
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxaliplatin and [Pt(R,R-DACH)(panobinostat-2H)] show nanomolar cytotoxicity towards diffuse intrinsic pontine glioma (DIPG)†
Marie H. C.
Boulet
a,
Laura K.
Marsh
a,
Alison
Howarth
b,
Alice
Woolman
a and
Nicola J.
Farrer
 *a
*a
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: Nicola.Farrer@chem.ox.ac.uk; Tel: +44 (0)1865 285131
bStructural Genomics Consortium, University of Oxford, Oxford, UK
First published on 16th April 2020
Abstract
We report the synthesis of two novel platinum(II) complexes which incorporate histone deacetylase (HDAC) inhibitors: [PtII(R,R-DACH)(Sub-H)] (1), [PtII(R,R-DACH)(panobinostat-2H)] (2), where SubH = suberoyl-bis-hydroxamic acid; DACH = (1R,2R)-(–)-1,2-diaminocyclohexane and panobinostat = (E)-N-hydroxy-3-[4-[[2-(2-methyl-1H-indol-3-yl)ethylamino]methyl]phenyl]prop-2-enamide. Complexes 1 and 2 were characterised by 1H, 13C, 195Pt NMR spectroscopy and ESI-MS. Whilst oxaliplatin demonstrated considerable cytotoxicity in two patient-derived low-passage paediatric glioma DIPG cell lines (IC50 values of 0.333 μM in SU-DIPG-IV, and 0.135 μM in SU-DIPG-XXI), complex 2 showed even greater cytotoxicities, with IC50 values of 0.021 μM (SU-DIPG-IV), 0.067 μM (BIOMEDE 194) and 0.009 μM (SU-DIPG-XXI). Complex 2 also demonstrated superior aqueous solubility in comparison to panobinostat. Complex 2 released free intact panobinostat under HPLC conditions, as determined by ESI-MS. Incubation of solutions of oxaliplatin (H2O) and panobinostat (DMF) resulted in instantaneous reactivity and precipitation of a panobinostat derivative which was not a platinum complex; the same reactivity was not observed between carboplatin and panobinostat.
Introduction
Brain tumours are the most common cause of cancer-related death for children and young people under 40.1 Diffuse midline glioma, H3 K27M-mutant (previously known as diffuse intrinsic pontine glioma (DIPG)) is highly aggressive, with a poor clinical prognosis, such that it is the leading cause of death from brain cancer in children.2 Surgical removal is not possible for DIPG, due to the brainstem location and diffuse tumour nature. Radiotherapy can be mildly effective in 70% of cases – prolonging survival by up to 6 months, after which period the tumour grows back, leading to death; the 5-year survival rate for DIPG is just 1%.2A key challenge for the therapeutic treatment of DIPG is the delivery of agents across the typically intact blood brain barrier (BBB).3 Combined therapies such as carboplatin and the histone deacetylase (HDAC) inhibitor sodium valproate are reportedly synergistic towards DIPG in cellulo4 and direct delivery of these agents to the brainstem by convection-enhanced delivery (CED) is used clinically.5 DIPG cell lines6 and xenograft models6,7 also demonstrate sensitivity to the potent broad-spectrum HDAC inhibitor panobinostat (Fig. 1),8 and panobinostat is being evaluated in several clinical trials for DIPG (e.g. NCT02717455, NCT03632317 and NCT03566199). CED delivery of panobinostat itself,6 a micellular formulation of panobinostat,9 and a PET-reporting 18F-panobinostat derivative10in vivo have also been reported. Panobinostat is only sparingly soluble in aqueous media, and initiatives to increase the solubility and cellular delivery are highly desirable. Co-ordination of bioactive ligands to platinum drugs is an established strategy, which can improve solubility, also having the potential to result in a synergistic anti-cancer effect, since the platinum fragment and bioactive ligand typically exert anti-cancer activity through different mechanisms.11–14 Several types of therapeutics have been coordinated to platinum complexes, including kinase inhibitors15 anti-inflammatories,16 and HDAC inhibitors (see Fig. 2). Several HDAC inhibitors (e.g. SubH, panobinostat) contain a hydroxamic acid group which chelates Zn2+ in the active site of certain HDAC enzymes; any HDAC delivery agent must therefore preserve the integrity of the hydroxamic functional group.17,18
 | ||
| Fig. 1 Structures of common histone deacetylase (HDAC) inhibitors in widespread use. | ||
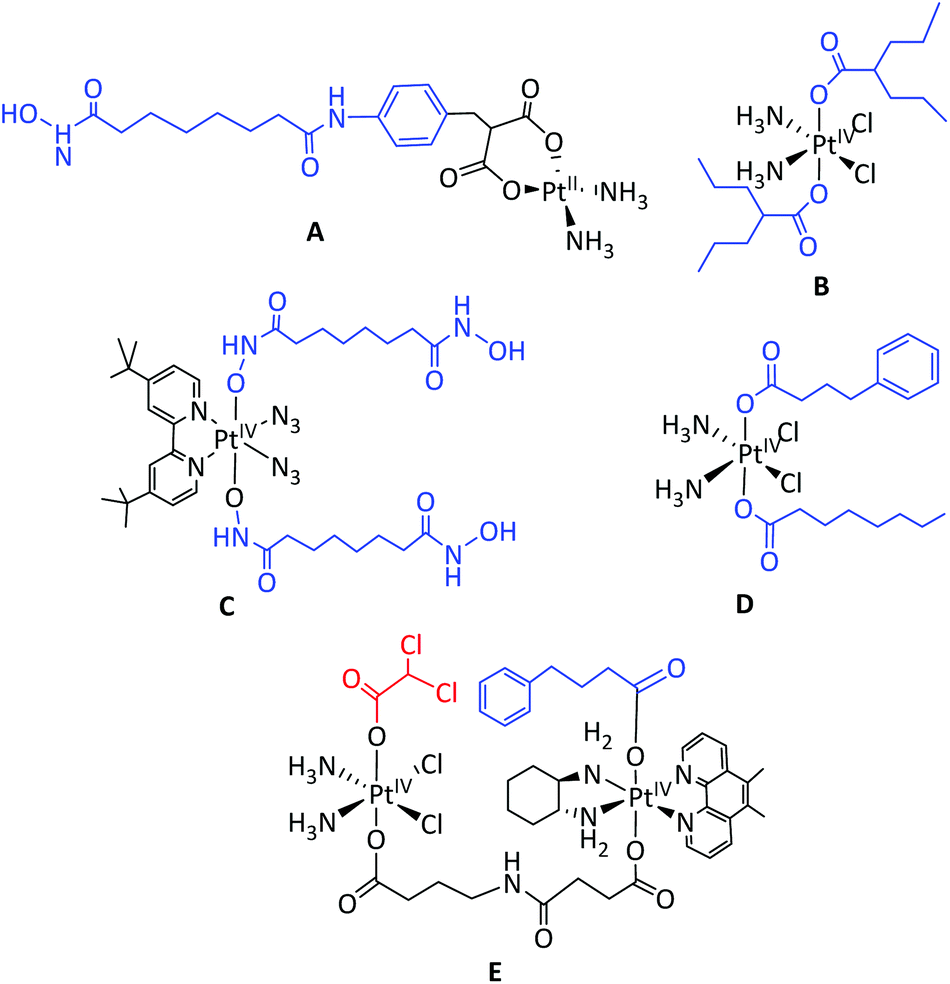 | ||
| Fig. 2 Pt HDAC inhibitor complexes. A: cis-[PtII(NH3)2(malSAHA-2H)] (where SAHA = suberoylanilide hydroxamic acid);25,26B: PtIV valproate complex;27–29C: PtIV Sub complex; D: cis,trans,cis-[Pt(NH3)2(OA)(PhB)Cl2] where OA = octanoate and PhB = phenylbutyrate.30E = dinuclear PtIV multi-modal complex. HDAC inhibitors are labelled in blue, kinase inhibitors (DCA) in red.15 | ||
For these hydroxamic acid HDAC inhibitors, the central region of the molecule occupies the histone deacetylase enzyme's narrow channel, and the terminal group interacts with residues on the enzyme surface. Histone deacetylase inhibition can lead to increased global acetylation levels and relaxation of nuclear DNA, followed by modification of gene regulation and protein recruitment. The precise mechanisms of anti-cancer activity of a given HDAC inhibitor is both cancer-specific and relatively complex.19,20
Established platinum(II) anti-cancer complexes exert their anti-cancer effect primarily by cross-linking DNA (e.g. cisplatin, carboplatin)21 or through alternative mechanisms such as the induction of ribosomal biogenesis stress (e.g. oxaliplatin).22 Oxaliplatin has also been shown to induce multi-faceted biological effects in murine glioma cells at sub-apoptotic drug concentrations.23 Once a platinum(II) compound is taken up into a cancer cell, the chelating spectator ligands (cyclobutane-1,1-dicarboxylate and oxalate, for carboplatin and oxaliplatin respectively) dissociate. For oxaliplatin, it is the oxalate ligand, rather than the aquated Pt(II)-1R,2R-diamminocyclohexane species which is implicated in the common treatment side-effect; peripheral sensory neuropathy (nerve damage).24
Both platinum-based drugs and hydroxamic acid-based HDAC inhibitors are in widespread clinical use for chemotherapy. The hydroxamic acid HDAC inhibitor vorinostat has previously been combined with cisplatin (NCT01045538, NCT00867178, NCT00106626) or carboplatin31,32 for a number of indications in cellulo and in clinical trials.33 Synergies have been reported when using vorinostat and oxaliplatin for hepatocellular carcinoma in vitro and in vivo.34 Several reports have detailed the coordination and co-delivery of hydroxamic acids with platinum(II) complexes,35 for either their HDAC inhibitory or NO releasing properties.36 Notably, the hydroxamic acid suberoylanilide hydroxamic acid (SAHA) was derivatised with malonate (malSAHA) at the terminal end, with (carboplatin-like) platinum(II) coordination through the malonate group rather than the hydroxamic acid (complex A, Fig. 2). Although this enabled successful platinum(II) coordination, the resulting complex exhibited no HDAC inhibitory activity in the cell line tested.25,26 Our preliminary cytotoxicity data (Table 1) indicated that oxaliplatin showed promising cytotoxicity – significantly higher than carboplatin – towards DIPG cell lines. We were therefore keen to evaluate the effect of combining both oxaliplatin and structural derivatives of oxaliplatin with additional therapeutics. Different strategies for incorporating the hydroxamic acid HDAC inhibitors SubH and panobinostat within the coordination sphere of platinum were investigated, including the replacement of the oxalate ligand of oxaliplatin. We characterised the resultant novel platinum complexes and compared their biological activity to that of established chemotherapeutics, and report our findings here.
| Compound | Diffuse intrinsic pontine glioma (DIPG) cell line | |||||
|---|---|---|---|---|---|---|
| SU-DIPG-IV | BIOMEDE 194 | SU-DIPG-XXI | ||||
| IC50 (μM) | 95% CI (μM) | IC50 (μM) | 95% CI (μM) | IC50 (μM) | 95% CI (μM) | |
| All experiments were conducted with n ≥ 3, except fora (n = 1) andb (n = 2). Three biological repeats were conducted for all experiments. | ||||||
| Carboplatin | 66.3 | 30–143 | 78.5b | 55–133 | >5 | — |
| Oxaliplatin | 0.333 | 0.108–1.043 | >5 | — | 0.135 | 0.101–0.179 |
| SubH | 210.7a | 123.1–370.7 | >5 | — | >5 | — |
| Panobinostat | 0.002 | 0.002–0.003 | 0.027 | 0.015–0.049 | 0.002 | 0.001–0.002 |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Oxaliplatin + Panobinostat :1 Oxaliplatin + Panobinostat |
0.006 | 0.004–0.009 | 0.023 | 0.0153–0.034 | 0.003 | 0.002–0.004 |
| 1:1 Oxaliplatin + SubH |
0.552 | 0.226.7–1.504 | >5 | — | 0.250 | 0.177–0.354 |
| Complex 1 | >5 | — | >5 | — | >5 | — |
| Complex 2 | 0.021 | 0.0114–0.039 | 0.067 | 0.046–0.095 | 0.009 | 0.007–0.013 |
Results and discussion
Synthesis and characterisation of novel platinum(II) complexes
Oxaliplatin was purchased from MedChemTronica and used as received (purity data in ESI†); [PtII(R,R-DACH)Cl2]37 was synthesised by literature methods (see ESI†). Treatment of [PtII(R,R-DACH)Cl2] with AgNO3 in H2O abstracted the chloride ligands, with AgCl removed by filtration. Following addition of the hydroxamic acid (SubH or panobinostat), the solution was stirred and then concentrated under reduced pressure. The compounds [PtII(R,R-DACH)(Sub-H)] [1] and [PtII(R,R-DACH)(panobinostat-2H)] [2] were isolated by mass-directed prep-HPLC (MeCN + 0.1% NH4OH/H2O + 0.1% NH4OH, pH 9) and lyophilised (Fig. 3). Complex 1 demonstrated good aqueous solubility (maximum solubility 344 mg mL−1, 672 mM), as determined by ICP-MS. Complex 2 was sparingly soluble in H2O (maximum solubility 0.047 mg mL−1, 0.072 mM), but to a greater extent than free panobinostat (maximum aqueous solubility: 0.000834 mg mL−1 (calculation – ALOGPS); 20 μM–50 μM (experimentally reported). Complex 2 showed good solubility in d7-DMF (49 mg mL−1, 74 mM). Complexes 1 and 2 were characterised by HRMS, NMR spectroscopy (1H, 13C, 195Pt) and UV-vis spectroscopy. HRMS of [1 + H]+ (C14H28N4O4PtH: 512.1829 m/z found; 512.18211 m/z calculated (1.5 ppm error) (Fig. S1†) and HRMS of [2 + H]+ (C27H35N5O2PtH: 657.2522 m/z found; 657.2511 m/z calculated (1.7 ppm error) (Fig. S2†) were in good agreement with theoretical values. | ||
| Fig. 3 Novel platinum(II) complexes: [PtII(R,R-DACH)(Sub-H)] (1), [PtII(R,R-DACH)(panobinostat-2H)] (2). | ||
195Pt NMR spectroscopic resonances were also observed in the expected spectral region; with resonances for complex 1 at −1973 ppm (D2O; −1947 ppm in d7-DMF) and complex 2 at −1918 ppm (DMF-d7) (Fig. S30†), in a similar region to the resonance reported for oxaliplatin (−1989 ppm, d7-DMF).38 The structures of 1 and 2 were both assigned as far as possible from 1D and 2D NMR spectra, and alternative coordination modes were also considered (see ESI†). A change in chemical shifts for the coordinated ligands compared to the free hydroxamic acids were observed by both 1H NMR and 13C NMR spectroscopy, with 1H integral ratios also supporting ligand complexation. For both 1 and 2, platinum coordination of the ligand induced inequivalence in the 13C NMR spectral resonances in the R,R-DACH ring, consistent with a reduction in symmetry of the platinum(II) complex, in comparison to oxaliplatin. For complex 1; small resonances which may correlate to DACH-NH2 protons were observed in D2O at 5.87 ppm and 5.19 ppm (Fig. S11†). Acquiring the 1H NMR spectra of 1 in DMF-d7 enabled unequivocal visualisation of the four DACH-NH2 proton environments, which were deshielded compared to oxaliplatin: being observed at 6.38 ppm and 6.34 ppm (HJ, HL), and 5.62 ppm and 5.55 ppm (HK, HM), and 6.35 ppm and 5.57 ppm in 1 (DMF-d7, Fig. S12†), in comparison to 6.15 ppm and 5.35 ppm in oxaliplatin (DMF-d7, Fig. S2†). These resonances showed correlations to one another and to the DACH HA′ and HA′′ protons (COSY, Fig. S13†). Resonances at higher field (12.56 ppm, 10.68 ppm and 9.08 ppm) all exchanged with the H2O resonance at 3.70 ppm. The resonance at 12.56 ppm showed an HMBC correlation to CO-2 (171.6 ppm) and may correspond with solution protonation of the platinum-coordinated hydroxamic acid group; the resonance at 10.68 ppm shows an HMBC correlation to CO-1 (170.7 ppm) and is fairly similar to the hydroxamic acid OH resonance in the free ligand at 10.32 ppm (d6-DMSO). The inequivalence of HI (2.34 ppm) and HD (2.07 ppm) is consistent with one end of the SubH ligand binding to the platinum centre to a greater extent than the other end of the ligand. For complex 2; multiple functional groups are present in panobinostat which could potentially coordinate to the platinum centre including the hydroxamic acid, the alkene (η2-coordination), the aliphatic amine and the indole amine. Comparing the 1H NMR spectrum of 2 to free panobinostat, there was a notable change in resonances assigned to the vinyl protons, which moved from 6.62 ppm to 6.91 ppm (HU) and from 7.53 ppm to 7.45 ppm (HT). This change could be indicative of either the alkene or hydroxamic acid group coordinating to platinum, however, no platinum satellites were detected in the 13C NMR spectrum for either CU or CT (anticipated coupling ca.1JPtC = 153 Hz reported for [PtCl2(COD)]),39 suggesting that a strong interaction between the alkene and platinum is unlikely, and therefore the deshielding is more likely to be due to coordination of at least part of the hydroxamic acid group. Whilst the 1H NMR spectral resonances of the indole ring were relatively unchanged, systematic deshielding was also observed for the resonances around the aliphatic amine of the panobinostat ligand, possibly indicative of an interaction of this amine with the platinum: HK was shifted from 2.93 ppm to 3.39 ppm, HJ from 2.84 ppm to 3.22 ppm and HM from 3.86 ppm to 4.50 ppm. The resonances at 6.52 ppm and 5.74 ppm which correspond to DACH amine protons showed weak HMBC correlations to the DACH CA′ and CA′′13C NMR spectral resonances. In comparison to free panobinostat, two new singlets were also observed for complex 2 at 13.26 ppm and 9.43 ppm, as well as the resonance at 10.90 ppm (possibly a hydroxamic acid OH group), similar to the coordination behaviour observed for complex 1. These new resonances could be NH groups, but are difficult to assign further, the resonance at 9.43 ppm showed no HMBC correlations (the HMBC experiment did not incorporate the 13.26 ppm resonance). If this is the case, it could imply protonation/retention of the hydroxamic acid OH group following platinum coordination (see Fig. S23†).
Stability of complexes 1 and 2
The solution stability of complexes 1 (D2O) and 2 (DMF-d7) was investigated by 1H NMR spectroscopy and HPLC. 1H NMR spectroscopy demonstrated that complexes 1 and 2 were unchanged in D2O and DMF-d7 respectively for at least 3 d at 18 °C, with no free hydroxamic acid ligand observed. When the complexes 1 and 2 were analysed by HPLC under either neutral or basic conditions, free ligand (205.15 m/z for [SubH + H]+ and 350.20 m/z for [panobinostat + H]+) was observed, suggesting that the hydroxamic ligands are partially labile under HPLC conditions. For both complexes, carboxylic acid derivatives of the hydroxamic acid ligands (suggested to have formed via nitrosocarbonyl intermediates40) were also detected in the HPLC trace (Fig. S34†); for complex 1, [“SubH − NH” + H]+ (F) was detected at 190.08 m/z (model 190.11 m/z, positive ionisation mode). For complex 2, [“pano − NH” + H]+ (G) was detected at 335.16 m/z (model 333.18 m/z, positive ionisation mode) and the corresponding species [“pano − NH2”]− (H) was detected at 333.12 m/z (model 333.16 m/z, negative ionisation mode) at the same retention time. However, these carboxylate derivatives were also observed during HPLC analysis of the free ligands (SubH and panobinostat) alone.Attempted oxidation of 1 and 2 to platinum(IV) complexes
The oxidation of complex 1 (20 mg in 5 ml H2O and 3 eq. H2O2) at ambient temperature was monitored over a period of 24 h by HPLC; formation of [PtII(R,R-DACH)(Sub)(OH)2] (3) (detected as [3 + H]+) was confirmed by HRMS (C14H30N4O6PtH: 546.1887 m/z found; 546.1886 m/z calculated, 0.18 ppm error) (Fig. S35†), but the reaction did not proceed to completion, despite extended reaction times (48 h). Several aggregation species and fragmentation products of 3 were also observed, supporting the identification of the oxidised product (Fig. S36†). Complex 3 was isolated by mass-directed HPLC as a pale yellow solid, however, following solvent removal (lyophilisation) 3 readily decomposed, forming a black Pt(0) precipitate. Use of more forcing conditions (excess H2O2, reaction temperatures of 45 °C, 55 °C or 75 °C) did not improve conversion to 3, as judged by HPLC. Despite repeated attempts to purify and analyse 3 (including isolation of the product by precipitation with diethyl ether from the reaction solution, as an alternative to HPLC purification), the complex was not sufficiently stable to enable further meaningful characterisation.The attempted oxidation of 2 was also monitored by HPLC: the starting complex 2 (detected as the [M + H]+ ion at 657.25 m/z) was gradually consumed over a period of 4h (50 °C, 30 eq. H2O2, DMF), but oxidised product [PtIV(R,R-DACH)(panobinostat-2H)(OH)2] was not detected under any conditions (anticipated at 691.26 m/z).
Compound interaction studies
We conducted brief investigations into the interactions between panobinostat (often administered orally)41 and carboplatin or oxaliplatin (administered intravenously) in solution, since it is typical for hydroxamic acid HDAC inhibitors (e.g. SAHA) and platinum(II) complexes (e.g. carboplatin) to be administered on the same day during clinical treatment regimes.31,32Solutions containing oxaliplatin (2 mg in 0.5 mL H2O) and panobinostat (2 mg in 0.5 mL DMF) were mixed. The vial became warm and a white precipitate formed within 1 min of mixing. This reactivity was not observed when the panobinostat solution (DMF) was mixed with H2O only (i.e. no oxaliplatin). The solution was filtered to remove the precipitate and the filtrate analysed by HPLC at regular timepoints: complex 2 was only detected (as [2 + H]+, 657.29 m/z, tR = 5.4 min) at a very low concentration in the filtrate 24 h after mixing. The experiment was repeated at a lower concentration (as above, but with 0.75 mg compound/0.5 mL solvent) resulting in minimal precipitate formation, such that the reaction could be monitored directly by NMR spectroscopy. 195Pt and 1H NMR spectral monitoring showed only oxaliplatin and panobinostat, with no change or evidence of 2 formation for at least 8 days. At 8 days, HPLC analysis of the NMR sample showed presence of 2 (in addition to panobinostat and oxaliplatin) but a simultaneous 1H NMR spectrum did not, suggesting that 2 is formed only at low concentrations, below the 1H NMR spectroscopic detection limit. To identify the precipitate, the experiment was repeated at higher concentration (as above, but 5 mg compound/0.5 mL solvent) and both solutions were syringe-filtered before mixing. The off-white precipitate (6 mg) was isolated by filtration and dried under vacuum before being re-suspended in DMF-d7. 1H, 195Pt and 13C NMR spectra showed a panobinostat-like compound, with no 195Pt resonances observed over the anticipated 195Pt spectral range (δ: −1204 ppm to −2395 ppm), and crucially no R,R-DACH resonances were detected in the 1H NMR spectrum. Although at higher chemical shifts (>4 ppm) the 1H NMR spectra of panobinostat and the precipitate were identical, a slight shielding of the aliphatic protons (M, K and J) around the aliphatic amine was observed for the precipitate in comparison to free panobinostat (Fig. S31†), possibly indicating a change in the protonation state of the amine group. HPLC analysis of the precipitate was consistent with this, showing predominantly panobinostat (350.06 m/z) and the panobinostat fragmentation product (G, Fig. S34,† at 335.05 m/z) with small contaminants of oxaliplatin ([M + H]+ at 398.94 m/z) and with no evidence of complex 2. The filtrate did not produce further precipitate on standing. HPLC and NMR spectral analysis of the commercial oxaliplatin sample used confirmed a high level of purity (see ESI†). Overall, this suggests that the white precipitate is a panobinostat derivative, rather than a panobinostat-oxaliplatin compound. The mixing was repeated with solutions of carboplatin (2 mg in 0.5 mL H2O) and panobinostat (2 mg in 0.5 mL DMF); no precipitate formed, and HPLC analysis showed no observable formation of a carboplatin adduct of panobinostat, monitored over a period of 3 d.
Biological studies
Complexes 1 and 2 were evaluated in cellulo alongside established complexes oxaliplatin, carboplatin, panobinostat and SubH, as well as 1:1 mixtures of oxaliplatin with SubH and panobinostat (Table 1 and Fig. S37†). Cytotoxicity values for carboplatin4,6 and panobinostat6 have previously been reported in DIPG cell lines, but we could find no reports evaluating either oxaliplatin or SubH for DIPG. We used three well-characterised, patient-derived, low passage DIPG cell lines with different mutational statuses in the key driver mutations (H3K27M and ALK2). H2O (carboplatin) or DMF (all other compounds) was used for solubilising compounds for cell testing, the toxicity of DMF itself was shown to be negligible at the concentrations used. Cisplatin, carboplatin and oxaliplatin are known to be unstable in DMSO:buffer mixtures, for this reason DMSO was avoided.42,43 SubH showed no observable cytotoxicity towards the cell lines, and mixtures of SubH and oxaliplatin were less cytotoxic than oxaliplatin alone. Panobinostat was potently cytotoxic towards all three cell lines, particularly SU-DIPG-IV; consistent with the value of ∼0.1 μM reported for a range of DIPG lines.6
Mixtures of panobinostat and oxaliplatin were slightly less potent than panobinostat alone. Carboplatin showed no observable toxicity towards the DIPG cell lines at the concentrations evaluated, consistent with reports of IC50 values >100 μM in seven DIPG lines, including SU-DIPG-IV, which was evaluated here.6 It is noted however that other glioma cell lines are known to demonstrate sensitivity to carboplatin (e.g. cell lines DUB D003: IC50 0.047 mg mL−1; SF8628 IC50 0.026 mg mL−1 and SF7761 IC50 0.0048 mg mL−1).4
Discussion
Synthesis, characterisation and stability of novel platinum complexes
Since several hydroxamic acid-chelated platinum(II) complexes have been previously reported,44 equatorial hydroxamic acid chelation was investigated, as an alternative route to complexation. Oxaliplatin was selected as the parent complex, due to encouraging preliminary cytotoxicity data (Table 1). It was envisaged that the platinum(II) complex could act as a “carrier” of the chelated hydroxamic acid – with the potential to improve the solubility in the case of panobinostat – with the hydroxamic acid being released from the platinum(II) complex in cellulo in a similar manner to dissociation of the oxalate ligand from oxaliplatin. The platinum(II) complexes of SubH and panobinostat (1 and 2 respectively) were successfully synthesised and characterised, with analytical data consistent with the proposed structures. For previously reported platinum(II) hydroxamic acid complexes, the hydroxamic acid ligand was demonstrated to have rearranged to a hydroximic acid through deprotonation of the nitrogen and delocalisation of the carbonyl double bond in the coordinated complex (Fig. 4, Complex F),35 and we suggest that this is the most likely coordination geometry in 1 and 2, although alternative binding modes are presented in the ESI.† Since the integrity of the released hydroxamic acid was also important for preserving potential HDAC inhibitory activity, this was investigated by HPLC.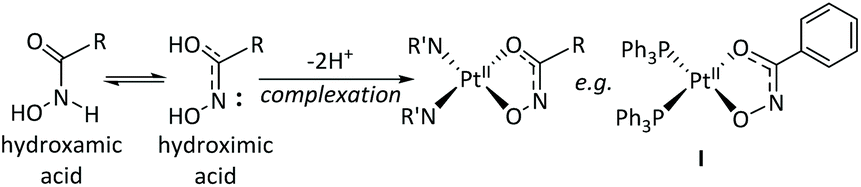 | ||
| Fig. 4 Previously reported hydroxamic acid complexation modes for Pt(II); complex I: [Pt(sha-H)(PPh3)2].35 | ||
Although a small amount of degraded hydroxamic acid (F and G/H) was detected, this was roughly constant with the intensities observed during HPLC analysis of solutions of the free hydroxamic ligands themselves. The majority of the ligand which was released from 1 and 2 was detected as the protonated intact hydroxamic acid – either free SubH or panobinostat – rather than the degraded forms, suggesting that the hydroxamic acid ligands are relatively labile from the complex under HPLC conditions, and that release from 1 and 2 does not result in any appreciable degradation of the hydroxamic acid.
Attempted oxidation of 1 and 2 to platinum(IV) complexes
Platinum(IV) complexes typically demonstrate greater kinetic inertness towards ligand substitution compared with platinum(II) complexes, due to their octahedral, d6 configuration.11 It was envisaged that such a platinum(IV) complex could retain coordination of the hydroxamic acid until the complex was reduced to platinum(II) in cellulo.Previous attempts to oxidise platinum(II) hydroxamate complexes have met with mixed success; attempted oxidation of a platinum(II) pyridinehydroxamate complex resulted in formation of a platinum(IV) complex with axial chlorido, rather than hydroxido ligands, and also resulted in destruction of the hydroxamic acid group to form a carboxylic acid.45 Furthermore, hydroxamic acids have been reported to facilitate reduction of platinum(IV) to platinum(II).46 However, we felt it was of interest to briefly investigate the possibility of accessing platinum(IV) derivatives of 1 and 2, since there was the possibility that hydroxamic chelation could be stabilised due to the hydroxamic acid favouring harder metal ions (PtIVvs. PtII), and the relative inertness of the d6-configuration. Whilst ESI-MS provided evidence that 1 had been successfully oxidised to 3, this compound was unstable and a purified sample could not be further analysed. No evidence of oxidation of complex 2 was observed.
Biological investigations
As a mutant histone phenotype is observed in up to 80% of DIPG cases, the use of HDAC inhibitors in combination with other agents is a promising line of investigation for DIPG.47 We determined that oxaliplatin, complex 2 and panobinostat showed good activity towards SU-DIPG-IV (HIST1H3B and ACVR1-R206H mutant) and SUDIPG-XXI (HIST1H3B and ACVR1-G328W mutant) lines. This is encouraging, since oxaliplatin has not previously been identified as a potential treatment for DIPG. There was however, no synergy observed for treatment with both oxaliplatin and panobinostat in a 1:1 ratio, under the conditions tested. Furthermore, the cytotoxicity of complex 2 is 3–10 fold lower than that of panobinostat itself. This demonstrates that complexation has impacted on the absolute cytotoxicity of panobinostat. However, this is not neccesarily concerning, partly because cell culture is often not the best indicator of the in vivo activity of a prodrug, and also because it is too early to comment on the specificity of activity of 2 towards DIPG in comparison to non-cancerous cells. Determining if complexation can reduce the broad spectrum toxicity of panobinostat is planned in subsequent work. All compounds, including the broad-spectrum HDAC inhibitor panobinostat were more effective in the two cell lines with driver mutations than the BIOMEDE 194 (wild type in both driver mutations) cell line. Since oxaliplatin is likely to exert its cytotoxic effect through a different mechanism of action to panobinostat, this suggests a global increase in sensitivity in the cells harbouring driver mutations, but further work is needed to elucidate the precise reasons and mechansims of cell death. In comparison to oxaliplatin, carboplatin showed only modest cytotoxicity towards the DIPG cell lines under the conditions reported here.6
Experimental
For Materials, Methods and Procedures see ESI.†Conclusions
Two novel PtII complexes 1 (SubH derivative) and 2 (panobinostat deriviative) have been synthesised and characterised. Complex 1 could be oxidised to the platinum(IV) complex 3 but this was insufficiently stable for analysis beyond ESI-MS, decomposing on isolation, and making further development of 3 as an anti-cancer Pt(IV) prodrug unlikely. We were unable to oxidise the panobinostat complex 2 to the corresponding dihydroxido Pt(IV) species.Complexes 1 and 2 were biologically evaluated in three low-passage patient-derived DIPG cell lines, alongside both established compounds and 1:1 mixtures of those compounds. Oxaliplatin showed good activity towards the three DIPG cell lines, whereas carboplatin showed modest cytotoxicity in these lines. To our knowledge, oxaliplatin has not been previously evaluated for DIPG, either in cellulo or in vivo. Complex 2 showed signficantly greater cytotoxicity than oxaliplatin towards all three DIPG cell lines. We observed no obvious synergies in cellulo for any of the mixtures of compounds. The unexpected immediate reactivity observed on mixing oxaliplatin and panobinostat solutions in vitro formed a precipitate which was determined to be largely panobinostat, with no platinum complexation observed. The same reactivity was not observed between panobinostat and carboplatin. Since 2 does not include the oxalate ligand, the potential for side-effects of peripheral neuropathy are anticipated to be reduced for 2, in comparison to oxaliplatin. Although complex 2 was moderately less cytotoxic than panobinostat towards all three DIPG cell lines, it exhibited greater aqueous solubility than panobinostat, such that it could potentially be infused by CED at a higher concentration, a key current limitation of panobinostat. Panobinostat is also known to exhibit broad-spectrum toxicity, including towards non-cancerous cells; it is therefore of interest to determine the selectivity of 2, in comparison to panobinostat. HPLC investigations suggested that panobinostat could be released intact (as a hydroxamic acid) from complex 2, such that it would be anticiptated to retain HDAC inhibitory activity in cellulo. Further investigations into the detailed biological effects of oxaliplatin and 2 – including HDAC inhibitory activity for 2 – in a wider panel of patient-derived low-passage DIPG cell lines and (non-cancerous) astrocytes are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Wellcome Trust (201406/Z/16/Z), Cancer Research UK (CR-UK) C5255/A18085, through the CRUK Oxford Centre and the John Fell Fund. We thank Prof. Chris Jones and Prof. Michelle Monje for the DIPG cell lines, Prof. Alex Bullock for assistance with cellular assays and Profs. Stephen Faulkner, Stuart Conway and Akane Kawamura for helpful discussions.References
- ONS 2015 Death Registration Summary Tables, England & Wales, Table 2 at: http://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/datasets/deathregistrationssummarytablesenglandandwalesreferencetables.
- Q. T. Ostrom, H. Gittleman, J. Xu, C. Kromer, Y. Wolinsky, C. Kruchko and J. S. Barnholtz-Sloan, Neuro-Oncol., 2016, 18, v1 CrossRef PubMed.
- H. S. Gwak and H. J. Park, Crit. Rev. Oncol. Hematol., 2017, 120, 111 CrossRef PubMed.
- C. L. Killick-Cole, W. G. B. Singleton, A. S. Bienemann, D. J. Asby, M. J. Wyatt, L. J. Boulter, N. U. Barua and S. S. Gill, PLoS One, 2017, 12, e0176855 CrossRef PubMed.
- N. U. Barua, S. P. Lowis, M. Woolley, S. O'Sullivan, R. Harrison and S. S. Gill, Acta Neurochir., 2013, 155, 1459 CrossRef CAS PubMed.
- C. S. Grasso, Y. Tang, N. Truffaux, N. E. Berlow, L. Liu, M. A. Debily, M. J. Quist, L. E. Davis, E. C. Huang, P. J. Woo, A. Ponnuswami, S. Chen, T. B. Johung, W. Sun, M. Kogiso, Y. Du, L. Qi, Y. Huang, M. Hütt-Cabezas, K. E. Warren, L. Le Dret, P. S. Meltzer, H. Mao, M. Quezado, D. G. Van Vuurden, J. Abraham, M. Fouladi, M. N. Svalina, N. Wang, C. Hawkins, J. Nazarian, M. M. Alonso, E. H. Raabe, E. Hulleman, P. T. Spellman, X. N. Li, C. Keller, R. Pal, J. Grill and M. Monje, Nat. Med., 2015, 21, 555 CrossRef CAS PubMed.
- W. G. B. Singleton, A. S. Bieneman, M. Woolley, D. Johnson, O. Lewis, M. J. Wyatt, S. J. P. Damment, L. J. Boulter, C. L. Killick-Cole, D. J. Asby and S. S. Gill, J. Neurosurg. Pediatr., 2018, 288 Search PubMed.
- T. Hennika, G. Hu, N. G. Olaciregui, K. L. Barton, A. Ehteda, A. Chitranjan, C. Chang, A. J. Gifford, M. Tsoli, D. S. Ziegler, A. M. Carcaboso and O. J. Becher, PLoS One, 2017, 12, 1 CrossRef PubMed.
- W. G. Singleton, A. M. Collins, A. S. Bienemann, C. L. Killick-Cole, H. R. Haynes, D. J. Asby, C. P. Butts, M. J. Wyatt, N. U. Barua and S. S. Gill, Int. J. Nanomed., 2017, 12, 1385 CrossRef CAS PubMed.
- H. Kommidi, U. Tosi, U. B. Maachani, H. Guo, C. S. Marnell, B. Law, M. M. Souweidane and R. Ting, ACS Med. Chem. Lett., 2018, 9, 114 CrossRef CAS PubMed.
- D. Gibson, J. Inorg. Biochem., 2019, 191, 77 CrossRef CAS PubMed.
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Chem. Rev., 2019, 119, 1138 CrossRef CAS PubMed.
- R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Eur. J. Inorg. Chem., 2017, 1596 CrossRef CAS.
- M. Ravera, E. Gabano, M. J. McGlinchey and D. Osella, Inorg. Chim. Acta, 2019, 492, 32 CrossRef CAS.
- E. Petruzzella, J. P. Braude, J. R. Aldrich-Wright, V. Gandin and D. Gibson, Angew. Chem., Int. Ed., 2017, 56, 11539 CrossRef CAS PubMed.
- F. P. Intini, J. Zajac, V. Novohradsky, T. Saltarella, C. Pacifico, V. Brabec, G. Natile and J. Kasparkova, Inorg. Chem., 2017, 56, 1483 CrossRef CAS PubMed.
- F. Yang, N. Zhao, D. Ge and Y. Chen, RSC Adv., 2019, 9, 19571 RSC.
- R. Codd, Coord. Chem. Rev., 2008, 252, 1387 CrossRef CAS.
- P. Bose, Y. Dai and S. Grant, Pharmacol. Ther., 2014, 143, 323 CrossRef CAS PubMed.
- T. Eckschlager, J. Plch, M. Stiborova and J. Hrabeta, Int. J. Mol. Sci., 2017, 18, 1 Search PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS PubMed.
- P. M. Bruno, Y. Liu, G. Y. Park, J. Murai, C. E. Koch, T. J. Eisen, J. R. Pritchard, Y. Pommier, S. J. Lippard and M. T. Hemann, Nat. Med., 2017, 23, 461 CrossRef CAS PubMed.
- N. B. Roberts, A. Alqazzaz, J. R. Hwang, X. Qi, A. D. Keegan, A. J. Kim, J. A. Winkles and G. F. Woodworth, J. Neurooncol., 2018, 140, 497 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645 RSC.
- D. Griffith, M. P. Morgan and C. J. Marmion, Chem. Commun., 2009, 6735 RSC.
- V. Brabec, D. M. Griffith, A. Kisova, H. Kostrhunova, L. Zerzankova, C. J. Marmion and J. Kasparkova, Mol. Pharm., 2012, 9, 1990 CrossRef CAS PubMed.
- E. Gabano, M. Ravera and D. Osella, Dalton Trans., 2014, 43, 9813 RSC.
- J. Yang, X. Sun, W. Mao, M. Sui, J. Tang and Y. Shen, Mol. Pharm., 2012, 9, 2793 CrossRef CAS PubMed.
- M. Alessio, I. Zanellato, I. Bonarrigo, E. Gabano, M. Ravera and D. Osella, J. Inorg. Biochem., 2013, 129, 52 CrossRef CAS PubMed.
- H. Kostrhunova, E. Petruzzella, D. Gibson, J. Kasparkova and V. Brabec, Chem. – Eur. J., 2019, 25, 5235 CrossRef CAS PubMed.
- S. S. Ramalingam, M. L. Maitland, P. Frankel, A. E. Argiris, M. Koczywas, B. Gitlitz, S. Thomas, I. Espinoza-Delgado, E. E. Vokes, D. R. Gandara and C. P. Belani, J. Clin. Oncol., 2010, 28, 56 CrossRef CAS PubMed.
- S. S. Ramalingam, R. A. Parise, R. K. Ramananthan, T. F. Lagattuta, L. A. Musguire, R. G. Stoller, D. M. Potter, A. E. Argiris, J. A. Zwiebel, M. J. Egorin and C. P. Belani, Clin. Cancer Res., 2007, 13, 3605 CrossRef CAS PubMed.
- H. V. K. Diyabalanage, M. L. Granda and J. M. Hooker, Cancer Lett., 2013, 329, 1 CrossRef CAS PubMed.
- B. Liao, Y. Zhang, Q. Sun and P. Jiang, Cancer Med., 2018, 7, 196 CrossRef CAS PubMed.
- M. D. Hall, T. W. Failes, D. E. Hibbs and T. W. Hambley, Inorg. Chem., 2002, 41, 1223 CrossRef CAS PubMed.
- D. Griffith, A. Bergamo, S. Pin, M. Vadori, H. Müller-Bunz, G. Sava and C. J. Marmion, Polyhedron, 2007, 26, 4697 CrossRef CAS.
- S. Kemp, N. J. Wheate, D. P. Buck, M. Nikac, J. G. Collins and J. R. Aldrich-Wright, J. Inorg. Biochem., 2007, 101, 1049 CrossRef CAS PubMed.
- A. C. Tsipis and I. N. Karapetsas, Dalton Trans., 2014, 43, 5409 RSC.
- M. Enders, B. Görling, A. B. Braun, J. E. Seltenreich, L. F. Reichenbach, K. Rissanen, M. Nieger, B. Luy, U. Schepers and S. Bräse, Organometallics, 2014, 33, 4027 CrossRef CAS.
- S. Nourian, R. P. Lesko, D. A. Guthrie and J. P. Toscano, Tetrahedron, 2016, 72, 6037 CrossRef CAS.
- S. F. Jones, J. R. Infante, D. S. Thompson, A. Mohyuddin, J. C. Bendell, D. A. Yardley and H. A. Burris, Cancer Chemother. Pharmacol., 2012, 70, 471 CrossRef CAS PubMed.
- M. D. Hall, K. A. Telma, K. E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913 CrossRef CAS PubMed.
- H. P. Varbanov, D. Ortiz, D. Höfer, L. Menin, M. Galanski, B. K. Keppler and P. J. Dyson, Dalton Trans., 2017, 46, 8929 RSC.
- T. W. Failes, M. D. Hall and T. W. Hambley, Dalton Trans., 2003, 2, 1596 RSC.
- D. Griffith, K. Lyssenko, P. Jensen, P. E. Kruger and C. J. Marmion, Dalton Trans., 2005, 956 RSC.
- D. Griffith, A. Chopra, H. Müller-Bunz and C. J. Marmion, Dalton Trans., 2008, 2, 6933 RSC.
- A. Mackay, A. Burford, D. Carvalho, E. Izquierdo, J. Fazal-Salom, K. R. Taylor, L. Bjerke, M. Clarke, M. Vinci, M. Nandhabalan, S. Temelso, S. Popov, V. Molinari, P. Raman, A. J. Waanders, H. J. Han, S. Gupta, L. Marshall, S. Zacharoulis, S. Vaidya, H. C. Mandeville, L. R. Bridges, A. J. Martin, S. Al-Sarraj, C. Chandler, H.-K. Ng, X. Li, K. Mu, S. Trabelsi, D. H.-B. Brahim, A. N. Kisljakov, D. M. Konovalov, A. S. Moore, A. M. Carcaboso, M. Sunol, C. de Torres, O. Cruz, J. Mora, L. I. Shats, J. N. Stavale, L. T. Bidinotto, R. M. Reis, N. Entz-Werle, M. Farrell, J. Cryan, D. Crimmins, J. Caird, J. Pears, M. Monje, M.-A. Debily, D. Castel, J. Grill, C. Hawkins, H. Nikbakht, N. Jabado, S. J. Baker, S. M. Pfister, D. T. W. Jones, M. Fouladi, A. O. von Bueren, M. Baudis, A. Resnick and C. Jones, Cancer Cell, 2017, 32, 520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, characterisation data and cytotoxicity data. See DOI: 10.1039/c9dt04862f |
| This journal is © The Royal Society of Chemistry 2020 |