
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric hydrogenation of an α-unsaturated carboxylic acid catalyzed by intact chiral transition metal carbonyl clusters – diastereomeric control of enantioselectivity†
Ahmed F.
Abdel-Magied
 a,
Yusuf
Theibich
a,
Amrendra K.
Singh
a,
Ahibur
Rahaman
a,
Isa
Doverbratt
b,
Arun K.
Raha
a,
Matti
Haukka
c,
Michael G.
Richmond
d and
Ebbe
Nordlander
*a
a,
Yusuf
Theibich
a,
Amrendra K.
Singh
a,
Ahibur
Rahaman
a,
Isa
Doverbratt
b,
Arun K.
Raha
a,
Matti
Haukka
c,
Michael G.
Richmond
d and
Ebbe
Nordlander
*a
aChemical Physics, Department of Chemistry, Lund University, Box 124, SE-221 00, Lund, Sweden. E-mail: Ebbe.Nordlander@chemphys.lu.se
bCentre for Analysis and Synthesis, Department of Chemistry, Lund University, Box 124, SE-221 00, Lund, Sweden
cDepartment of Chemistry, University of Jyväskylä, Box 35, FI-400 14, Jyväskylä, Finland
dDepartment of Chemistry, The University of North Texas, Denton, Texas 76203, USA
First published on 27th February 2020
Abstract
Twenty clusters of the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-P–P*)] (P–P* = chiral diphosphine of the ferrocene-based Walphos or Josiphos families) have been synthesised and characterised. The clusters have been tested as catalysts for asymmetric hydrogenation of tiglic acid [trans-2-methyl-2-butenoic acid]. The observed enantioselectivities and conversion rates strongly support catalysis by intact Ru3 clusters. A catalytic mechanism involving an active Ru3 catalyst generated by CO loss from [(μ-H)2Ru3(μ3-S)(CO)7(μ-P–P*)] has been investigated by DFT calculations.
Introduction
Chalcogenide-bridged derivatives of transition metal carbonyl clusters have been widely studied.1–7 Examples of such compounds include the triruthenium hydrido clusters [(μ-H)2Ru3(μ3-E)(CO)9-2x(dppm)x] (E = O, x = 2; E = S, x = 1, 2), which have been shown to be efficient catalyst precursors for olefin hydrogenation reactions.7,8 In these clusters, the triply bridging chalcogenide ligands may function as “clamps” that maintain an intact cluster framework throughout reactions. It is, however, difficult to clearly identify clusters as active catalysts in homogeneous systems,9–11 and there has been considerable debate about whether clusters fragment to form either mononuclear or colloidal species, aggregate to form nanoparticles, or remain as intact clusters during the catalytic cycle. A number of convincing examples of homogeneous cluster catalysis based on indirect evidence have been reported.12 For example, [H4Pt3Ru6(CO)21] catalyses alkyne hydrosilylation and hydrogenation without appearing to undergo fragmentation.13,14 The face-capped trinuclear cluster [HRu3(CO)9(μ3-η2-NMePy)] appears to be the active catalyst in the hydroformylation of diphenylacetylene to α-phenyl-cinnamaldehyde although conversion of [HRu3(CO)9(μ3-η2-NMePy)] to the catalytically inactive dinuclear species [Ru2(CO)6(η3-NMePyCO)] occurs after several cycles.15 Parahydrogen (p-H2) NMR methods have been used to identify active catalysts in hydrogenation reactions.16,17 By using such techniques, Blazina and co-workers have demonstrated that the clusters [H2Ru3(CO)10(L)2] (L = PMe2Ph or PPh3) function as homogeneous catalysts for the hydrogenation of alkynes (diphenylacetylene).18 The active catalysts that were identified are derived from the dihydride species [HRu3(μ-H)(CO)9(L)2].19,20In previous studies, we have investigated asymmetric hydrogenation of α-unsaturated carboxylic acids using catalytic systems based on [(μ-H)4Ru4(CO)12] clusters derivatised with chiral diphosphines. We have shown that such diphosphine ligands may effect strong chiral induction,21,22 and that good conversions and enantioselectivities may be achieved. While catalysis by fragmentation products cannot be excluded, the recovery of unaltered starting clusters after catalytic reactions, recycling of catalysts/catalyst precursors with identical catalytic results, and mercury poisoning tests all implicate the presence of an active cluster catalyst.23
Norton24 has identified a criterion which, if fulfilled, provides incontrovertible evidence for a cluster species acting as an active catalyst, viz. the observation of asymmetric induction in a reaction that is catalysed by a cluster that is chiral by virtue of the cluster framework only (i.e. excluding chiral ligands). Based on this concept, the silylation of acetophenone using chiral tetrahedrane clusters has been established, but cluster racemization was found to occur faster than productive catalysis.24 Here we wish to present the fulfilment of a corollary to Norton's criterion, i.e. proof of diastereomeric control of enantioselectivity when diastereomeric pairs of [(μ-H)2Ru3(μ-S)(CO)7(μ-P–P*)] clusters are used as (asymmetric) hydrogenation catalysts and the chirality of the diphosphine ligand is maintained intact while that of the cluster framework changes. Part of these results have been published in an earlier communication.25
Results and discussion
The syntheses of derivatives of [(μ-H)2Ru3(μ3-S)(CO)9] containing chiral diphosphine ligands were based on oxidative decarbonylation of the starting cluster (using Me3NO) in dichloromethane solution at ambient temperature, in the presence of the relevant diphosphine. The clusters were identified via IR, 1H and 31P NMR spectroscopies, mass spectrometry and, wherever possible, X-ray crystallography.Synthesis and characterization of sulfide-capped triruthenium hydrido clusters containing Walphos ligands
Using the synthetic methodology described above, clusters 3–17 with the common empirical formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-P–P*)], where the diphosphine P–P* is one of the chiral Walphos ligands 1a–1h (Scheme 1), were prepared. Coordination of the heterobidentate ligand to adjacent metals in a bridging mode leads to an intrinsically chiral metallic framework unless there is complete planarity, and the coordination of the enantiomerically pure diphosphine ligand thus yields a mixture of diastereomers due to different connectivities of the heterobidentate ligand (cf.Scheme 1). The diastereomers were separated and isolated using thin layer chromatography, and they were identified on the basis of comparison of their IR and 1H/31P NMR spectral data with those of [(μ-H)2Ru3(μ3-S)(CO)7(dppm)].7 Clusters 3–17 were completely characterized by spectroscopic methods (cf. Experimental section). The empirical formulae of all clusters were confirmed by mass spectrometry and in the cases of clusters 3–6 and 8, their solid-state structures were determined by X-ray crystallography. The 1H and 31P NMR data confirmed the difference in the bridging mode of the diphosphine ligands in 3–17.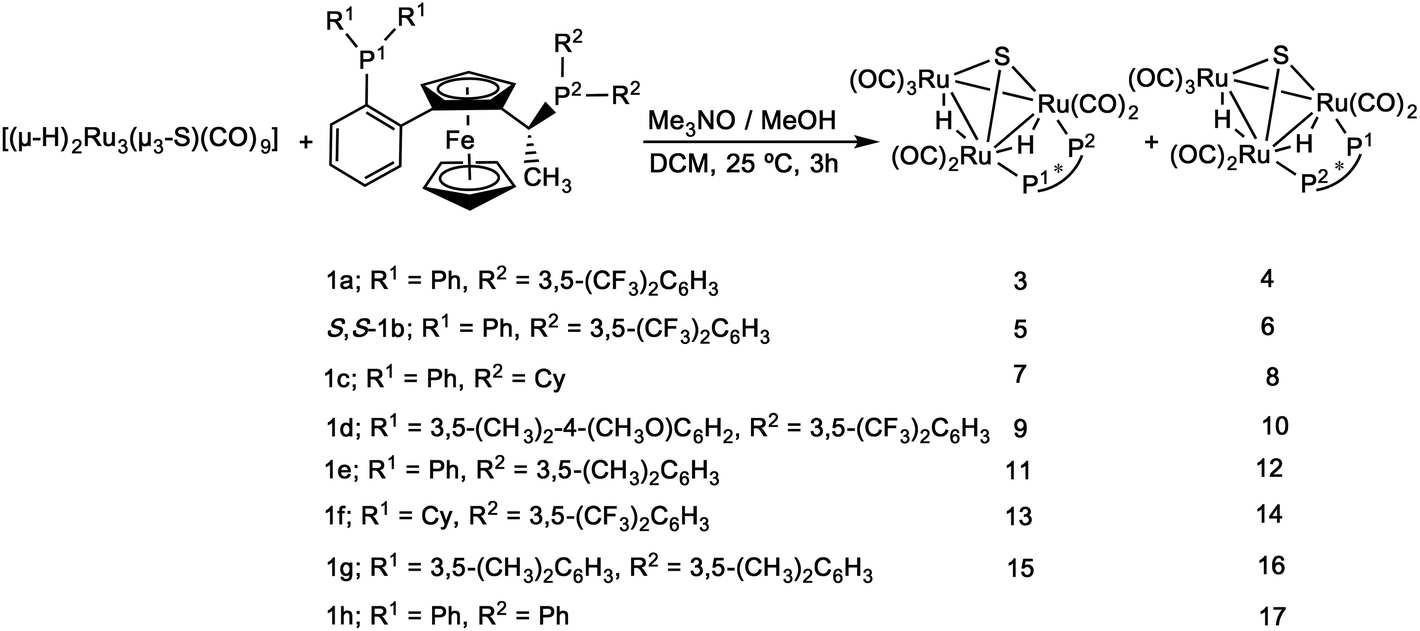 | ||
| Scheme 1 Synthesis of clusters 3–17 containing Walphos ligands 1a–1h. | ||
For 3, the 1H NMR in the hydride region shows two signals, a doublet of doublet of doublets at δ −18.13 (JH–P = 12.4, JH–P = 11.2, JH–H = 2.9 Hz) and an (apparent) doublet of triplets at δ −18.40 (JH–P = 9.8, JH–H = 2.9 Hz), while for the corresponding diastereomer 4, the 1H NMR in the hydride region shows two signals at δ −17.82 (ddd, JH–P = 12.2, JH–P = 8.8, JH–H = 3.5 Hz) and δ −18.63 (ddd, JH–P = 7.5, JH–P = 3.6, JH–H = 3.5 Hz).
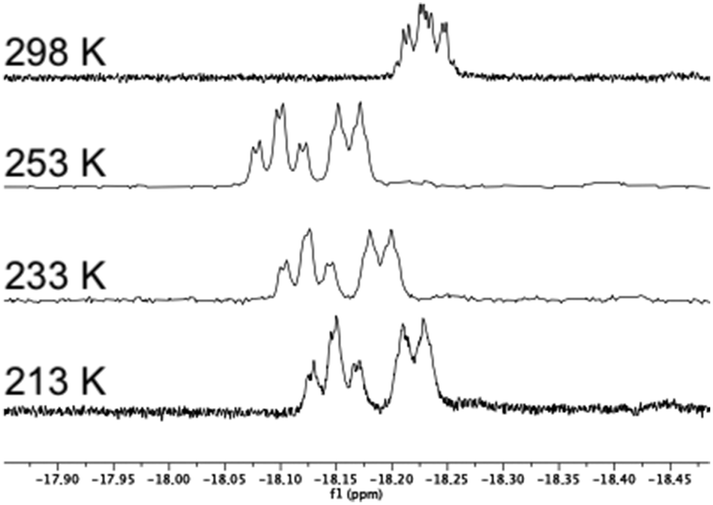
In the 31P NMR spectrum of 3, the signal for phosphorus P1 (cf.Scheme 1) appears as a triplet, indicating coupling to both hydrides, while the signal for P2 is a doublet. In contrast, the signal for P1 in 4 appears as a doublet while that of P2 is a doublet of doublets. The identities of the two diastereomers were further confirmed by the determination of their crystal structures (Fig. 2 and 3). The analogous diastereomeric pair based on S,S-1b, viz.5 (with diphosphine connectivity corresponding to 3) and 6 (connectivity corresponding to 4) were prepared, and characterized by comparing their spectroscopic data with those of 3 and 4. The 1H NMR spectrum of 5 in the hydride region shows two signals at δ −18.12 (ddd, JH–P = 12.6, JH–P = 11.1, JH–H = 3.0 Hz) and at δ −18.40 (d′t′, JH–P = 9.9, JH–H = 3.0 Hz), while for 6 the corresponding signals are found at δ −17.84 (ddd, JH–P = 12.4, JH–P = 9.1, JH–H = 3.0 Hz) and δ −18.65 (ddd, JH–P = 7.1, JH–P = 3.2, JH–H = 3.0 Hz). Similarly, in the 31P NMR spectrum of 5, the P1 signal (cf.Scheme 1) appears as a doublet of doublets, while the signal for P2 is a triplet, indicating coupling to both hydrides. In contrast, the signal for P1 in the corresponding diastereomer 6 appears as a doublet of doublets while that of P2 is a doublet. Again, the identities of the two diastereomers were further confirmed by the determination of their crystal structures (Fig. 2 and 3).
 | ||
| Fig. 1 Variable-temperature 1H NMR spectra (hydride region) of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1g)] 16 recorded over the temperature range 298–213 K (top to bottom). | ||
 | ||
| Fig. 2 Molecular structures of four diastereomers (two diastereomeric pairs) representing different combinations of cluster framework and ligand chiralities, using pure enantiomeric forms R,R-1a or S,S-1b ligands; (a) [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1a)] 3, CCDC 993899,† selected bond distances [Å] and angles [°]: Ru1–Ru2 2.8952(9), Ru1–Ru3 2.9022(9), Ru2–Ru3 2.7537(8), Ru1–P1 2.359(2), Ru2–P2 2.3332(19), Ru1–S1 2.368(2), Ru2–S1 2.375(2), Ru3–S1 2.346(2), Ru1–H1 1.77(7), Ru1–H2 1.70(6), Ru2–H2 1.75(6), Ru3–H1 1.77(7); (b) [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1a)] 4, CCDC 993900,† selected bond distances [Å] and angles [°]: Ru1–Ru2 2.915(2), Ru1–Ru3 2.746(2), Ru2–Ru3 2.894(2), Ru1–P2 2.335(5), Ru2–P1 2.349(5), Ru1–S1 2.361(5), Ru2–S1 2.358(4), Ru3–S1 2.351(5). Ru1–H1 1.569, Ru1–H2 1.57, Ru2–H2 1.528, Ru3–H1 1.674 (riding on Ru atoms); (c) [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)] 5, CCDC 1043616,† selected bond distances [Å] and angles [°]: Ru1–Ru2 2.9054(2), Ru1–Ru3 2.7518(2), Ru2–Ru3 2.8818(2), Ru1–P1 2.3331(6), Ru2–P2 2.3502(6), Ru1–S1 2.3811(5), Ru2–S1 2.3728(5), Ru3–S1 2.35568(5), Ru1–H1 1.71(4), Ru2–H1 1.74(4), Ru2–H2 1.84(4), Ru3–H2 1.69(4), and (d) [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)] 6, CCDC 1043617,† selected bond distances [Å] and angles [°]: Ru1–Ru2 2.9210 (12), Ru1–Ru3 2.893(3), Ru2–Ru3 2.7657(12), Ru1–P2 2.341(2), Ru2–P1 2.361(2), Ru1–S1 2.359(2), Ru2–S1 2.357(2), Ru3–S1 2.355(3), Ru1–H1 1.72(5), Ru1–H2 1.678(17), Ru2–H2 1.57(5), Ru3–H1 1.57(4). Thermal ellipsoids are drawn at the 50% probability level and C–H hydrogen atoms have been omitted for the sake of clarity. | ||
 | ||
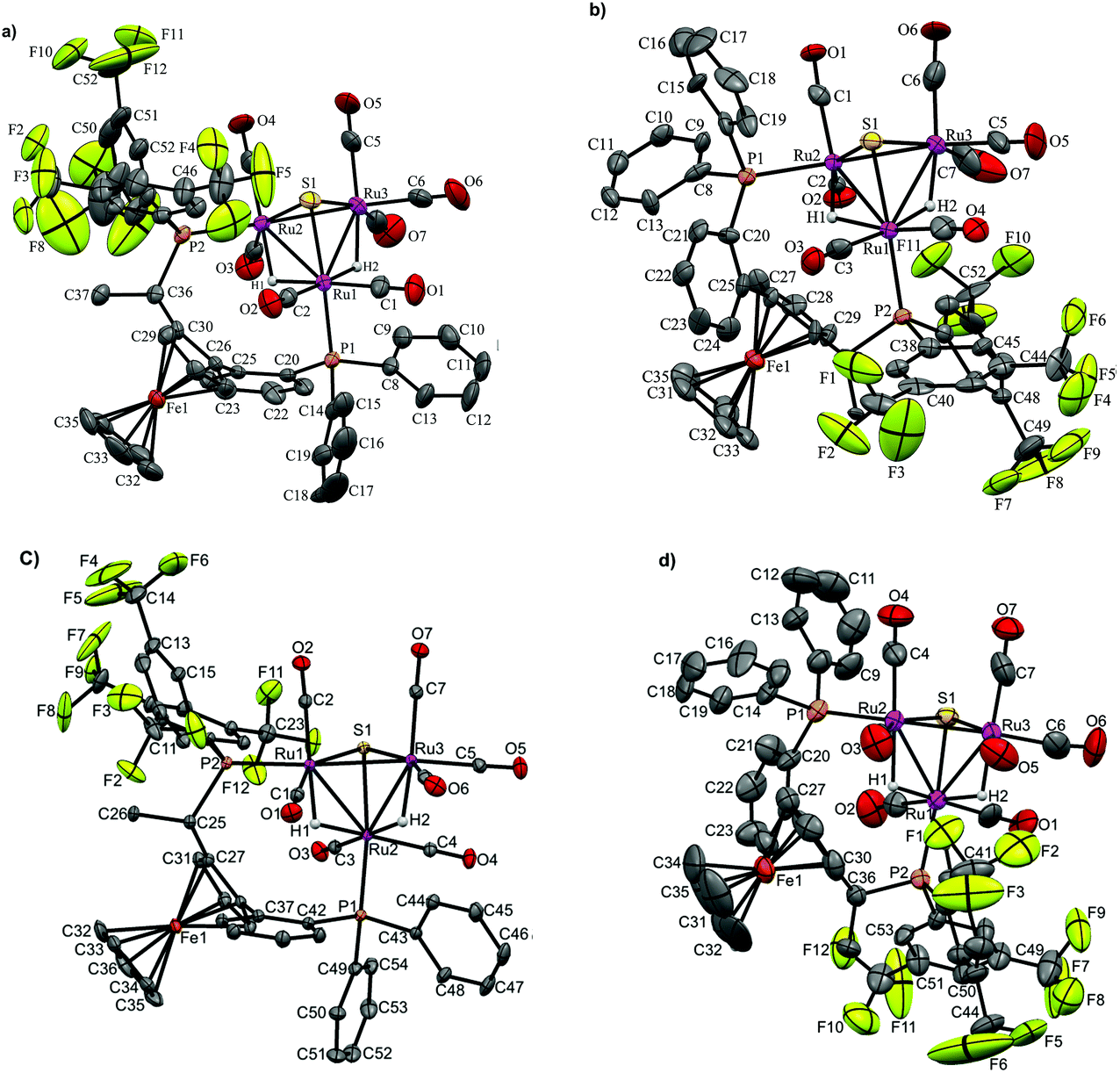
| Fig. 3 Molecular structure of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1c)] 8 (CCDC 1043618†) with thermal ellipsoids drawn at the 50% probability level. C–H hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°]: Ru1–Ru2 2.9276(6), Ru1–Ru3 2.7511(6), Ru2–Ru3 2.9123(6), Ru1–P1 2.3376(13), Ru2–P2 2.3833(13), Ru1–S1 2.3649(14), Ru2–S1 2.3653(13), Ru3–S1 2.3635(13), Ru1–H1 1.90(4), Ru2–H2 1.76(7), Ru2–H1 1.78(4), Ru3–H2 1.94(7). | ||
The 1H NMR of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1g)] 16 in the hydride region shows a multiplet signal at δ −18.23, which indicates fluxionality of the hydrides at ambient temperature. Variable-temperature 1H NMR spectra in the hydride region for 16 show that the fluxionality may be frozen out at approximately 253 K to give two signals, an apparent triplet of doublets at δ −18.12 (d′t′ = ddd, JH–P = 10.9, JH–H = 3.0 Hz) and a doublet of ‘triplets’ (ddd) at δ −18.19 (JH–P = 9.4 Hz) (Fig. 1).
Crystal and molecular structures of clusters 3–6, 8 and 17
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 25 and [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)] 5 and 6.
The molecular structures of 3–6 are shown in Fig. 2 and selected bond distances and angles are listed in the caption. Details regarding data collection and reduction, and structure refinement are found in the Experimental section, and relevant crystallographic data are collated in Table S1, ESI.†
25 and [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)] 5 and 6.
The molecular structures of 3–6 are shown in Fig. 2 and selected bond distances and angles are listed in the caption. Details regarding data collection and reduction, and structure refinement are found in the Experimental section, and relevant crystallographic data are collated in Table S1, ESI.†
As may be expected, the different diastereomers crystallize in non-centrosymmetric space groups (3, 4 and 6: P212121). For complex 5, the polar space group Cc was identified. It was found that this structure contained two different isomers of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)] with opposite chiralities of the diphosphine ligand 1b (cf.Scheme 1), i.e. both S,S-1b and R,R-1b were identified in the structure even though enantiopure S,S-1b had been used in the synthesis of cluster 5. Our explanation for this surprising result is that a small amount of R,R-1b was present in the ligand batch used, and the resultant isomer of 5 co-crystallized with the (majority) product [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-S,S-1b)]. Compound 5 was resynthesized several times and several crystallization attempts were made. A second crystal structure of what appears to be diastereomerically pure [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-S,S-1b)] 5 was obtained (CCDC 1982115). For this structure, the non-centrosymmetric space group P21 was identified, but the quality of the diffraction data was unfortunately so poor that this assignment could not be made with absolute certainty. For this reason, the discussion below relates to the data for [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-S,S-1b)] taken from the crystal structure in the polar space group Cc.
The structures of the two diastereomers based on ligand 1a (3 and 4) are discussed in detail below. The molecules consist of triangular ruthenium frameworks involving Ru–Ru bond lengths that are divided into two distinctive classes, the two hydride-bridged Ru–Ru bonds are “long” [3: Ru(1)–Ru(2) 2.8952(9), Ru(1)–Ru(3) 2.9022(9) Å]; 4: [Ru(1)–Ru(2) 2.915(2), Ru(1)–Ru(3) 2.746(2) Å] and the non-bridged Ru–Ru bond is “short” [3: Ru(2)–Ru(3) 2.7537(8); 4: Ru(2)–Ru(3) 2.894(2) Å]. The triply bridging sulfur atom caps the ruthenium triangle quite symmetrically [Ru–S 2.375(2)–2.368(2) Å for 3, and 2.361(5)–2.358(5) Å for 4]. All the carbonyl ligands are coordinated in a terminal monodentate coordination mode. The diphosphine ligands were found to exclusively coordinate in a bridging fashion, giving rise to nine-membered “dimetallacycles”. As previously observed for related tetraruthenium tetrahydrido clusters,22 the Walphos ligand coordinates in an axial-equatorial mode. In the case of 3, P1 is coordinated in an axial position to Ru1, the metal that is coordinated to both of the hydrides, while P2 is coordinated in an equatorial position to Ru2, which is coordinated by one hydride. For the diastereomer 4, P1 is coordinated in an equatorial position to Ru2, which is coordinated to H1, while P2 is coordinated in an axial position to Ru1, which is coordinated to H1 and H2.
In the molecular structures of the analogous diastereomers based on 1b, 5 and 6, the same difference in the coordination mode of the diphosphine ligand S,S-1b is observed – P1 is coordinated in an axial position to Ru2 which is coordinated to both hydrides, in case of 5, and for 6, P1 is coordinated in an equatorial position to Ru2 which is bridged only with one hydride H1.
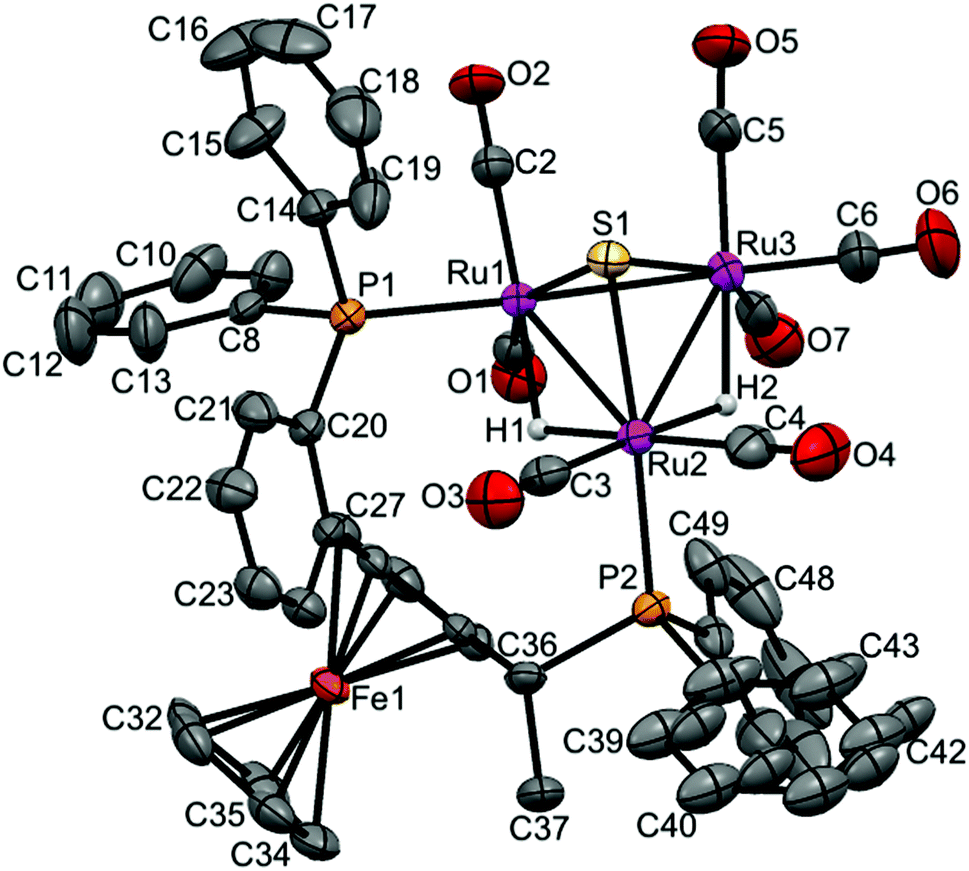 | ||
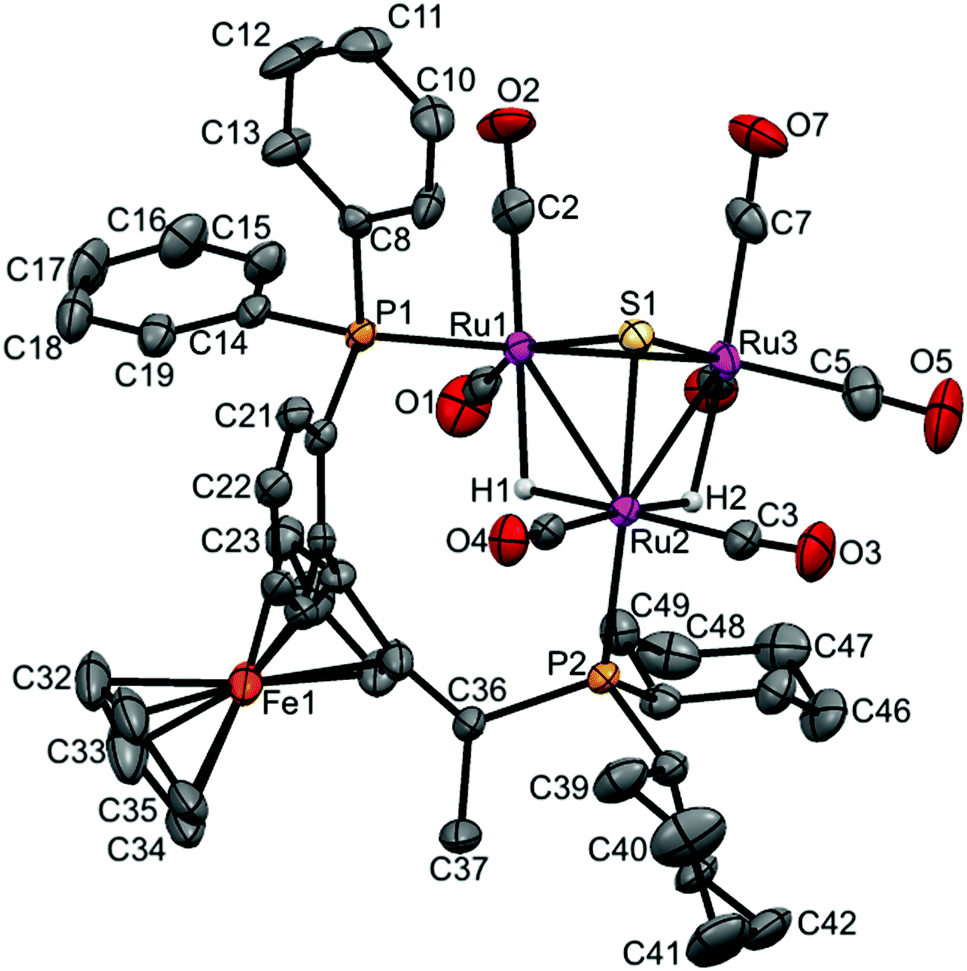
| Fig. 4 Molecular structure of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1h)] 17 (CCDC 1043619†) with thermal ellipsoids drawn at the 50% probability level. C–H hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°]: Ru1–Ru2 2.9173(8), Ru1–Ru3 2.7359(8), Ru2–Ru3 2.8921(8), Ru1–P1 2.339(19), Ru2–P2 2.3558(19), Ru1–S1 2.359(2), Ru2–S1 2.365(2), Ru3–S1 2.358(2), Ru1–H1 1.7945, Ru2–H2 1.7324, Ru2–H1 1.7347, Ru3–H2 1.7658 (riding on Ru atoms). | ||
Again, two different distinctive classes of Ru–Ru bond lengths can be observed, the two hydrido-bridged Ru–Ru bonds are “long” [Ru(1)–Ru(2) 2.9276(6), and Ru(2)–Ru(3) 2.9123(6)] and the “shortest” [Ru(1)–Ru(3) 2.7511(6)]. A longer P–P distance is observed for 8 (5.61 Å) with a torsion angle of 87.70, in comparison to 5.48 Å and torsion angle of 83.98 for 4. As expected, the angles in 8 are a bit different than 4, where P1–Ru1–Ru2 = 106.85, P2–Ru2–Ru1 = 118.59 and P2–Ru2–Ru3 = 120.54 in 8, and for 4 it is 103.05, 120.06 and 123.84, respectively.
In case of 17, the shortest Ru–Ru distance is the Ru1–Ru3 edge (2.7359(8) Å), which is not bridged by any hydride, while the two Ru–Ru edges that bridged with two hydrides have slightly symmetric longer distances (2.9173(8) Å for Ru1–Ru2 and 2.8921(8) Å for Ru2–Ru3). The two phosphorus atoms of the coordinated ligand are separated by 5.512 Å with a torsion angle of 87.36(8). The Ru–P bonds are symmetric in lengths (2.3539(19) Å for Ru1–P1 and 2.3558(19) Å for Ru2–P2), and the angles are P1–Ru1–Ru2 = 105.41(5), P2–Ru2–Ru1 = 116.98(5) and P2–Ru2–Ru3 = 121.39(5).
Synthesis and characterization of sulfide-capped triruthenium hydrido clusters containing the Josiphos ligands 2a–2e
The coordination of the Josiphos ligands 2a–2e to the parent cluster [(μ-H)2Ru3(μ3-S)(CO)9] was performed by oxidative decarbonylation as discussed above (cf.Scheme 2). The bridging coordination mode and the different connectivities of the heterobidentate ligands in the diastereomers 18–22 have been identified by spectroscopic methods and, in the case of 20, by X-ray crystallography. | ||
| Scheme 2 Synthesis of clusters 18–22 by using Josiphos family ligands. | ||
The 1H and 31P NMR data confirmed the difference in the bridging mode of the diphosphine ligand 2a in clusters 18 and 19. For 18, the 1H NMR in the hydride region shows two signals, a doublet of ‘triplets’ at δ −18.29 (dt, JH–P = 36.5, JH–H = 2.5 Hz) and a doublet of doublet of doublets at δ −19.46 (ddd, JH–P = 13.3, JH–P = 7.1, JH–H = 2.5 Hz), while the hydride signals for 19 consist of a doublet of doublet of doublets at δ −17.84 (JH–P = 12.7, JH–P = 9.0, JH–H = 3.6 Hz) and a doublet of doublet of doublets at δ −18.65 (JH–P = 7.5, JH–P = 3.6 Hz). In the 31P NMR spectrum of 18, the P1 signal (cf.Scheme 2) appears as a doublet of doublets, indicating coupling to both hydrides, while the signal for P2 is a doublet. In contrast, the signal for P1 in 19 appears as a doublet while that for P2 is a doublet of doublets. The ES+ mass spectrometric data match with the assigned molecular formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-P–P)] (m/z = 1401 [M + H]+) for both 18 and 19. The empirical formulae of clusters 20–22 were confirmed by mass spectrometry, and their structures in solution and the solid state were identified by IR and NMR spectroscopy (cf. Experimental section and ESI†). Due to the high degree of hydride fluxionality exhibited by [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2d)] 22 at ambient temperature, only one sharp multiplet resonance could be detected for the hydride in the 1H NMR. Variable-temperature (VT) 1H NMR of cluster 22 shows that the fluxionality may be frozen out at 253 K, to reveal the two hydride signals that were expected for the cluster (Fig. 5).
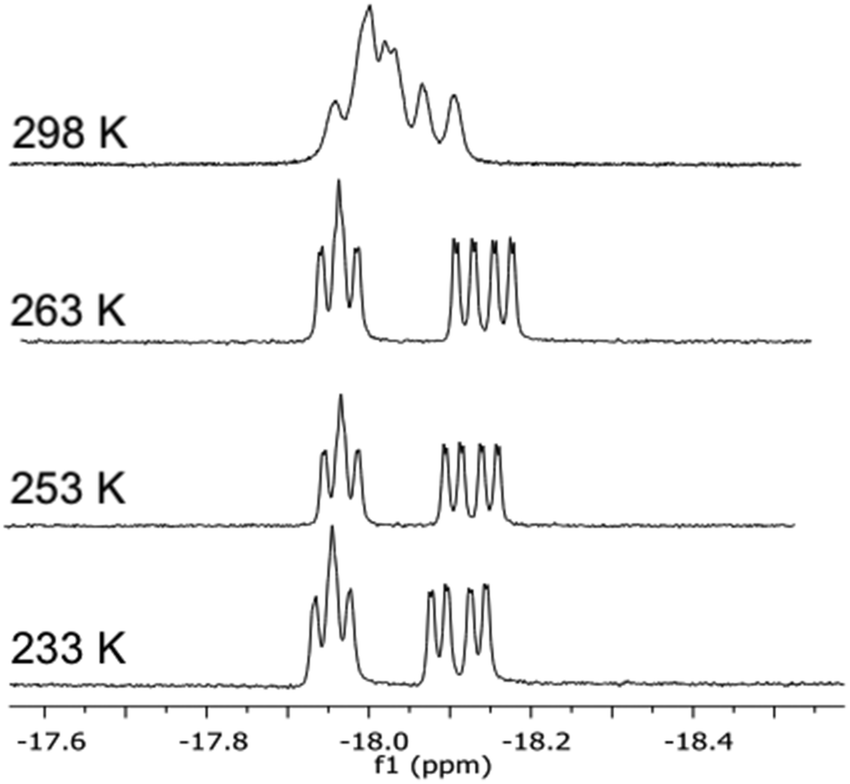 | ||
| Fig. 5 Variable-temperature 1H NMR spectra in the hydride region of cluster [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2d)] 22 recorded over the temperature range 298–233 K (top to bottom). | ||
Crystal and molecular structure of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2b)] 20
The crystal structure of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2b)] 20 was determined by X-ray diffraction analysis. A view of the molecular structure of 20 is shown in Fig. 6 and selected bond distances and angles are listed in the caption. Relevant crystallographic data are listed in Table S1, ESI.† The cluster is very similar to 3–6, 8 and 17 (vide supra), with one exception: the ligand 2b bridges Ru1 and Ru2 atoms, but the two phosphine moieties bind in the more common cis-eq,eq coordination mode that is expected for diphosphines with a relatively short backbone, rather than the slightly distorted cis-eq,ax coordination mode that is observed for the above-mentioned Walphos derivatives. The latter coordination mode is presumably enforced by the relatively long backbone of the Walphos ligands. We have previously observed exactly the same types of coordination modes for Josiphos and Walphos ligands coordinated to tetraruthenium tetrahydride clusters.22 The two metal–metal bonds in 20 that are bridged by hydrides are slightly elongated in comparison to the non-bridged Ru1–Ru3 edge (cf caption, Fig. 6). Whereas the average P–P distance in the Walphos complexes 3–6, 8 and 17 is 5.50 Å, it is 4.43 Å in 20 and significant differences are seen for the Ru–P bonds lengths (2.3295(8) Å for Ru1–P1 and 2.3887(7) Å for Ru2–P2).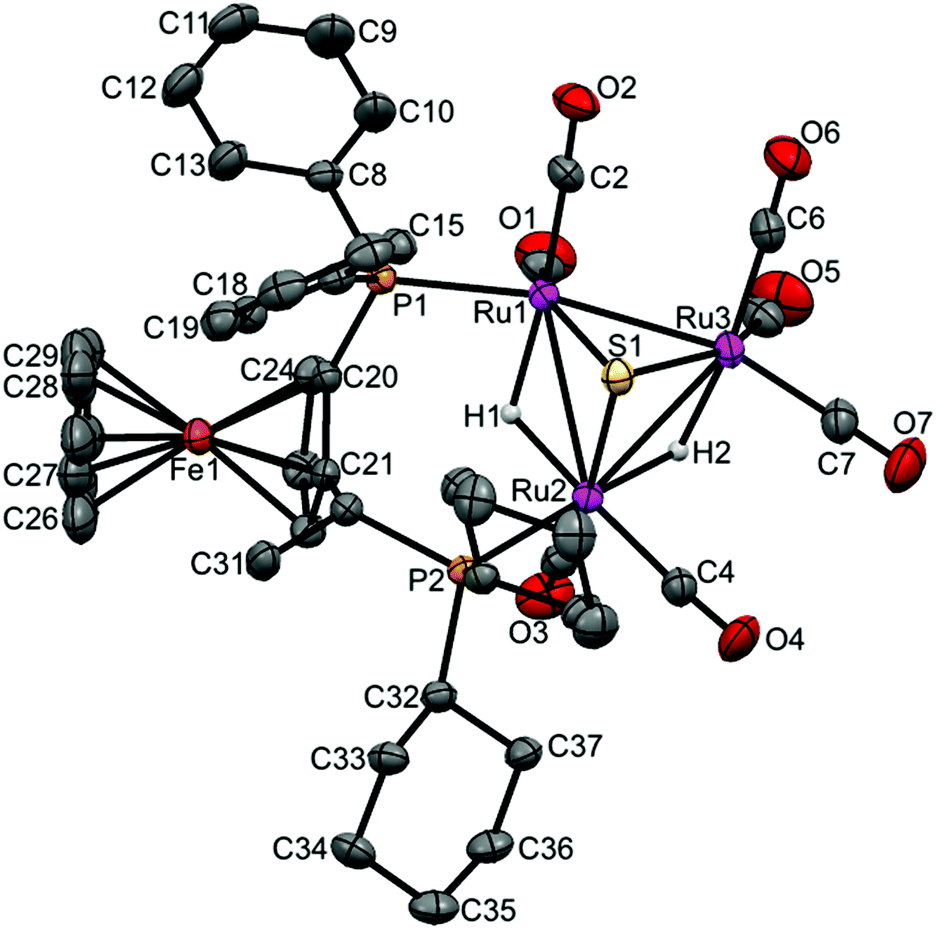 | ||
| Fig. 6 Molecular structure of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2b)] 20 (CCDC 1043620†) with thermal ellipsoids drawn at the 50% probability level. C–H hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and angles [°]: Ru1–Ru2 2.9272(3), Ru1–Ru3 2.7314(3), Ru2–Ru3 2.8947(3), Ru1–P1 2.3295(8), Ru2–P2 2.3887(7), Ru1–S1 2.3701(8), Ru2–S1 2.3871(8), Ru3–S1 2.3667(7), Ru1–H1 1.82(4), Ru2–H2 1.79(4), Ru2–H1 1.76(4), Ru3–H2 1.86(4). | ||
Catalytic activities
In previous studies, we have shown that the chiral diphosphine ligands play an important role in asymmetric hydrogenation21,22,26 and that the chiral configurations of the hydrogenated products are dependent on the chiral ligand used.27 The ability of the new clusters 3–22 to function as catalysts for asymmetric hydrogenation was investigated. Tiglic acid [trans-2-methyl-2-butenoic acid] was chosen as a substrate because (i) it has been used as a benchmark substrate in earlier assessments of asymmetric hydrogenation effected by transition metal carbonyl clusters, facilitating comparison with other work,21,26,28–31 (ii) there is a well-established and reliable protocol for the evaluation of enantiomeric excess, and (iii) asymmetric hydrogenation of α-unsaturated carboxylic acids and their substituted derivatives, which are essential pharmaceuticals or chiral building blocks for the synthesis of biologically active compounds, is of considerable importance.32,33 Instead of the high hydrogen pressure that has been commonly used in previous cluster-based catalysis investigations (130 bar),34 a hydrogen pressure of 50 bar was used. Lower hydrogen pressure was expected to enhance the chiral induction, as demonstrated by us in a previous investigation.27The catalysis results for clusters 3–22 are summarized in Table 1. The clusters show moderate catalytic activity in terms of conversion. Furthermore, the enantioselectivities are moderate in comparison to mononuclear catalysts, but relatively high by the standards of cluster-based systems. The enantioselectivites reflect an inherent weakness in the chiral induction effected by clusters – the substrate is likely to bind at a metal site with minimum steric hindrance (vide infra) and the chiral induction effected by bulky chiral ligands is therefore diminished. However, we have observed significantly higher enantioselectivities in other cluster-based systems,21,22,28 and it should be borne in mind that ee's exceeding 40% were unprecedented30,31,34–36 in cluster-based asymmetric hydrogenation before we began our studies. Unfortunately, the separation of the hydrogenated products from the organic phase after using clusters 13 and 16 as catalysts (Table 1, entries 11, 14 and 20, respectively) turned out to be unsuccessful in our hands, so we could not determine the enantiomeric excess for these reactions, albeit with full conversion in case of 16. From Table 1, it is clear that in these catalytic systems the stereochemistry and the bridging mode of the coordinated chiral diphosphine ligands strongly affect the outcome of the hydrogenation reactions, both in terms of conversion and enantioselectivity. In earlier work, we have been able to demonstrate that reversal of enantioselectivity can be effected by reversal of phosphine chirality in cluster-based systems.27 It may be noted that the diastereomeric pairs 3/4, 5/6, 7/8, 9/10, 11/12, and 18/19, within which the chirality of the diphosphine remains the same, but the chirality of the cluster framework changes, all show a reversal in enantioselectivity when the chirality of the cluster framework changes. Furthermore, the pairs 3/5 and 4/6 are diastereomers where the chirality of the cluster framework remains intact while that of the ligand changes (cf.Fig. 2), and for these pairs the enantioselectivity is reversed with reversal in chirality of the ligands. It may thus be concluded that both the cluster framework and the ligand chiralities influence the enantioselectivity for a given cluster. These observations provide compelling evidence for the clusters, or immediate derivatives of the clusters with maintained cluster framework chiralities,37 being the active catalysts.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Catalyst | Conv.a [%] | ee [%] | Config.b |
a The amount of substrate consumed in the catalytic experiment, assed by 1H NMR spectroscopy, p(H2) = 50 bar, T = 100 °C, solvent = EtOH–toluene 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (5 mL), n(substrate)/n(catalyst) = 100.
b Favoured enantiomer. ND: not detected. :1 (5 mL), n(substrate)/n(catalyst) = 100.
b Favoured enantiomer. ND: not detected.
|
|||||
| 1 | 1a | 3 | 49 | 23 | R |
| 2 | 1a | 4 | 79 | 56 | S |
| 3 | S,S-1b | 5 | 55 | 24 | S |
| 4 | S,S-1b | 6 | 64 | 52 | R |
| 5 | 1c | 7 | 83 | 33 | R |
| 6 | 1c | 8 | 80 | 21 | S |
| 7 | 1d | 9 | 94 | 9 | R |
| 8 | 1d | 10 | 77 | 7 | S |
| 9 | 1e | 11 | 66 | 17 | S |
| 10 | 1e | 12 | 7 | 8 | R |
| 11 | 1f | 13 | 14 | ND | ND |
| 12 | 1f | 14 | 23 | 48 | S |
| 13 | 1g | 15 | 47 | 21 | S |
| 14 | 1g | 16 | 100 | ND | ND |
| 15 | 1h | 17 | 43 | 13 | R |
| 16 | 2a | 18 | 88 | 31 | R |
| 17 | 2a | 19 | 66 | 13 | S |
| 18 | 2b | 20 | 41 | 17 | S |
| 19 | 2c | 21 | 68 | 37 | S |
| 20 | 2d | 22 | 38 | 26 | S |
| 21 | 1a | 3 + Hg | 51 | 26 | R |
A mechanism involving cluster decomposition during catalysis and regeneration of the intact cluster at the end of the catalytic cycle would be accompanied by formation of both diastereomers,38 even if only one form of the cluster was initially used. However, no trace of such racemization was observed at the end of catalytic cycles employing all clusters. All catalysts were recovered in good yields (≈70%) after a complete catalytic experiment, with one exception: only trace amounts (at best) of catalyst 20 could be recovered. For the other catalysts, the recovery may be considered to be quantitative, when the small amount of catalyst used is taken into account. Furthermore, recyclability was proven for cluster 3 – after completion of the reaction (Table 1, entry 1), the cluster was recovered, purified by TLC and reused under identical experimental conditions. Identical conversion rates and enantiomeric excesses for the two catalytic runs were obtained.
Nature of the active catalyst
:1 v/v) in the absence of any potential catalyst and heated at 100 °C under 50 bar of H2. No hydrogenated products were detected, which strongly support that the temperature and the hydrogen pressure used in our catalytic experiments are not forcing enough for the hydrogenation of tiglic acid and that no metallic residues or similar contaminations that may cause (catalyze) hydrogenation were present.
Mercury poisoning
It is well known that forcing conditions, such as high temperature and high pressure of hydrogen gas, increase the possibility of formation of colloidal metal particles that may function as catalysts.39,40 The ability of Hg(0) to poison metal colloids or heterogeneous catalysts has been used for more than 80 years.41 Therefore, a mercury poisoning test42 was performed in order to investigate the potential formation of (catalytically active) metal colloids in our hydrogenation experiments. Hydrogenation of tiglic acid using 3 was carried out under the typical reaction conditions described above. The reaction was allowed to proceed to ∼50% completion, followed by release of H2 pressure and the addition of a 2000-fold excess of Hg(0) to the reaction mixture. The solution was stirred for 30 min to enable Hg(0) to react with any colloidal particles present. As previously observed for analogous reactions utilizing tetraruthenium clusters as catalysts/catalyst precursors,22 the mercury poisoning experiment appeared to give an increase in conversion and enantioselectivity by 2–3 percentage units (Table 1, entries 1 vs. 21) but this difference lies within experimental error. The obtained result and the fact that the yield and enantioselectivity do not decrease suggest that little or no colloidal particles are formed during the catalytic reaction and that the effect of any such colloidal particles is very limited for the hydrogenation reaction.Proposed catalytic cycle
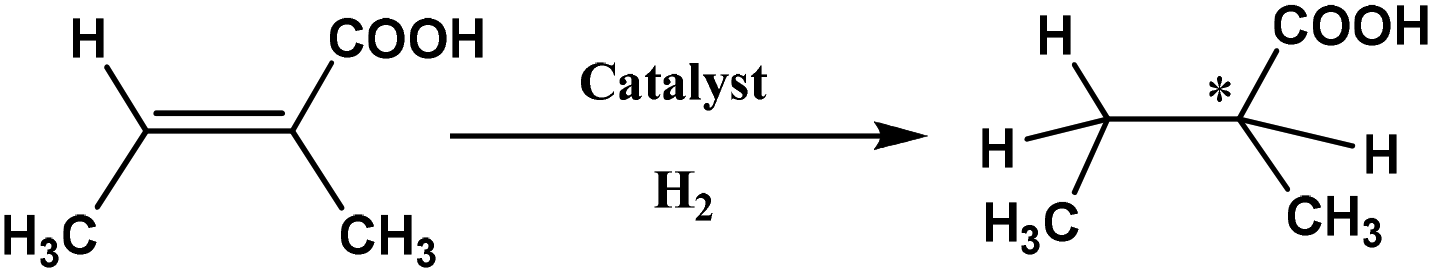
In an earlier study,27 we proposed two plausible mechanisms for the activation of tiglic acid by intact Ru4 clusters having the composition [H4Ru4(CO)10(L)] (where L = chiral diphosphine ligand) – either dissociation of a carbonyl ligand to yield a vacant coordination site or metal–metal bond scission resulting in an open butterfly structure. To help elucidate the catalytic cycle(s) associated with the present Ru3 clusters, we have modelled the course of the hydrogenation reaction using density functional theory calculations. Cluster 4, which gives good conversion and enantioselectivity by the standards of these catalysts, was chosen as the model catalyst for comprehensive DFT evaluation. The computed catalytic cycle is depicted in Scheme 3, and the free energy surface for the hydrogenation reaction is shown in Fig. 7. The cycle is initiated by a site-selective dissociation of an equatorial CO at the Ru(CO)3 moiety; of the two equatorial CO groups here, the one that is proximally located to the bridging hydride is preferentially labilized. The formation of the coordinatively unsaturated species B, which proceeds without cluster fragmentation, serves as the bifurcation point leading to the 2-methylbutyric acid product. Si-face coordination of tiglic acid to B affords the alkene-substituted cluster D. Regiospecific insertion of the alkene into the proximal bridging hydride occurs via transition structure TSDE, which lies 29.6 kcal mol−1 above B and C. The alternative alkene insertion route that involves the most substituted alkene carbon atom and that would directly yield the (S) stereogenic center, lies 6.6 kcal mol−1 above TSDE (not shown). The resulting agostic alkyl cluster E reacts with H2 to furnish the transient trihydride cluster F. Reductive elimination in F gives (S)-2-methylbutyric acid (G) and regenerates cluster B, completing the catalytic cycle with a net release of 13.2 kcal mol−1. The geometry-optimized structures for these species are shown in Fig. 8. Re-face alkene coordination at B gives the diastereomeric alkene cluster D_alt, from which the hydrogenation reaction proceeds through a series of identical steps, all of which lie higher in energy than the si-face route.43 The energetics computed for the reaction profile from D_alt depicted in Fig. 7 (red energy surface) are in concert with the observed preference for the (S) enantiomer when cluster 4 is employed as the catalyst precursor. The mechanistic steps computed involve a fixed stereochemistry at the cluster, whose chirality directly influences the asymmetric induction observed in the hydrogenation product. The computed scheme is in agreement with the proposed catalytic cycle for the hydrogenation of ethane by [H4Ru4(CO)12]. On the basis of kinetic data, a mechanism, where suppression of catalysis by the addition of CO and the evolution of CO on exposure of a heptane solution of [H4Ru4(CO)12] to hydrogen, has been suggested to involve an initial dissociation of CO ligand and formation of [H4Ru4(CO)11(alkene)], followed by an alkene insertion reaction to furnish the Ru4 cluster [H3Ru4(CO)11(alkyl)] as an intermediate.44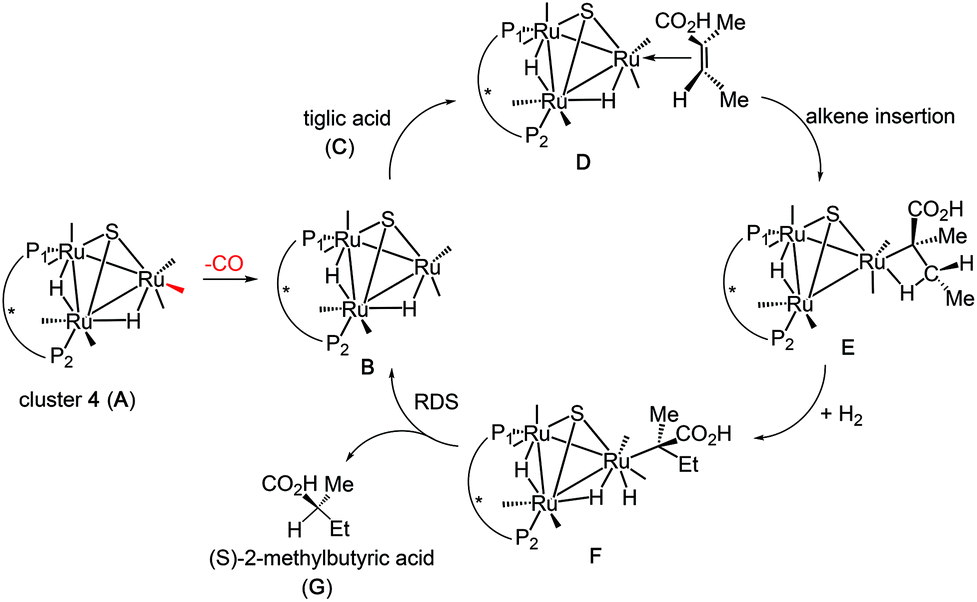 | ||
| Scheme 3 Computed catalytic hydrogenation cycle of tiglic acid by cluster 4 to give (S)-2-methylbutyric acid. | ||
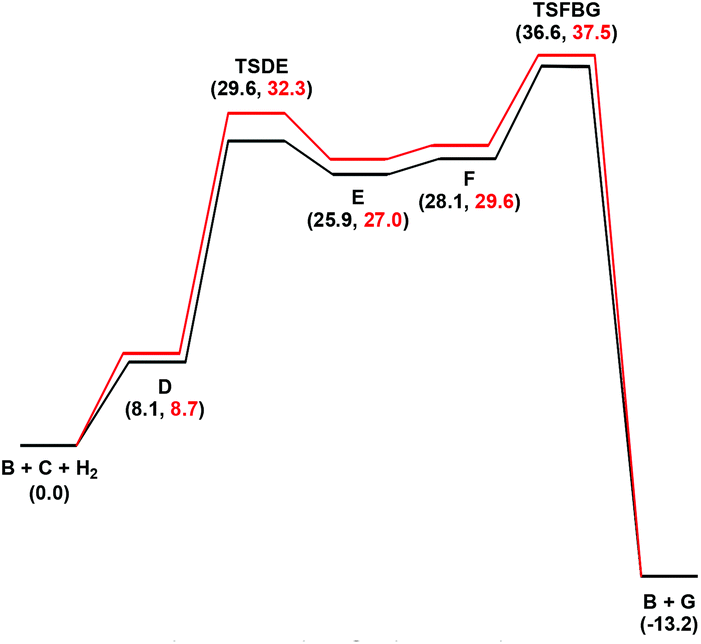 | ||
| Fig. 7 Free energy profiles for the hydrogenation of tiglic acid catalyzed by cluster 4 (A) starting from the unsaturated cluster B. Black and red profiles are for the si- and re-face alkene coordination routes, respectively. | ||
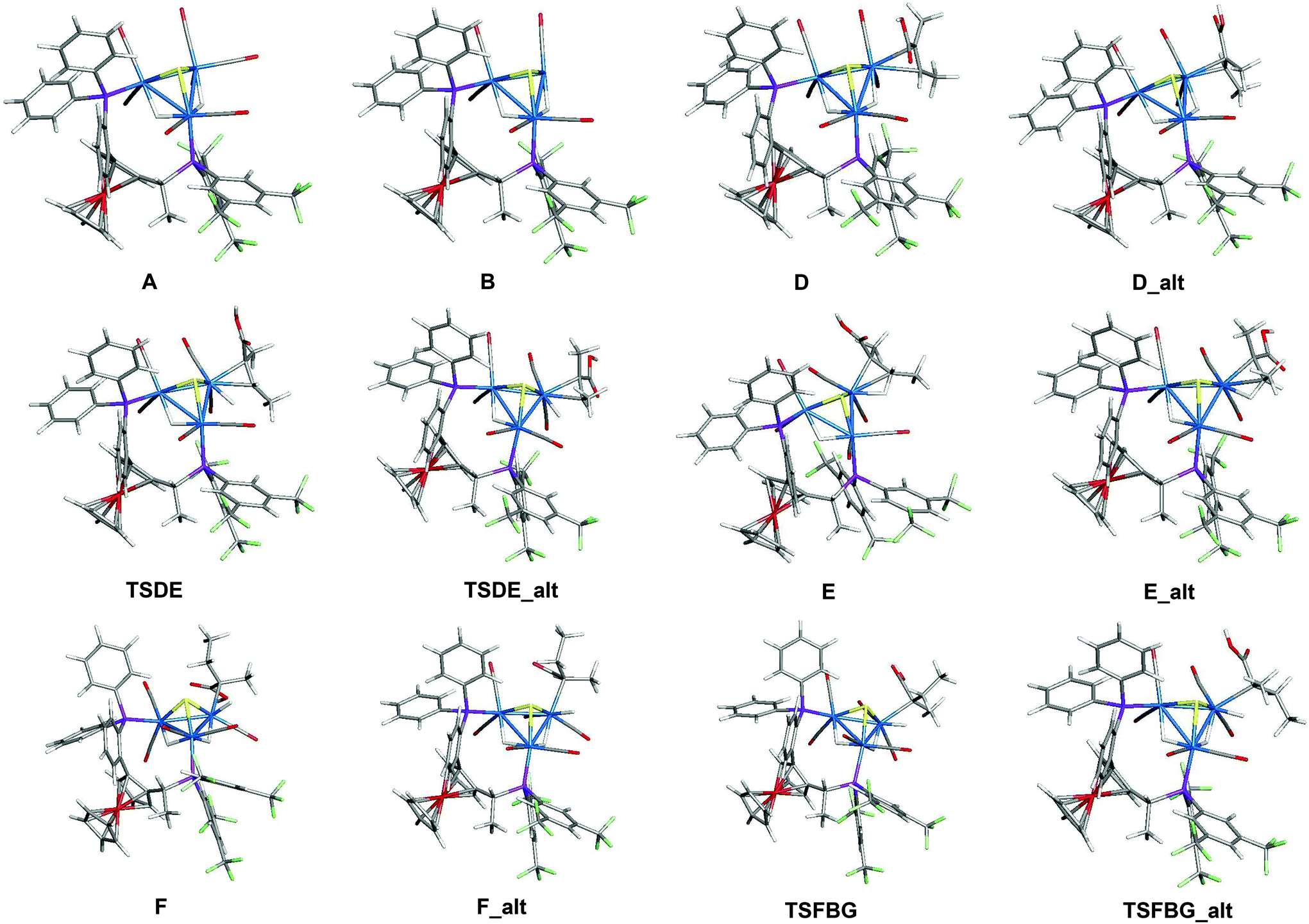 | ||
| Fig. 8 Geometry-optimized structures associated with the hydrogenation of tiglic acid to 2-methylbutyric acid catalyzed by cluster precursor 4. The optimized structures of CO, H2, tiglic acid (C), and 2-methylbutyric acid (G) are not shown. | ||
Conclusions
In summary, twenty clusters of the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-L)] (L = chiral diphosphine ligand), containing chiral clusters frameworks, have been prepared. All cluster diastereomers show different catalytic activities in terms of conversion and enantioselectivity in the asymmetric hydrogenation of tiglic acid under relatively mild conditions. The reversal in enantioselectivity of the hydrogenation reaction when the chirality of the cluster framework is changed, while the chirality of the ligand remains intact, strongly implicates the involvement of intact Ru3 clusters as the active hydrogenation catalysts. The conversion rates are relatively good but the enantioselectivites are low, albeit good when compared to most other cluster-based catalytic systems for asymmetric reactions.30,31,34–36Experimental section
General procedures
All reactions and other manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques. All solvents were dried and distilled under a nitrogen atmosphere prior to use. Infra-red spectra were recorded as solutions in 0.5 mm NaCl cells on a Nicolet Avatar 360 FT-IR-spectrometer.1H and 31P NMR spectra were recorded on a Varian Unity 500 MHz NMR spectrometer; 31P NMR shifts were referenced to external H3PO4 (85%). The parent cluster [(μ-H)2Ru3(μ3-S)(CO)9] was prepared according to a literature procedure and its purity was assessed using thin-layer chromatography (TLC) and IR spectroscopy.45 The chiral phosphines, 2-methylbutyric acid, S-methyl mandelate and tiglic acid (trans-2-methyl-2-butenoic acid) were purchased from Sigma Aldrich. Separations were carried out by preparative thin-layer chromatography on glass plates (20 × 20 cm2) coated with silica gel (Merck, 0.5 mm thick). Catalysis experiments were carried out using a 45 mL Parr autoclave with a PTFE reaction vessel.General method for the synthesis of sulfide-capped triruthenium hydrido clusters containing chiral Walphos (1a–1h) and Josiphos (2a–2e) ligands
In all preparations, a solution of Me3NO (13 mg, 173 μmol) in methanol (5 mL) was added dropwise to a stirred solution of [(μ-H)2Ru3(μ3-S)(CO)9] (42 mg, 71 μmol) and the proper stoichiometric ratio of the ligand (vide infra) in dichloromethane (20 mL) under nitrogen atmosphere over a period of 20 minutes. The reaction mixture was stirred for 3 h and filtered through silica. The solvent was removed in vacuo and the resultant residue was subjected to thin layer chromatography using glass plates coated with silica gel F254, using a dichloromethane/hexane mixture (3:7 v/v) as eluent (for 11 and 12, dichloromethane/hexane (3:2) was used as eluent). Clusters 3 and 4 were obtained as two closely spaced yellow bands. Clusters 5–6, 7–8, 9–10, 11–12, 13–14, 15–16, 17–18, 19, 20–21, 22 and finally 23, were prepared via identical procedures using 1a, S,S-1b, 1c, 1d, 1e, 1f, 1g, 1h, 2a, 2b, 2c, 2d, 2e, respectively. Crystallization of 3–6, 8, 17 and 20 from CH2Cl2–hexane solutions at 4 °C gave red crystals suitable for X-ray diffraction analysis. For the 1H NMR spectra reported below, only hydride signals are listed. Spectra with all non-hydride signals are found in the ESI.†
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1a)], 3 and 4
A total of 66 mg (71 μmol) of 1a was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1a)].[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1a)] 3, yield 32 mg (31%); (Rf = 0.6); IR (ν(CO)/cm−1, cyclohexane) 2066(s), 2046(vs), 2010(vs), 1994(s), 1979(m), 1951(m); 1H NMR (500 MHz, CDCl3) δ −18.13 (ddd, J = 12.4, 11.2, 2.9 Hz, 1H), −18.40 (dt, J = 9.8, 2.9 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 49.75 (d, J = 10.6 Hz), 37.74 (t, J = 9.1 Hz), ES+ MS (m/z): 1464 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1a)] 4, yield 39 mg (38%); (Rf = 0.4); IR (ν(CO)/cm−1, cyclohexane) 2066(s), 2048(vs), 2008(vs), 1995(s), 1982(w), 1951(m); 1H NMR (500 MHz, CDCl3) δ −17.82 (ddd, J = 12.2, 8.8, 3.5 Hz, 1H), −18.63 (ddd, J = 7.5, 3.5 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 48.52 (dd, J = 11.1, 6.6 Hz), 34.41 (d, J = 8.2 Hz). ES+ MS (m/z): 1464 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)], 5 and 6
A total of 66 mg (71 μmol) of S,S-1b was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)].[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)] 5, yield 29 mg (28%); (Rf = 0.6); IR (ν(CO)/cm−1, cyclohexane) 2066(s), 2048(vs), 2008(vs), 1995(s), 1979(m), 1951(m); 1H NMR (500 MHz, CDCl3) δ −18.12 (ddd, J = 12.6, 11.1, 3.0 Hz, 1H), −18.40 (dt, J = 9.6, 3.1, 3.0 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 48.68 (dd, J = 11.8, 2.0 Hz), 36.60 (t, J = 9.8 Hz). ES+ MS (m/z): 1464 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1b)] 6, yield 40 mg (38%); (Rf = 0.5); IR (ν(CO)/cm−1, cyclohexane) 2066(s), 2046(vs), 2010(vs), 1994(s), 1979(m), 1951(m); 1H NMR (500 MHz, CDCl3) δ −17.84 (ddd, J = 12.4, 9.1, 3.0 Hz, 1H), −18.65 (ddd, J = 7.1, 3.2, 3.0 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 48.59 (dd, J = 11.3, 5.9 Hz), 35.55 (d, J = 8.4 Hz). ES+ MS (m/z): 1464 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1c)], 7 and 8
A total of 48 mg (71 μmol) of 1c was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1c)].[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1c)] 7, yield 27 mg (32%); (Rf = 0.5); IR (ν(CO)/cm−1, cyclohexane) 2057(s), 2038(vs), 1995(vs), 1984(s), 1965(m), 1932(m); 1H NMR (500 MHz, CDCl3) δ −18.51 (ddd, J = 11.0, 9.7, 3.0 Hz, 1H), −18.64 (dt, J = 11.8, 3.0 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 56.47 (dd, J = 9.6, 5.0 Hz), 37.16 (t, J = 9.7 Hz). ES+ MS (m/z): 1228 [M + Na]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1c)] 8, yield 33 mg (39%); (Rf = 0.4); IR (ν(CO)/cm−1, cyclohexane) 2057(s), 2034(vs), 1998(vs), 1985(s), 1969(m), 1942(m); 1H NMR (500 MHz, CDCl3) δ −18.08 (ddd, J = 10.3, 9.0, 3.0 Hz, 1H), −19.02 (‘m’, ddd, J = not resolved 1H, cf. ESI†); 31P NMR (202 MHz, CDCl3) δ 59.08 (dd, J = 9.6, 6.2 Hz), 35.24 (dd, J = 8.6, 2.7 Hz); ES+ MS (m/z): 1228 [M + Na]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1d)], 9 and 10
A total of 74 mg (71 μmol) of 1d was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1d)].[(μ-H)2Ru3(μ3-S)((CO)7(μ-1,2-1d)] 9, yield 26 mg (23%); (Rf = 0.5); IR (ν(CO)/cm−1, cyclohexane) 2063(s), 2044(vs), 2008(vs), 1993(s), 1976(m), 1951(m); 1H NMR (500 MHz, CDCl3) δ −18.09 (dt, J = 9.6, 2.8 Hz, 1H), −18.39 (ddd, J = 12.8, 10.2, 2.9 1H); 31P NMR (202 MHz, CDCl3) δ 48.89 (d, J = 11.8 Hz), 35.65 (t, J = 9.6 Hz). ES+ MS (m/z): 1580 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1d)] 10, yield 37 mg (33%); (Rf = 0.4); IR (ν(CO)/cm−1, cyclohexane) 2064(s), 2047(vs), 2006(vs), 1992(s), 1980(m), 1949(m); 1H NMR (500 MHz, CDCl3) δ −17.82 (ddd, J = 12.2, 8.8, 3.4 Hz, 1H), −18.63 (ddd, J = 7.3, 5.0, 3.4 Hz 1H); 31P NMR (202 MHz, CDCl3) δ 48.49 (dd, J = 11.3, 6.5 Hz), 34.37 (d, J = 8.4 Hz). ES+ MS (m/z): 1603 [M + Na]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1e)], 11 and 12
A total of 51 mg (71 μmol) of 1e was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1e)].[(μ-H)2Ru3(μ3-S)((CO)7(μ-1,2-1e)] 11, yield 29 mg (33%); (Rf = 0.6); IR (ν(CO)/cm−1, cyclohexane) 2059(s), 2042(vs), 2000(vs), 1985(m), 1970(w), 1939(w); 1H NMR (500 MHz, CDCl3) δ −17.61 (td, J = 9.8, 3.0 Hz, 1H), −18.17 (‘m’, ddd, J = not resolved 1H, cf. ESI†); 31P NMR (202 MHz, CDCl3) δ 43.88 (dd, J = 9.5, 6.2 Hz), 35.63 (dd, J = 8.9, 2.4 Hz). ES+ MS (m/z): 1248 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1e)] 12, yield 38 mg (43%); (Rf = 0.5); IR (ν(CO)/cm−1, cyclohexane) 2059(s), 2042(vs), 2000(vs), 1985(m), 1970(w), 1939(w); 1H NMR (500 MHz, CDCl3) δ −17.61 (ddd, J = 10.0, 3.0 Hz, 1H), −18.16 (‘m’, ddd, J = not resolved 1H, cf. ESI†); 31P NMR (202 MHz, CDCl3 49.09 (dd, J = 10.2, 1.7 Hz), 36.60 (t, J = 9.5 Hz). ES+ MS (m/z): 1248 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1f)], 13 and 14
A total of 70 mg (71 μmol) of 1f was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1f)].[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1f)] 13, yield 33 mg (31%); (Rf = 0.6); IR (ν(CO)/cm−1, cyclohexane) 2064(vs), 2047(m), 2038(s), 2003(vs), 1992(s), 1983(m), 1976(w), 1945(m), 1938(w); 1H NMR (500 MHz, CDCl3) δ −18.14 (ddd, J = 13.8, 4.5, 2.5 Hz, 1H), −18.31 (‘m’, ddd, J = not resolved 1H, cf. ESI†); 31P NMR (202 MHz, CDCl3) δ 52.62 (m), 49.49 (dd, J = 11.1, 1.3 Hz). ES+ MS (m/z): 1502 [M + Na]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1f)] 14, yield 45 mg (43%); (Rf = 0.6); IR (ν(CO)/cm−1, cyclohexane) 2064(vs), 2047(s), 2037(m), 2003(vs), 1992(m), 1977(w), 1946(w), 1939(w); 1H NMR (500 MHz, CDCl3) δ −18.17 (ddd, J = 11.9, 7.0, 3.6 Hz, 1H), −18.83 (ddd, J = 4.8, 3.6 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 48.49 (dd, J = 11.3, 6.5 Hz), 34.37 (d, J = 8.4 Hz). ES+ MS (m/z): 1479 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1g)], 15 and 16
A total of 55 mg (71 μmol) of 1g was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1g)].[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1g)] 15, yield 36 mg (39%); (Rf = 0.6); IR (ν(CO)/cm−1, cyclohexane) 2059(s), 2042(vs), 1999(vs), 1983(s), 1970(m), 1938(m); 1H NMR (500 MHz, CDCl3) δ −17.69 (td, J = 9.7, 3.0 Hz, 1H), −18.25 (dt, J = 6.4, 3.0 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 44.05 (dd, J = 9.5, 6.4 Hz), 36.51 (dd, J = 8.8, 2.6 Hz). ES+ MS (m/z): 1327 [M + Na]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1g)] 16, yield 48 mg (52%); (Rf = 0.5); IR (ν(CO)/cm−1, cyclohexane) 2057(s), 2039(s), 2003(s), 2003(vs), 1984(s), 1967(m), 1940(w); 1H NMR (500 MHz, CDCl3) δ −18.16 (m, 2H); (500 MHz, acetone-d6, 253 K) δ −18.12 (dt, J = 11.0, 3.0 Hz, 1H), −18.19 (‘m’, ddd, J = not resolved 1H, cf. ESI†); 31P NMR (202 MHz, CDCl3) δ 49.32 (dd, J = 7.1, 5.1 Hz), 38.10 (t, J = 9.6 Hz). ES+ MS (m/z): 1305 [M + H]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1h)], 17
A total of 47 mg (71 μmol) of 1h was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1h)] 17.[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1h)] 17, yield 36 mg (42%); (Rf = 0.5); IR (ν(CO)/cm−1, cyclohexane) 2060(s), 2043(vs), 2001(vs), 1986(s), 1972(m), 1941(m); 1H NMR (500 MHz, CDCl3) δ −17.75 (ddd, J = 10.1, 9.1, 3 Hz, 1H), −18.47 (dt, J = 6.1, 3.0 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 44.95 (dd, J = 9.9, 5.7 Hz), 35.39 (dd, J = 9.1, 1.8 Hz). ES+ MS (m/z): 1191 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2a)], 18 and 19
A total of 62 mg (71 μmol) of 2e was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give two diastereomers with the general formula [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2a)].[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2a)] 18, yield 14 mg (14%); (Rf = 0.6): IR (ν(CO)/cm−1, cyclohexane) 2066(vs), 2036(s), 2005(vs), 1996(s), 1987(m), 1951(m); 1H NMR (500 MHz, CDCl3) δ −18.29 (dt, J = 36.5, 2.5 Hz, 1H), −19.46 (ddd, J = 13.3, 7.1, 2.5 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 56.94 (dd, J = 34.7, 5.9 Hz), 25.71 (d, J = 11.8 Hz). ES+ MS (m/z): 1400 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2a)] 19, yield 25 mg (25%); (Rf = 0.5): IR (ν(CO)/cm−1, cyclohexane) 2064(vs), 2039(ms, 2032(w), 2007(vs), 1993(m), 1980(m), 1967(w), 1951(w); 1H NMR (500 MHz, CDCl3) δ −17.84 (ddd, J = 12.7, 9.0, 3.6 Hz, 1H), −18.65 (ddd, J = 7.5, 3.6 Hz, 1H); 31P NMR (202 MHz, CDCl3) δ 48.59 (dd, J = 10.4, 4.7 Hz), 35.54 (d, J = 8.4 Hz). ES+ MS (m/z): 1400 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2b)], 20
A total of 41 mg (71 μmol) of 2b was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2b)] 20.[(μ-H)2Ru3(μ3-S)((CO)7(μ-1,2-2b)] 20, yield 33 mg (44%); (Rf = 0.6): IR (ν(CO)/cm−1, cyclohexane) 2059(vs), 2031(s), 2016(vs), 1998(vs), 1970(w), 1941(m); 1H NMR (500 MHz, CDCl3) δ −18.31 (dd, J = 32.6, 2.0 Hz, 1H), −19.13 (‘t’, ddd, J = not resolved 1H, cf. ESI†); 31P NMR (202 MHz, CDCl3) δ 57.29 (m), 25.41 (m). ES+ MS (m/z): 1174 [M]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2c)], 21
A total of 39 mg (71 μmol) of 2c was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2c)] 21.[(μ-H)2Ru3(μ3-S)((CO)7(μ-1,2-2c)] 21, yield 44 mg (59%); (Rf = 0.6): IR (ν(CO)/cm−1, cyclohexane) 2057(vs), 2033(s), 1994(vs), 1980(m), 1969(w), 1926(w); 1H NMR (500 MHz, CDCl3) δ −18.77 (br, 1H), −19.09 (m, 1H); 31P NMR (202 MHz, CDCl3) δ 65.05 (m), 35.49 (m). ES+ MS (m/z): 1163 [M + Na]+.
[(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2d)], 22
A total of 41 mg (71 μmol) of 2d was reacted with [(μ-H)2Ru3(μ3-S)(CO)9] to give [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2d)] 22.[(μ-H)2Ru3(μ3-S)((CO)7(μ-1,2-2d)] 22, yield, 36 mg (48%); (Rf = 0.6): IR (ν(CO)/cm−1, cyclohexane) 2059(vs), 2038(s), 2001(vs), 1988(m), 1970(w), 1943(w); 1H NMR (500 MHz, CDCl3) δ −18.04 (m, 1H); (500 MHz, acetone-d6, 253 K) δ −17.93 (ddd, J = 11.8, 2.0 Hz, 1H), −18.10 (ddd, J = 23.4, 10.4, 2.0 Hz); 31P NMR (202 MHz, CDCl3) δ 41.66 (m), 20.65 (m). ES+ MS (m/z): 1172 [M]+.
Homogeneous catalytic experiments
In the catalysis experiments, the catalyst and substrate were loaded into the autoclave under N2, and the degassed solvent mixture was added (2.5 mL of EtOH/2.5 mL of toluene). The reaction vessel was closed and purged three times with hydrogen before final pressurizing to 50 bar. The reaction mixture was continuously stirred with a magnetic stirrer (ca. 750 rpm) and heated at 100 °C for 24 h. After a cooling period of approximately 45 min, the reaction vessel was depressurized and opened. The homogeneous reaction mixture was transferred to a 50 mL flask and concentrated under vacuum. The conversions for the catalysis runs were calculated on the basis of NMR analyses.Precautions were taken to avoid the possibility of catalytic activity due to contamination of the reaction vessel or the magnetic stir bars: the reaction vessel and the magnetic stir bars were washed with acetone and rinsed with dichloromethane, followed by a careful visual examination. Whenever the magnetic stir bars appeared to be contaminated, they were discarded.
To separate the carboxylic acid from the cluster, the reaction residue was dissolved in 10 mL of diethyl ether and the carboxylic acid was extracted with aqueous sodium hydroxide solution (1 M, 3 × 10 mL) and washed with diethyl ether (3 × 5 mL), leaving the cluster in the organic solvent. The carboxylate was protonated with sulfuric acid and extracted with diethyl ether (3 × 10 mL), washed with water (2 × 5 mL) and dried over magnesium sulfate. Filtration, followed by evaporation of the ether under vacuum, yielded the carboxylic acid quantitatively. The original ether phase, from which the carboxylic acid was extracted, was concentrated under vacuum to recover the remaining cluster. In certain cases, where ester formation was obtained during the catalytic experiment, the recovered catalyst was dissolved in a minimum quantity of dichloromethane and the products were separated by preparative TLC, eluting with dichloromethane/petroleum ether (1:2). Usually 60–70% of the cluster was recovered after a catalytic experiment and it was analyzed by IR and NMR spectroscopies. The enantiomeric excess of the product was detected by derivatizing 2-methylbutyric acid with S-methyl mandelate and analyzing the diastereomeric product mixture by NMR, as fully described by Tyrrell et al.46 It was found that flash chromatography of the final products was not necessary.
Catalyst poisoning test using mercury
The experimental setup followed the procedure described above for catalytic experiments, except that after 4 h the reaction was stopped and the reaction vessel was disconnected and approximately 2 grams of metallic mercury was added to the reaction mixture before the autoclave was sealed and pressurized and a second hydrogenation reaction was started. After a complete catalytic run, the products were separated and analyzed as described above.VT 1H NMR studies
All NMR solvents (Aldrich) were used as received. Approximately 0.5 mL of a saturated acetone-d6 solution of [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-1h)] 16 and [(μ-H)2Ru3(μ3-S)(CO)7(μ-1,2-2d)] 22 were added to a NMR tube, which was subsequently sealed under N2 atmosphere. The sample tube was placed into the probe using a ceramic spinner. Dry air (for measurements above RT) or liquid nitrogen was used as the VT control gas. The sample was allowed to equilibrate at each desired temperature for 5–10 minutes before the start of shimming and data acquisition. The temperature ranges were 25 to −40 °C for 16 and 25 to −70 °C for 22. For each sample 32 scans were acquired to reach sufficient signal intensity. The residual proton signal of acetone (2.04 ppm rel. to TMS) was used as a chemical shift reference.X-ray structure determinations
The crystals were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 100 K for 5, 170 K for 3, 4, 8, 17 and 20, and finally 293 K for 6. The X-ray diffraction data were collected on a Rigaku Supernova or a Bruker AXS Kappa ApexII Duo or Oxford Diffraction Excalibur EOS diffractometer using Mo Kα radiation (λ = 0.71073 Å). The CrysAlisPro47 or APEXII48 program packages were used for cell refinements and data reductions. A detailed discussion of the refinements of the structures and a table of selected crystallographic details (Table S1†) are found in the ESI.† All data for the structures reported here have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers 993899 (complex 3), 993900 (4), 1043616(5), 1043617 (6), 1043618 (8), 1043619 (17), and 1043620 (20).†Computational modelling and DFT calculations
The DFT calculations were carried out with the Gaussian 09 package of programs.49 The different species in Scheme 3 were examined computationally using Morokuma'a ONIOM method,50 using a two-layered ONIOM (B3LYP/genecp:PM6) approach. Here the Walphos ligand, except for the phosphorus and iron atoms, was confined to the lower layer. The Ru and Fe atoms were described with the Stuttgart-Dresden effective core potential (ecp) and an SDD basis set, and all of the other high-level atoms were described by a 6-31G(d′) basis set.
All reported geometries were fully optimized, and the analytical Hessian was evaluated at each stationary point to determine whether the geometry was an energy minimum (no negative eigenvalues) or a transition structure (one negative eigenvalue). Unscaled vibrational frequencies were used to make zero-point and thermal corrections to the electronic energies. The resulting free energies are reported in kcal mol−1 relative to the specified standard. Standard state corrections were applied to all species to convert concentrations from 1 atm to 1 M according to the treatise of Cramer.51 Internal reaction coordinate (IRC) calculations were performed in order to establish the reactant and product species associated with each transition-state structure. The geometry-optimized structures have been drawn with the JIMP2 molecular visualization and manipulation program.52
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AFA thanks the EU Erasmus Mundus program for a predoctoral fellowship. AKS thanks the Carl Trygger Foundation for a postdoctoral fellowship. MGR thanks the Robert A. Welch Foundation (grant B-1093) for financial support and acknowledges computational resources through UNT's High Performance Computing Services and CASCaM. We thank Dr David Hrovat and Prof. Xinzheng Yang for helpful ONIOM-based discussions. We are indebted to Dr Thomas Brimert at Red Glead Discovery for assistance with chiral HPLC measurements.Notes and references
- A. J. Deeming and M. Underhill, J. Organomet. Chem., 1972, 42, C60–C62 CrossRef CAS.
- H. Vahrenkamp, Angew. Chem., Int. Ed. Engl., 1975, 14, 322–329 CrossRef.
- T. A. Cresswell, J. A. K. Howard, F. G. Kennedy, S. A. R. Knox and H. Wadepohl, J. Chem. Soc., Dalton Trans., 1981, 2220–2229 RSC.
- K. A. Azam, G. M. Golzar Hossain, S. E. Kabir, K. M. Abdul Malik, M. A. Mottalib, S. Perven and N. C. Sarker, Polyhedron, 2002, 21, 381–387 CrossRef CAS.
- A. J. Deeming, M. M. Hassan, S. E. Kabir, E. Nordlander and D. A. Tocher, Dalton Trans., 2004, 3709–3714 RSC.
- H. Akter, A. J. Deeming, G. M. G. Hossain, S. E. Kabir, D. N. Mondol, E. Nordlander, A. Sharmin and D. A. Tocher, J. Organomet. Chem., 2005, 690, 4628–4639 CrossRef CAS.
- S. J. Ahmed, M. I. Hyder, S. E. Kabir, M. A. Miah, A. J. Deeming and E. Nordlander, J. Organomet. Chem., 2006, 691, 309–322 CrossRef CAS.
- C. Bergounhou, P. Fompeyrine, G. Commenges and J. J. Bonnet, J. Mol. Catal., 1988, 48, 285–312 CrossRef CAS.
- Y. Lin and R. G. Finke, Inorg. Chem., 1994, 33, 4891–4910 CrossRef CAS.
- C. M. Hagen, L. Vieille-Petit, G. Laurenczy, G. Süss-Fink and R. G. Finke, Organometallics, 2005, 24, 1819–1831 CrossRef CAS.
- L. Vieille-Petit, G. Süss-Fink, B. Therrien, T. R. Ward, H. Stœckli-Evans, G. Labat, L. Karmazin-Brelot, A. Neels, T. Bürgi, R. G. Finke and C. M. Hagen, Organometallics, 2005, 24, 6104–6119 CrossRef CAS.
- M. T. Nielsen, R. M. Padilla Paz and M. Nielsen, J. Cluster Sci., 2020, 31, 11–61 CrossRef.
- R. D. Adams and T. S. Barnard, Organometallics, 1998, 17, 2567–2573 CrossRef CAS.
- R. D. Adams and T. S. Barnard, Organometallics, 1998, 17, 2885–2890 CrossRef CAS.
- P. Nombel, N. Lugan, B. Donnadieu and G. Lavigne, Organometallics, 1998, 18, 187–196 CrossRef.
- S. Aime, R. Gobetto and D. Canet, J. Am. Chem. Soc., 1998, 120, 6770–6773 CrossRef CAS.
- B. Bergman, E. Rosenberg, R. Gobetto, S. Aime, L. Milone and F. Reineri, Organometallics, 2002, 21, 1508–1511 CrossRef CAS.
- D. Blazina, S. B. Duckett, P. J. Dyson, B. F. G. Johnson, J. A. B. Lohman and C. J. Sleigh, J. Am. Chem. Soc., 2001, 123, 9760–9768 CrossRef CAS PubMed.
- D. Blazina, S. B. Duckett, P. J. Dyson and J. A. B. Lohman, Chem. – Eur. J., 2003, 9, 1045–1061 CrossRef PubMed.
- D. Blazina, S. B. Duckett, P. J. Dyson and J. A. B. Lohman, Angew. Chem., Int. Ed., 2001, 40, 3874–3877 CrossRef CAS PubMed.
- V. Moberg, M. Haukka, I. O. Koshevoy, R. Ortiz and E. Nordlander, Organometallics, 2007, 26, 4090–4093 CrossRef CAS.
- V. Moberg, R. Duquesne, S. Contaldi, O. Rohrs, J. Nachtigall, L. Damoense, A. T. Hutton, M. Green, M. Monari, D. Santelia, M. Haukka and E. Nordlander, Chem. – Eur. J., 2012, 18, 12458–12478 CrossRef CAS PubMed.
- J. Norton, in Fundamental Research in Homogeneous Catalysis, ed. M. Tsutsui and R. Ugo, Springer US, 1977, ch. 4, pp. 99–114, DOI:10.1007/978-1-4615-7038-7_4.
- C. U. Pittman, M. G. Richmond, M. M. Absi-Halabi, H. Beurich, F. Richter and H. Vahrenkamp, Angew. Chem., Int. Ed. Engl., 1982, 94, 805–806 CAS.
- A. F. Abdel-Magied, A. K. Singh, M. Haukka, M. G. Richmond and E. Nordlander, Chem. Commun., 2014, 50, 7705–7708 RSC.
- V. Moberg, P. Homanen, S. Selva, R. Persson, M. Haukka, T. A. Pakkanen, M. Monari and E. Nordlander, Dalton Trans., 2006, 279–288 RSC.
- P. Homanen, R. Persson, M. Haukka, T. A. Pakkanen and E. Nordlander, Organometallics, 2000, 19, 5568–5574 CrossRef CAS.
- V. Moberg, R. Duquesne, S. Contaldi, O. Röhrs, J. Nachtigall, L. Damoense, A. T. Hutton, M. Green, M. Monari, D. Santelia, M. Haukka and E. Nordlander, Chem. – Eur. J., 2012, 18, 12458–12478 CrossRef CAS PubMed.
- M. J. Stchedroff, V. Moberg, E. Rodriguez, A. E. Aliev, J. Bottcher, J. W. Steed, E. Nordlander, M. Monari and A. J. Deeming, Inorg. Chim. Acta, 2006, 359, 926–937 CrossRef CAS.
- C. Botteghi, S. Gladiali, M. Bianchi, U. Matteoli, P. Frediani, P. G. Vergamini and E. Benedetti, J. Organomet. Chem., 1977, 140, 221–228 CrossRef CAS.
- M. Bianchi, U. Matteoli, G. Menchi, P. Frediani, F. Piacenti and C. Botteghi, J. Organomet. Chem., 1980, 195, 337–346 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- H.-U. Blaser, B. Pugin and F. Spindler, J. Mol. Catal. A: Chem., 2005, 231, 1–20 CrossRef CAS.
- U. Matteoli, V. Beghetto and A. Scrivanti, J. Mol. Catal. A: Chem., 1996, 109, 45–50 CrossRef CAS.
- U. Matteoli, M. Bianchi, P. Frediani, G. Menchi, C. Botteghi and M. Marchetti, J. Organomet. Chem., 1984, 263, 243–246 CrossRef CAS.
- U. Matteoli, G. Menchi, P. Frediani, M. Bianchi and F. Piacenti, J. Organomet. Chem., 1985, 285, 281–292 CrossRef CAS.
- Formation of a Noyori-type catalyst of the general formula [Ru(1)(O2CR)2] is in principle possible but cannot explain a reversal of enantioselectivity. Formation of a [Ru(1)2X2] (X = arbitrary monodentate anion) complex is even less likely, but could in principle form Λ and Δ-isomers; however, a racemic mixture would then expected to be formed, and the reversal of enantioselectivty would not be effected.
- This is especially true with cluster 4, whose DFT-computed ΔG° lies 2.0 kcal mol−1 above that of cluster 3. Any fragmentation of 4 during catalysis, followed by cluster reassembly, would thermodynamically afford 3, for more information, A. F. Abdel-Magied, A. K. Singh, M. Haukka, M. G. Richmond and E. Nordlander, Chem. Commun., 2014, 50, 7705–7708 RSC.
- J. G. de Vries, Dalton Trans., 2006, 421–429 RSC.
- L. N. Lewis, J. Am. Chem. Soc., 1986, 108, 743–749 CrossRef CAS.
- G. M. Whitesides, M. Hackett, R. L. Brainard, J.-P. P. M. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staudt, Organometallics, 1985, 4, 1819–1830 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- “Re” and “Si” denominations refer to Cahn-Ingold-Prelog (CIP) rules and nomenclature used to identify enantiotopic (prochiral) planes. The substitution priorities are the same as for S and R nomenclature – “Re” refers to clockwise orientation of the different substituents in order of priority, “Si” refers to anticlockwise orientation.
- Y. Doi, K. Koshizuka and T. Keii, Inorg. Chem., 1982, 21, 2732–2736 CrossRef CAS.
- R. D. Adams and D. A. Katahira, Organometallics, 1982, 1, 53–59 CrossRef CAS.
- E. Tyrrell, M. W. H. Tsang, G. A. Skinner and J. Fawcett, Tetrahedron, 1996, 52, 9841–9852 CrossRef CAS.
- Rigaku Oxford Diffraction, CrysAlisPro, Rigaku Oxford Diffraction Inc., Yarnton, Oxfordshire, U.K., 2013 Search PubMed.
- APEX2, SAINT-Plus and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber and K. Morokuma, J. Phys. Chem., 1996, 100, 19357–19363 CrossRef CAS.
- C. J. Cramer, Essentials of Computational Chemistry, Wiley, Chichester, UK, 2nd edn, 2004 Search PubMed.
- (a) JIMP2, version 0.091, a free program for the visualization and manipulation of molecules: M. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768–775 CrossRef CAS; (b) J. Manson, C. E. Webster and M. B. Hall, Texas A&M University, College Station, TX, 2006, http://www.chem.tamu.edu/jimp2/index.html.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic details, FT-IR and NMR spectra. Atomic coordinates of all optimized stationary points and transition states. CIF files for complexes 3 and 4. CCDC 993899, 993900 and 1046316–1046320. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04799a |
| This journal is © The Royal Society of Chemistry 2020 |