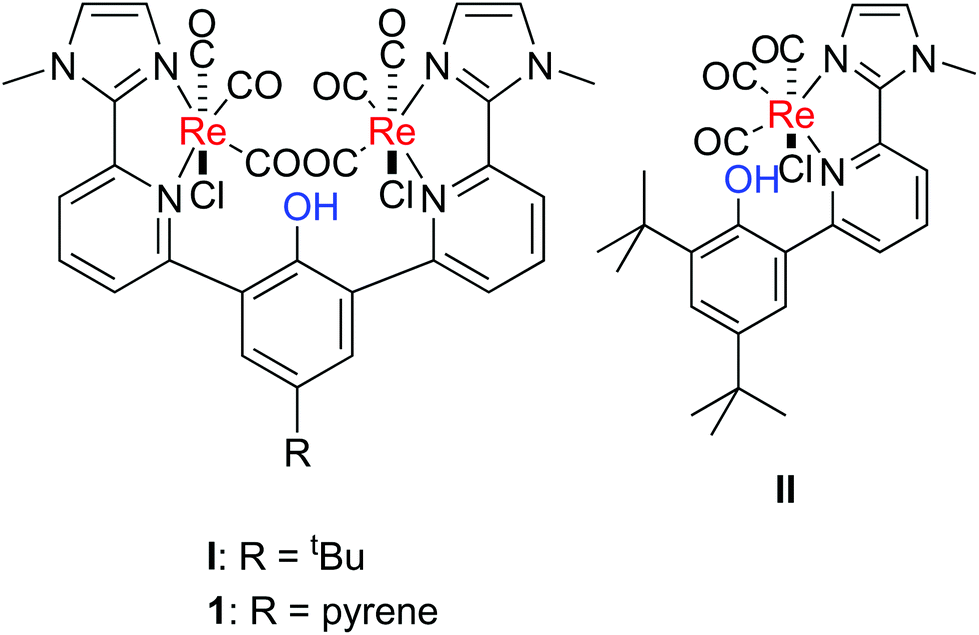 | ||
| Fig. 1 Selected Re(CO)3 complexes with proton responsive units, which have been used in the electrochemical CO2-to-CO conversion.5d | ||
The introduction of a phenol unit as in II led, indeed, to a lower overpotential in the electrochemical CO2 reduction.5d However, the maximum observed rate constant for catalysis was lower with II than for the complex with an anisole unit, since an irreversible O–H bond breaking step competes with catalysis. The complex releases H2 and Cl− after reduction in a competitive pathway forming a phenolate unit, which binds to the metal centre and blocks it for CO2 binding. However, the dinuclear complex I exhibits superior catalytic activity over Lehn's complex and II. It exhibits a much higher reaction rate and the same overpotential in comparison to II.5d
Some effort has been made to immobilise α-diimine-Re(CO)3 complexes on electrode surfaces. Such materials can then be used as devices in the CO2-to-CO reduction catalysis. One of the earliest reports for α-diimine-Re(CO)3 complexes exploiting this strategy appeared in 1989.4c Meyer and co-workers electropolymerized (vbpy)Re(CO)3 on a GC electrode (vbpy = 4-methyl-4′-vinyl-2,2′-bipyridine) and the material was moderately active in the electrochemical CO2 conversion in acetonitrile. A similar approach has been pursued with redox-active thiophene units in the backbone.6 Recently, a different strategy has been investigated: pyrene groups, which undergo π-interactions with carbonic materials, have been introduced in the ligand backbone in order to adsorb the catalyst onto the surface of carbon black7 or edge-plane graphite8 electrodes. The modified carbon black material was moderately active in MeCN with a faradaic yield of 70% for CO.7 Immobilized catalysts on carbonous materials offer the advantage to use water, in which α-diimine-Re(CO)3 complexes are usually not soluble, instead of organic solvents.8,9 Warren and co-workers showed that such modified electrodes are active for CO2-to-CO reduction in water and exhibit high TON.8 This was rather unexpected as the complexes were inactive in organic solvent mixtures and thus, the heterogenisation turned an inactive complex into an active catalysts. These results prompted us to investigate the immobilisation of the dinuclear Re complex with a proton responsive ligand via a pyrene group in the ligand backbone, since the complex exhibits an enhanced catalytic rate for the CO2-to-CO conversion with regard to Lehn's complex,5d and forms H2 in a competitive side reaction to give valuable CO/H2 (syngas) mixtures.
Results and discussion
Complex synthesis and characterisation
The ligand was synthesised in a ten step procedure similar to the one described for I. The pyrene group was introduced in the second to the last step in a coupling reaction between the boronic acid pyrene group and the ligand precursor with a bromo function in the backbone. This allows for minimum modification of the ligand synthesis10 and maximum flexibility in the anchor group as it is introduced late in the synthesis. The pyridine-imidazole sidearm was prepared in a Negishi coupling reaction as described previously.10 As second building block, 2,6-diiodophenol was used. The phenol unit was protected with ethoxymethyl, converted in a Miyaura-borylation reaction with bis(pinacolato)diboron to yield the pinacolboryl derivative, and subsequently coupled with the sidearm to form the protected ligand precursor (Scheme S1†). Subsequently, the phenol was deprotected and the reaction with bromine in pyridine, which serves as a solvent and base, led to selective bromination in the 4-position of the phenol unit. Protection with ethoxymethyl was necessary prior to the coupling of the two subunits, i.e. the pyrene and the ligand precursor. The pyrene group was finally introduced in a Suzuki coupling reaction and the ligand was obtained after deprotection in diluted HCl solution. The ligand was fully characterised by the usual methods.The reaction of the ligand L with Re(CO)5Cl in toluene at elevated temperature led to the formation of the desired complex [Re2(CO)6(L)Cl2], 1. The IR spectrum exhibits the typical CO vibrations as expected for a facial tricarbonyl complex11 and the frequencies are very similar to those of the parent complex I,5d which indicates that the pyrene anchor has only very little influence on the electronic structure at the Re centres. Microanalysis proved the purity of the material. Single crystals suitable for X-ray diffraction were obtained by diffusion of diethyl ether into a saturated solution of 1 in dmf. The result of the refinement is depicted in Fig. 2, Table 1, Fig. S1 and Tables S1, S2.† As expected from IR spectroscopy, the three carbonyl ligands occupy one face of the octahedron. The pyridine units and the phenol unit are highly twisted as previously observed in I (Table S2†).5b The distance between the Re atoms and the N donor atoms are similar to the respective distances in the corresponding mono and dinuclear Re complexes I and II,5b,d and the same holds for the other structural parameters (Table 1). In the crystal structure two pyrene groups interact via π-stacking, which leads to a dimeric structure of 1 in the solid state.12 The distance between the parallel planes defined by the pyrene groups is 3.52 Å.
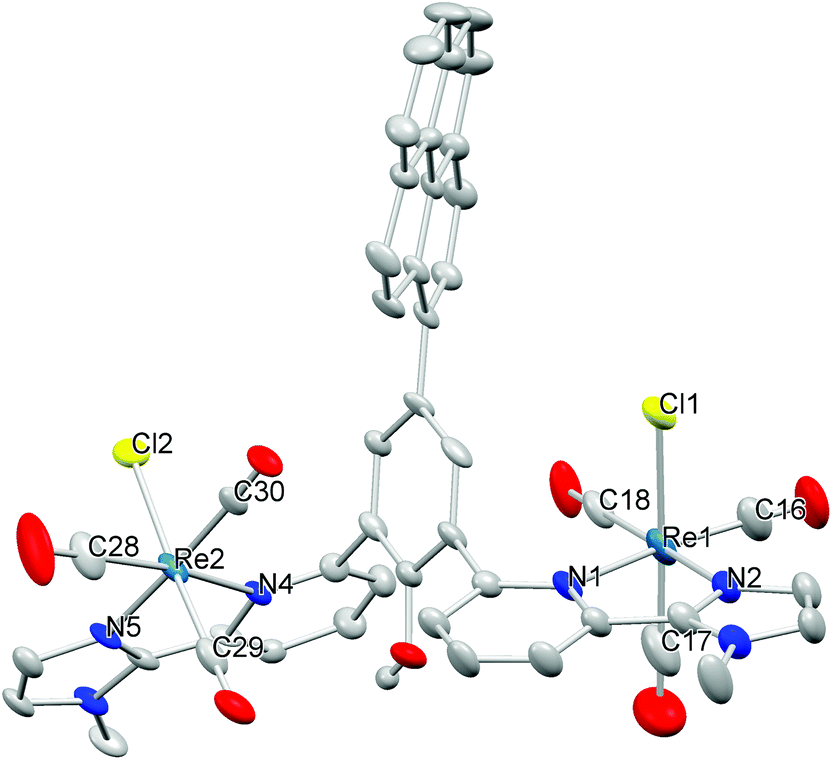 | ||
| Fig. 2 Molecular structure of 1. Most hydrogen atoms were omitted for clarity. Thermal ellipsoids were set at the 50% level. | ||
| Atoms | Distance | Atoms | Distance |
|---|---|---|---|
| Re1–N1 | 2.255(4) | Re2–N5 | 2.135(3) |
| Re1–N2 | 2.134(3) | Re2–N4 | 2.233(4) |
| Re1–Cl1 | 2.475(2) | Re2–Cl2 | 2.482(1) |
| Re1–C16 | 1.897(6) | Re2–C28 | 1.906(6) |
| Re1–C17 | 1.900(7) | Re2–C29 | 1.911(5) |
| Re1–C18 | 1.941(5) | Re2–C30 | 1.918(4) |
In dmf-d7, however, the monomer is present as determined by diffusion ordered NMR spectroscopy. The diffusion coefficient of 1 is the same as the one of I (cf.1: D = 3.2 × 10−10 m s−1 and I: D = 3.2 × 10−10 m s−1 in dmf-d7). As in I and II, isomers of 1 exist in solution as confirmed by 1H NMR spectroscopy. Once the OH group points to the CO ligand and once to the chlorido ligand establishing a hydrogen bond (Scheme S2†), which leads to two sets of signals in the proton NMR spectrum (Fig. S35†). The isomer ratio of the two subunits in dmf-d7 is around ∼4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is slightly different than in I (cf. 2:1 in dmf-d7).
:1, which is slightly different than in I (cf. 2:1 in dmf-d7).
Electrochemical characterisation in solution
We utilised a GC working electrode and a Pt counter electrode for the measurements. All cyclic voltammetry (CV) data in organic solvent (mixtures) were referenced internally vs. the redox couple of ferrocene (Fc+/0) and data in water vs. the saturated calomel electrode (SCE).13 The CV data of 1 in dmf show four reduction waves at Ep,c,1 = −2.10 V, Ep,c,2 = −2.35 V, Ep,c,3 = −2.46 V, and Ep,c,4 = −2.70 V, ν = 0.1 V s−1 (Fig. 3 and Fig. S2†). In the reverse scan, four re-oxidation waves appear at Ep,a,1 = −1.90 V, Ep,a,2 = −2.12 V, Ep,a,3 = −2.40 V, and Ep,a,4 = −2.56 V. With increasing scan rate, the CV data change, which indicates that the reduction processes are coupled to chemical reactions (Fig. S2†). At scan rates of 1 V s−1, the first wave becomes slightly reversible, that is a re-oxidation wave appears, and a further redox process at Ep,a = −2.20 V appears. The second process Ep,c,2 at −2.35 V shifts with increasing scan rates, Ep,a,3 becomes more prominent and shifts to −2.52 V. The process at −2.70 V does not shift and remains reversible. The peak potentials of the first three processes are very similar to the ones in I indicating similar reduction chemistry of 1 and I and only minor influences of the anchor group (Fig. S3†). However, the redox process at Ep,c,4 at −2.70 V is not present in the CV data of I and thus, it likely belongs to the reduction of pyrene. Pyrene exhibits a reduction process at −2.60 V in acetonitrile.14,15 The appearance of further reduction waves with increasing scan rates indicates chemical reactions such as chloride dissociation or OH bond cleavage following the reduction processes. Similar observations were made previously for I and II.5b,d Thus, IR-SEC experiments were pursued to identify the reduced species.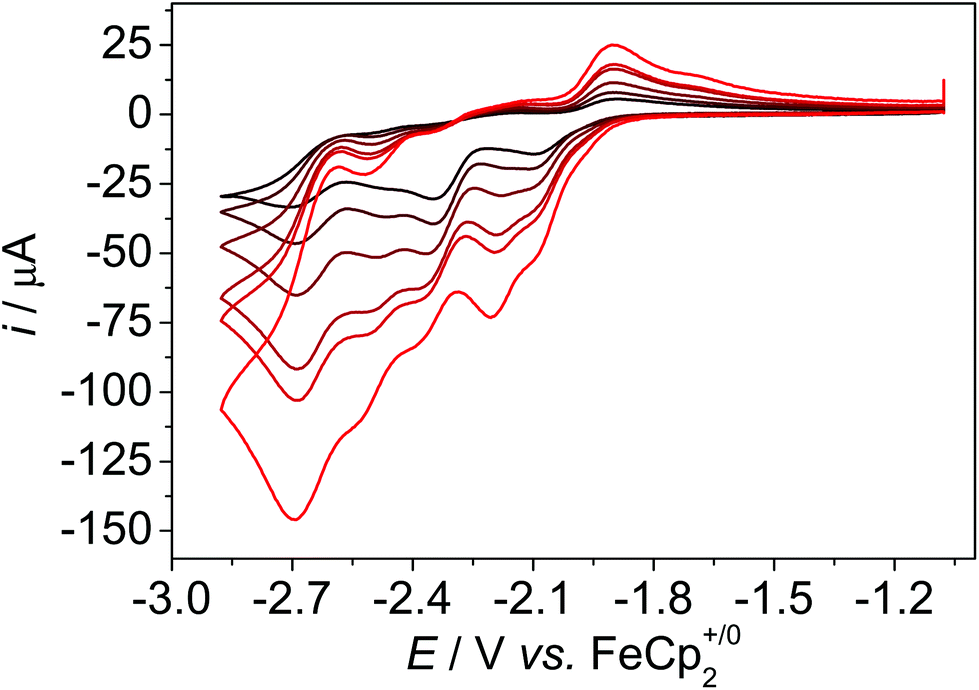 | ||
| Fig. 3 Scan rate dependent CV data of 1 under N2 atmosphere, dmf, c = 1 mM, 0.1 M nBu4NPF6, ν = 0.1, 0.2, 0.4, 0.8, 1, 2 V s−1. Further potential ranges can be found in Fig. S2.† | ||
Initial reduction leads to the formation of a species 1A, which exhibits CO absorption frequencies that are only slightly shifted with regard to 1 (Fig. 4, Table 2). The small shift indicates a reductive O–H bond cleavage, formation of 0.5 equiv. of H2 and chloride ion loss as observed previously in I and similar complexes (Scheme S3†).5a–d Thus the overall process is redox neutral for the complex and the net reaction for 1 represents HCl elimination. Indeed, the reaction of 1 with LDA led to a species, which exhibits CO absorption frequencies at the same frequencies as 1A, which supports this assignment (Fig. S28†). We assume that phenolate binds to the metal centre as the CO stretching frequencies of 1A are typical for facial tricarbonyl rhenium complexes with a X-type ligand in trans position to one CO ligand, that are chloride and phenolate, respectively. In parallel, a further species 1B appears in small amounts, which exhibits CO absorption frequencies shifted in average by −35 cm−1 with regard to 1, which indicates the formation of a complex with a {Re(CO)3L}-entity, that is one entity in the complex is reduced and the chlorido ligand is ejected.16 These two parallel reaction pathways have been observed previously in II.5d Since chloride ion loss is a reversible reaction whereas OH bond cleavage and H2 formation is an irreversible step, 1 is converted to 1A over time.
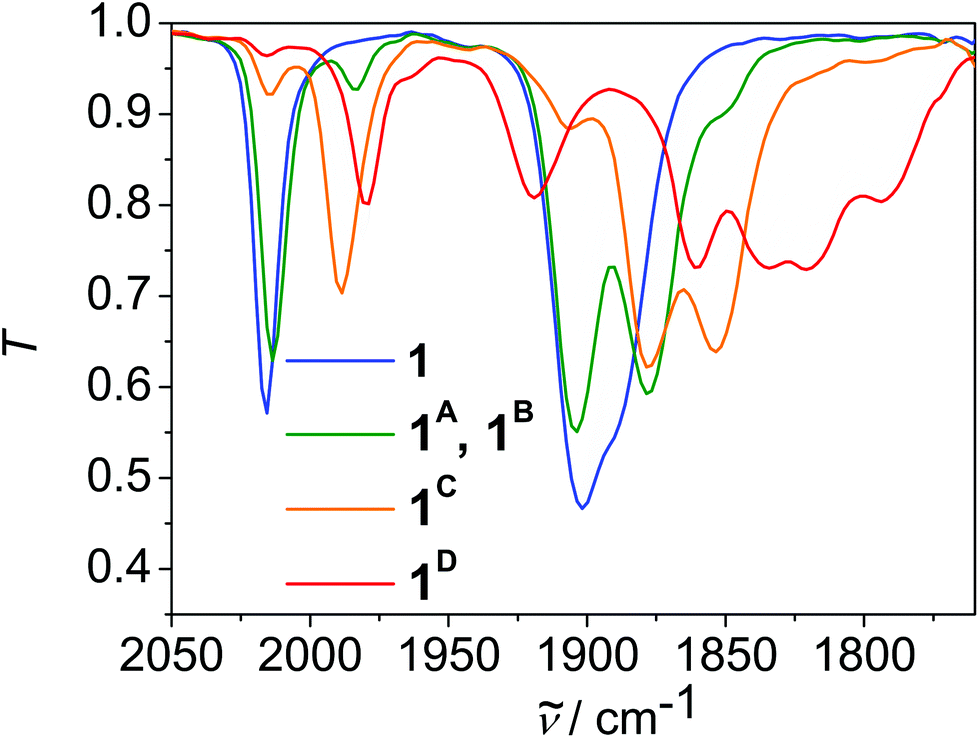 | ||
| Fig. 4 Linear sweep IR-SEC of 1 in dmf, 0.1 M nBu4NPF6. | ||
Further reduction of 1 leads to a species 1C with three CO vibrations, which are shifted in average by about −25 cm−1 with regard to 1A. This indicates the formation of a species, in which each diimine-ligand entity is reduced once and each ReI ion is coordinated by one X-type ligand, that is chloride and phenolate, respectively. Further reduction leads to the final species 1D, which exhibits 6 CO absorption frequencies indicating the formation of an asymmetric species. Likely this is the threefold reduced species [Re2(CO)6(LH−1)]2− in which one Re entity is reduced twice, one Re entity once, and both chlorido ligands are dissociated (Scheme S3†). We assume that the reduction equivalents are localised on one Re-unit on the time scale of the IR, since the ligand is highly twisted.
CO2 reduction catalysis in solution
Prior to anchoring of the complex, we investigated the catalytic activity of 1 in solution. Purging a solution of 1 in dmf with CO2, led to an increased current, which is indicative for a catalytic event (Fig. 5). In the presence of 6% or 10% of water, the current increase is less prominent, however, the onset potential of the catalysis is shifted to higher potentials. No current increase is observed, when water is added under argon atmosphere (Fig. S4†). Since dmf/water exhibits a pH of about 10, which decreases upon purging with CO2, these experiments indicate that the catalytic event is connected to H+/CO2 reduction catalysis under less basic conditions.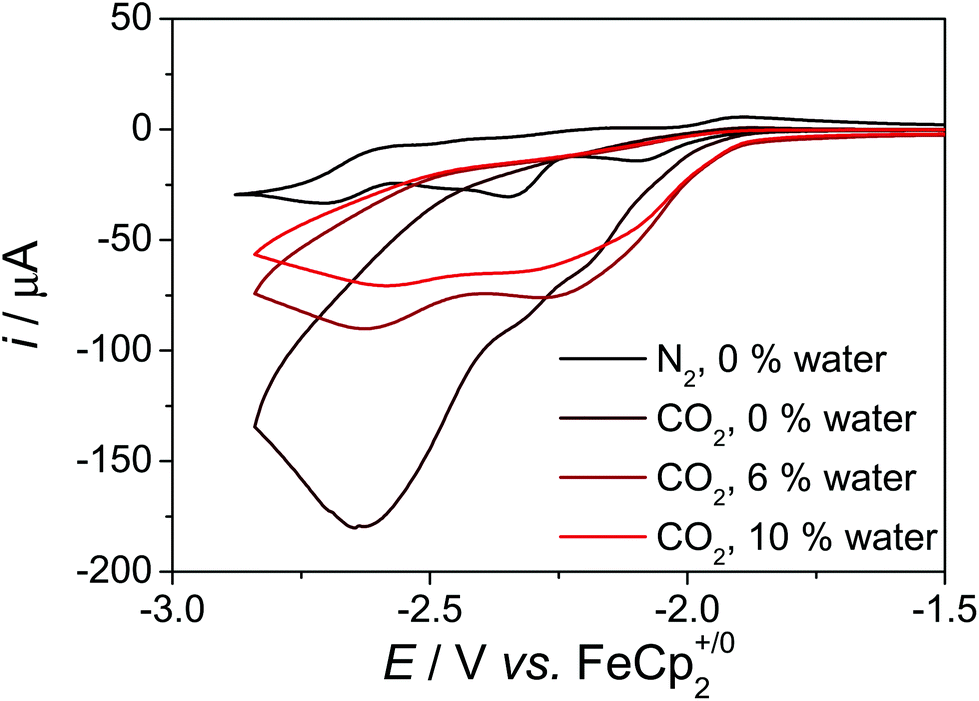 | ||
| Fig. 5 CV data of 1 the presence and absence of CO2 in dmf with various amounts of water, c = 1 mM, 0.1 M nBu4NPF6, ν = 0.1 V s−1. | ||
Controlled potential electrolysis (CPE) experiments at an applied potential of −2.20 V in dmf + 10% water led to the formation of CO and H2 in Faraday efficiencies (FE) of 45% and 35%, respectively (Table S3†). The charge builds up steadily over the course of the electrolysis experiment, which indicates good stability of 1 under catalytic conditions (Fig. S23†). The amount of H2 formed is much larger than in the experiments with I and II,5b,d which indicates that 1 catalyses the CO and H2 evolution reaction in competitive reaction pathways with similar rates. The applied potential has only minor influence on the rates of H2 and CO formation, though larger non-productive currents arise under more reductive conditions (Fig. S24†). CPE experiments at an applied potential of −2.50 V led to the formation of CO and H2 in Faraday efficiencies (FE) of 30% and 20%, respectively (Table S3†). Since the redox chemistry of 1 and I is very similar up to the applied potential of the catalysis (Fig. S3†), we hypothesize that this could be rationalised by different H-bonding properties, pKa values and OH bond dissociation free energies (BDFE) in 1 and I due to the conjugated pyrene anchor. The H-bonding properties of the phenol OH proton in 1 and I are different as reflected by the 1H resonance of the OH-proton in dmso,17 which is shifted downfield in 1 in comparison to I (1: cf. δ = 9.79 and 9.17 ppm (dmso-d6) (Fig. S35†), 10.39 and 9.25 ppm (dmf-d7); I: δ = 9.11 and 8.57 ppm (dmso-d6)5b and 9.71 and 8.79 (dmf-d7)). Furthermore, the pKa of the phenol in 1 should be lower than in I due the extended π-system in 4 position as this increases the stability of the anion, i.e.1 is more acidic than I. Since pyrene, which is in conjugation with the phenol, exhibits a reduction process at −2.60 V and an oxidation process at 0.93 V,14 the BDFE of the reduced and protonated5a as well as the oxidised phenol-pyrene unit in 1 should be slightly different in comparison to 2,4,6-tri-tert-butyl-phenol (cf. oxidation of phenol in dmso: 1.00 V (ref. 18)). Assuming free energy relationships for the H2 formation pathways in 1 and I, these small differences may lead to different reaction rates for H2 formation.
Scan rate dependent CV data showed, that the catalytic waves do not exhibit an S-shape, that the currents are not fully scan rate independent, and that all CVs have a wave at the foot of the catalytic wave (Fig. S5–S7†). Thus, the catalytic rate constant kobs can only be estimated from the limiting current according to eqn (1).19 Dividing icat (catalytic current) by ip (current of a non-catalytic event) eliminates several constants (eqn (2)).20 The plots of icat/ipvs. ν−1/2 gave straight lines with reasonable intercepts (Fig. S8–S10†). ip was estimated from the first reduction process in the scan rate dependent CV data of 1 in the presence of LDA as this process represents a reversible 1 e− reduction event (Fig. S11†).
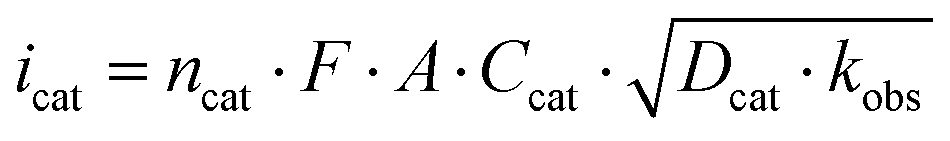 | (1) |
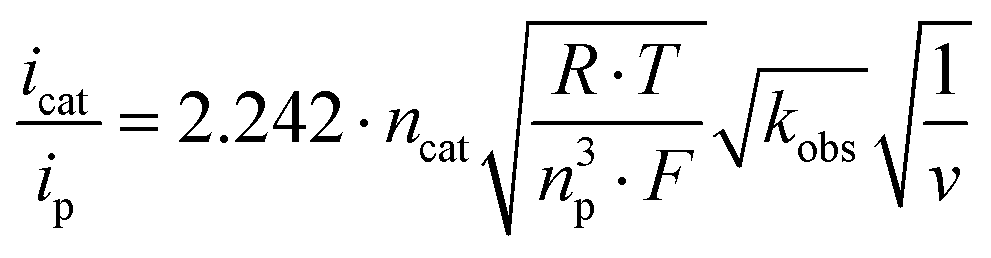 | (2) |
The overpotentials η were estimated from the half-peak potentials of the catalytic waves.21 The CO2|CO reduction potential is −0.73 V (ref. 2) and the H+|H2 reduction potential is −0.66 V (ref. 2 and 13) in dmf. The CO2|CO reduction potential in dmf/water has been determined to be −1.4 ± 0.1 V and the H+|H2 reduction potential is also −1.38 V based on the pKa value of 12.2 ± 1 for H2CO3 in dmf.22
In the presence of water, the half peak potential of the catalytic waves of I and 1 are the same, and less negative than in the absence of water, however, the rate constant with 1 is lower than with I.5b In the absence of water, the rate constant is higher in 1 than in I, though, the overpotential is higher. The lower overpotential of 1 in the presence of water may be rationalised – as previously for I and II – by the faster chloride ion loss in the protic environment due to better stabilisation of the anion.5d Chloride loss represents the first step in the catalytic cycle. However, the overall catalytic rate constant is smaller in 1 than in I, which can be rationalised by the OH-bond breaking reaction, which is considerably faster in 1 than in I.5b Only traces of H2 were observed with I, while considerable amounts of H2 were formed in the presence of 1 (vide infra). OH bond breaking results in the formation of the anionic rhenium species in which the 6th coordination side is blocked by the phenolate (vide supra, formation of 1A). Further protonation re-forms the starting material.
Anchoring of the complex on MWCNT
Subsequently, we anchored the complex on multi-walled carbon nanotubes (MWCNT). Pre-treated MWCNTs (1 mg mL−1) were suspended in thf and the suspension was dip coated several times on GC electrodes. Scanning electron microscopy (SEM) pictures of the GC electrode surface showed that the electrode surface was covered successfully with carbon nanotubes (Fig. S29†). Subsequently, the electrode was soaked in a saturated acetonitrile solution of 1 overnight. SEM pictures indicate that this does not change the surface morphology, and the carbon nanotubes are still present (Fig. S30†). High resolution XPS of the electrode surface confirmed the adsorption of the complex on the surface as the rhenium 4f7/2 and 4f5/2 peaks were present at 41.6 eV and 43.8 eV, respectively, which is characteristic for an α-diimine-Re(CO)3Cl complex (Fig. S32†).7Electrochemical measurements of 1@MWCNT have initially been conducted in dmf/water mixtures 10/1. CV measurements of 1@MWCNT showed two reduction processes with peak potentials Ep,c at ∼−1.97 V and at ∼−2.57 V (ν = 0.1 V s−1, Fig. S12†). The catalyst loading was estimated from the first scan under N2. Assuming that the first redox wave in 1@MWCNT belongs to a 2 electrons reduction process as it does for 1 in solution (vide supra), we estimated a catalyst loading of 8 × 10−13 mol on the electrode surface from the charge injected in the first wave (Q = 1.5 × 10−7 C, Øgeometric = 3 mm, Fig. S13†). The potential of the initial reduction of 1 shifted to slightly higher potentials with regard to solution, likely due to the differences in the solvation free energies and ion paring of a surface bound species vs. solution species.23 However, the current decreases quickly with increasing number of cycles and the redox waves of 1 disappear, indicating that the catalyst is detached upon successive reductive scanning (Fig. S12†). Blank measurements show that the current also decreases with increasing cycles in the absence of 1 due to smaller capacitive currents (Fig. S17†). However, the currents of the first scans of bare MWCNT are lower than of 1@MWCNT and thus, the decrease in 1@MWCNT cannot be related only to changes in the capacitive currents.
Using a freshly prepared sample and purging the solution with CO2 leads to an increased current with regard to the N2 measurement with two distinct plateaus at around −2.25 V and around −2.55 V (Fig. 6 and Fig. S14†). However, as under N2 atmosphere, the current decreases upon successive cycling and reaches the current as under N2 at the ∼15th cycle (Fig. S15 and S16). In the CV experiment with bare MWCNT under CO2 atmosphere, the current in the CV is not larger than the current in the CV experiments under N2, and the absolute current is lower than in the experiments with 1@MWCNT (Fig. S17†). These results suggest that the catalyst is not bound strongly to the surface under reductive conditions and dissolves in dmf/water. This was substantiated in a CPE experiment at an applied potential of −2.20 V for 30 minutes: the current decreases quickly within the first 50 seconds, indicating a rapid loss of activity of the hybrid (Fig. S25†). Indeed, XPS measurements of the electrode surface after electrolysis with 1@MWCNT showed the absence of rhenium atoms, which confirms the hypothesis that the catalyst dissolves under catalytic conditions (Fig. S33†). SEM pictures of the electrode surface after extended electrolysis, however, showed that the MWCNT are still present (Fig. S31†). This means that the anchor group is the weak point and the π-interactions between the pyrene group and the MWCNT are not sufficiently strong to prevent desorption. Since catalysis was conducted at a potential that is less negative than the potential required for the reduction of pyrene,14i.e. at −2.20 V, we assume that it is related to the rather good solubility of the reduced catalyst in the solvent mixture rather than to the redox activity of the pyrene anchor.
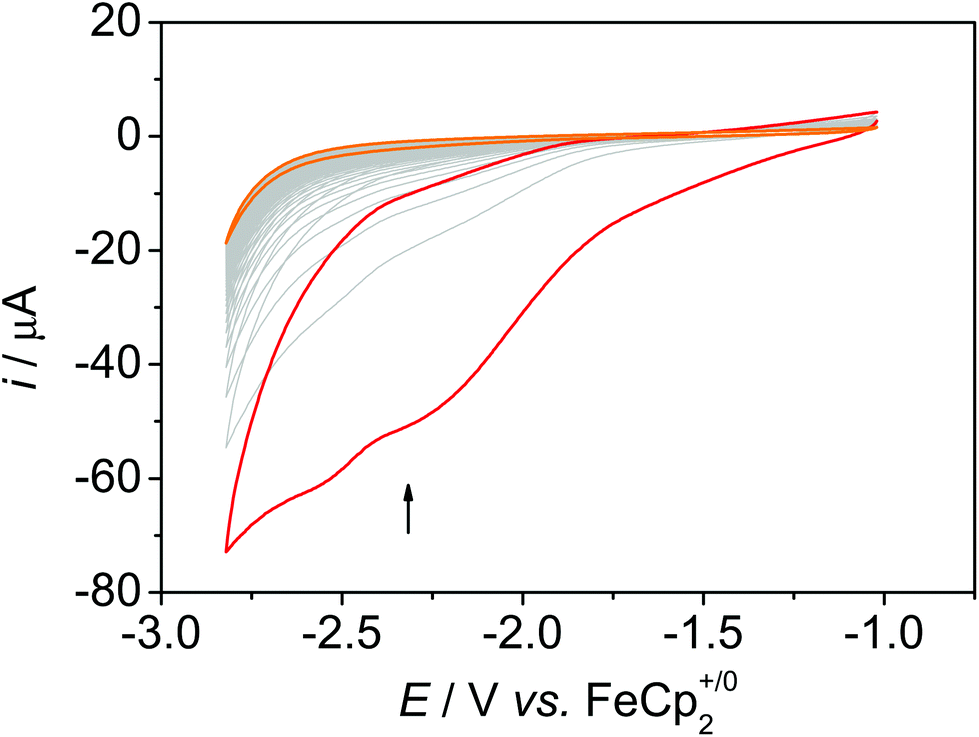 | ||
| Fig. 6 50 CV scans of 1@MWCNT under CO2 atmosphere in dmf + 10% water, 0.1 M nBu4NPF6, ν = 0.1 V s−1. | ||
Since the catalyst precursor 1 is not soluble in water, we investigated the hybrid in 100% water in order to prevent desorption. The current of 1@MWCNT under N2 atmosphere in water with 0.5 M NaHCO3 as electrolyte is much larger than in dmf/water. The CV data do not show pronounced redox processes, and only one small, broad redox feature at Ep,c of ∼−1.03 V vs. SCE13,15 was observed (Fig. S18†). The current quickly decreases with increasing number of cycles due to the decrease in the capacitive current (Fig. S18 and S22†), though in contrast to dmf/water the redox wave at −1.03 V, which we assigned to 1, did not vanish. Under CO2 atmosphere the current is larger than under N2, which indicates some catalytic activity, though the current increase is less distinct than in dmf/water (Fig. S19†). The current decreases with the number of scans, and after ∼15 cycles the current is similar to the current observed under N2 (Fig. S20 and S21†). The current of bare MWCNT decreases slightly under CO2 atmosphere (Fig. S22†). CPE experiments at −1.37 V vs. SCE confirmed the drop of the current within the first 100 s of the experiment (Fig. S26†). No CO was produced during the bulk electrolysis experiment, but instead H2 was formed with a Faraday yield of about 20%. However, high resolution XPS after 30 minutes of electrolysis confirmed the presence of the catalyst on the electrode surface as the rhenium 4f7/2 and 4f5/2 peaks were still present (Fig. S34†). The binding energies are shifted to higher energies (42.1 and 44.5 eV), which indicates that the complex is not reduced, as this would lead to a shift to lower energies. This suggests that the lack of CO production in 0.5 M aqueous NaHCO3 is not associated with catalyst desorption as in dmf/water. In the blank CPE experiment of a GC electrode just coated with MWCNT, the current also drops within the first minutes, however, it stabilises at a lower level than in the experiment of 1@MWCNT. Though, H2 was formed in a higher Faraday yield of about 64% (Table S4 and Fig. S27†). In both experiments the current drops, which we attributed to an increase of the pH near the electrode surface upon consumption of protons due to HER and a decrease in the capacitive current. However, solely H2 is formed in both experiments, which indicates that the Re complex has virtually no influence on the product distribution. This could be rationalised by, e.g., the differences in the proton (and CO2) concentrations in dmf/water mixtures, which is basic (pH ∼ 10) in comparison to water/HCO3−, which is less basic (pH ∼ 8). Since the respective reaction rates depend on the proton and CO2 concentrations, these differences could alter product formation and/or reduce the overpotential for H+ reduction. In contrast to the work of Warren and co-workers on similar systems, 1 turned to an inactive catalyst in water upon heterogenisation, which supports their statement that medium effects can have an important impact on the activity.8
Conclusion
The dinuclear Re complex 1 is an active catalyst in the electrochemical syngas formation, that is it catalyses the CO2-to-CO conversion and H+-to-H2 conversion in dmf/water with similar rates. Since the related complex I without a pyrene unit in conjugation to the proton responsive phenol unit catalyses selectively the CO formation, we assume that the differences in the thermodynamics of the phenol-pyrene versus the tert-butyl-phenol unit may give rise to the small differences in the reaction rates for H2 formation. The complexes exhibit different H-donor properties, the acidity of the OH unit in 1 is larger than in I, and the redox activity of the pyrene unit may lead to changes in the BDFE of the OH unit. In other words, although, the electronic structure and the redox properties of the metal centre are virtually unaffected by the pyrene unit, the pyrene unit in the backbone has an influence on the reactivity. Immobilisation of the complex on MWCNT on a GC electrode was successful, however, the complex quickly desorbs under reductive conditions in dmf/water. Changing to 0.5 M aqueous NaHCO3 as solvent leads to a loss of activity for CO2-to-CO conversion and only H2 formation was observed, as in the blank reaction. XPS confirmed a high stability of the complex on the electrode surface in 0.5 M aqueous NaHCO3. This underscores that medium effects have a large impact on the catalytic activity.23 The large differences in proton concentration in water and water/dmf mixtures may alter the reaction rates for H2 and CO formation and/or lower the overpotential for H2 evolution in water.Experimental section
ESI† includes experimental details for the ligand synthesis, all details on X-Ray crystallography, further electrochemical measurements, IR data, as well as SEM pictures and XPS data.General information
Manipulations of air-sensitive reagents were carried out by means of common Schlenk-type techniques involving the use of a dry argon or nitrogen atmosphere or performed in an MBraun glovebox. If not otherwise stated, the potentials in this paper are given vs. Fc+/0 in organic solvents and vs. SCE in water. MWCNTs were purified according to a literature procedure prior to use.241 was adsorbed by soaking MWCNT coated electrodes in a saturated solution of 1 in MeCN overnight. The soaked electrodes were rinsed with small amounts of fresh MeCN to remove unbound 1 and subsequently dried under a gentle nitrogen stream.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the DFG [SI 1577-2 (I. S.), SFB 1073 (I. S., F. M., C. J. project C01)], the Fonds der Chemischen Industrie, the Georg-August-Universität Göttingen, and the Otto-Röhm-Gedächtnisstiftung. We thank Dr C. Volkmann for measuring the X-Ray structure and Christian Höhn, Helmholtz-Zentrum Berlin für Materialien und Energie, for measuring the XPS. We thank Igor Fokin for conducting some CPE experiments. Support by Göttingen University is acknowledged.References
- A. Tatin, J. Bonin and M. Robert, ACS Energy Lett., 2016, 1, 1062–1064 CrossRef CAS; R. Foit, I. C. Vinke, L. G. J. de Haart and R. A. Eichel, Angew. Chem., Int. Ed., 2017, 56, 5402–5411 CrossRef PubMed.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 984, 328–330 RSC.
- (a) B. P. Sullivan, C. M. Bolinger, D. Conrad, W. J. Vining and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 985, 1414–1416 RSC; (b) J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS; (c) T. R. O'Toole, B. P. Sullivan, M. R.-M. Bruce, L. D. Margerum, R. W. Murray and T. J. Meyer, J. Electroanal. Chem. Interfacial Electrochem., 1989, 259, 217–239 CrossRef; (d) Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976–11987 CrossRef CAS PubMed; (e) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed; (f) J. Agarwal, E. Fujita, H. F. Schaefer 3rd and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef CAS PubMed; (g) J. A. Keith, K. A. Grice, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2013, 135, 15823–15829 CrossRef CAS PubMed ; and references therein.
- (a) G. F. Manbeck, J. T. Muckerman, D. J. Szalda, Y. Himeda and E. Fujita, J. Phys. Chem. B, 2015, 119, 7457–7466 CrossRef CAS PubMed; (b) A. Wilting, T. Stolper, R. A. Mata and I. Siewert, Inorg. Chem., 2017, 56, 4176–4185 CrossRef CAS PubMed; (c) A. Wilting and I. Siewert, ChemistrySelect, 2018, 3, 4593–4597 CrossRef CAS; (d) J.-P. Du, A. Wilting and I. Siewert, Chem. – Eur. J., 2019, 25, 5555–5564 CrossRef CAS PubMed; (e) L. Rotundo, C. Garino, E. Priola, D. Sassone, H. Rao, B. Ma, M. Robert, J. Fiedler, R. Gobetto and C. Nervi, Organometallics, 2019, 38, 1351–1360 CrossRef CAS; (f) S. A. Roell, B. R. Schrage, C. J. Ziegler and T. A. White, Inorg. Chim. Acta, 2020, 503, 119397 CrossRef; (g) J. O. Taylor, G. Neri, L. Banerji, A. J. Cowan and F. Hartl, Inorg. Chem., 2020, 59, 5564–5578 CrossRef CAS PubMed.
- C. Sun, S. Prosperini, P. Quagliotto, G. Viscardi, S. S. Yoon, R. Gobetto and C. Nervi, Dalton Trans., 2016, 45, 14678–14688 RSC.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- S. Sinha, A. Sonea, W. Shen, S. S. Hanson and J. J. Warren, Inorg. Chem., 2019, 58, 10454–10461 CrossRef CAS PubMed.
- A. Zhanaidarova, S. C. Jones, E. Despagnet-Ayoub, B. R. Pimentel and C. P. Kubiak, J. Am. Chem. Soc., 2019, 141, 17270–17277 CrossRef CAS PubMed.
- A. Wilting, M. Kügler and I. Siewert, Z. Anorg. Allg. Chem., 2015, 641, 2498–2505 CrossRef CAS.
- F. A. Cotton and C. S. Kraihanzel, J. Am. Chem. Soc., 1962, 84, 4432–4438 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- I. M. Kolthoff and M. K. Chantooni, et al., report a slightly different value of −0.69 V for the H+|H2 redox couple vs. Fc+/0 in dmf. Fc+/0 exhibits a standard reduction potential of 0.40 V vs. H+|H2 in water, J. Phys. Chem., 1972, 76, 2024–2034 CrossRef CAS.
- M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff, N. Blanchard and J.-P. Fouassier, Macromolecules, 2012, 45, 1746–1752 CrossRef CAS.
- For the conversion of the potential from Fc+/0 to SCE see: N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- B. Bernet and A. Vasella, Helv. Chim. Acta, 2000, 83, 995–1021 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- J. M. Saveant and E. Vianello, Electrochim. Acta, 1965, 10, 905–920 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- A. M. Appel and M. L. Helm, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
- S. Sinha and J. J. Warren, Inorg. Chem., 2018, 57, 12650–12656 CrossRef CAS PubMed.
- G. Gritzner, J. Electroanal. Chem. Interfacial Electrochem., 1983, 144, 259–277 CrossRef CAS; M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef; D. Acevedo and H. D. Abruna, J. Phys. Chem., 1991, 95, 9590–9594 CrossRef; H. Svith, H. Jensen, J. Almstedt, P. Andersson, T. Lundbäck, K. Daasbjerg and M. Jonsson, J. Phys. Chem. A, 2004, 108, 4805–4811 CrossRef.
- A. Ali Moosa, M. Ibrahim and R. F. Salloom, Int. J. Nano Dimens., 2014, 5, 97–104 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1973247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt00381f |
| This journal is © The Royal Society of Chemistry 2020 |