
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of tellurium nano- and microstructures by trace HCl in ionic liquids†
Tao
Zhang
ab,
Thomas
Doert
 a,
Kai
Schwedtmann
a,
Jan J.
Weigand
a and
Michael
Ruck
*ac
a,
Kai
Schwedtmann
a,
Jan J.
Weigand
a and
Michael
Ruck
*ac
aFaculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany. E-mail: michael.ruck@tu-dresden.de
bBeijing Key Laboratory of Ionic Liquids Clean Process, CAS Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, 100190 Beijing, China
cMax Planck Institute for Chemical Physics of Solids, 01187 Dresden, Germany
First published on 13th January 2020
Abstract
Ionic liquids (ILs) are widely used as versatile solvents for the synthesis of nanomaterials. However, the effect of IL impurities on the formation of nanomaterials is often neglected. Herein, we report on the formation of tellurium (Te) nanoparticles from the reaction of trialkylphosphane tellurides, formed by reactive dissolution of Te in dried commercial trihexyltetradecylphosphonium chloride ([P66614]Cl) at high temperatures, with common polar protic solvents (e.g. water, alcohols, or amides). Highly homogeneous Te nano- and microstructures with various sizes and morphologies including three-dimensional (3D) Te fusiform assemblies and 3D aloe-like Te microarchitectures are obtained. Our investigation shows that trace amounts of HCl impurities in [P66614]Cl tend to remain as [P66614][HCl2] due to the strong interaction with Cl−. The addition of a polar, protic solvent liberates active HCl from the HCl2− anion which we found to play an essential role in the formation of Te particles due to the accelerating effect of P–Te bond cleavage. This approach presents a general and convenient synthetic strategy for the preparation of Te nano- and microstructures.
1. Introduction
Ionic liquids (ILs) are, by definition, a family of molten salts with melting points below 100 °C.1 In the past few decades, ILs have become alternative solvents to traditional organic solvents and brought a revolution in both academic and industrial areas including catalysis, separation, organic synthesis, and material sciences.2–7 The unique properties such as excellent thermal stability, low vapor pressure, wide liquid range and electrochemical window, and tunable solvent properties are certainly the most highlighted and well-studied advantages of ILs in literature and the reasons for their broad application.8–12Toxicity, chemical reactivity, and impurity of ILs are rarely reported,13–15 and are typically considered as negative aspects of ILs. For instance, impurities in ILs may significantly influence their chemical and physical properties, which may cause non-uniform experimental data. Some examples: Suranna and co-workers reported that the presence of halide impurities in 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIm][BF4]) has a detrimental effect on the activity of organometallic catalysts towards Michael additions.16 Brutchey and co-workers demonstrated that trace amounts of water, chloride, and 1-methylimidazole in [BMIm][BF4] cause polydisperse and irregularly shaped ensembles of silver nanoparticles, and also negatively influence the stabilization of the resulting particles.15
On the other hand, impurities of ILs can play a positive role in reactions. We previously showed that HCl impurity in [P66614]Cl leads to a surface activation of elemental copper, promoting the formation of Cu3−xP with a high product yield.17 Another investigation revealed that trace amounts of metal ion impurities in 1-ethyl-3-methylimidazolium chloride ([EMIm]Cl), particularly Cu and Cr ions, promote reactions of cellulose depolymerization drastically due to the significant synergistic effect of metal halides and ILs.18,19 This stimulated us to continue exploring cases in which trace impurities commonly found in ILs have positive impacts on reactions.
Elemental tellurium (Te) is a typical p-type semiconductor with a narrow band gap (0.35 eV) at room temperature, exhibiting attractive physical and chemical properties with potential applications in a variety of different areas such as gas sensing, ion detection and removal, photoconductivity, and lithium–tellurium batteries.20–24 Te nano- and microstructures are commonly synthesized using solution-based strategies, typically by direct reduction of a Te precursor (e.g. a tellurite or tellurate) in the presence of a surfactant like polyvinylpyrrolidone (PVP).25–27 Hence, the reaction systems become complicated. In the present work, we developed a new and convenient synthetic method for the creation of novel, complex 3D Te microstructures by exploiting the reaction of trialkylphosphane tellurides, formed by reactive dissolution of Te in dried [P66614]Cl at high temperatures, with polar protic solvents. The reaction can be triggered by the amount of HCl in [P66614]Cl. According to our scanning electron microscopy (SEM) investigations, the sizes and morphologies of Te particles can be tuned by varying the polar protic solvents.
2. Experimental
2.1. Chemicals
Trihexyltetradecylphosphonium chloride ([P66614]Cl, >95%), trihexyltetradecylphosphonium dicyanamide ([P66614][N(CN)2], >95%), and trihexyltetradecylphosphonium decanoate ([P66614][decanoate], >95%) were purchased from io-li-tec, Germany. Te powder (Te, −325 mesh, 99.99%) was purchased from ABCR GmbH, Germany. All the organic solvents were purchased from Sigma-Aldrich. The typical drying of ILs was performed under dynamic vacuum at 110 °C overnight. The HCl impurity in commercial [P66614]Cl was removed by neutralization with a sodium hydroxide solution, as previously described.17 The Te powder was reduced in an H2 atmosphere at 400 °C for 7 h. All other chemicals were used as received without further purification.2.2. Preparation of Te solution in [P66614]Cl, [P66614][N(CN)2], and [P66614][decanoate]
The Te solution in ILs was prepared according to our previously reported method.14 Taking Te solution in [P66614]Cl as an example, 12 mg Te powder was added to 4 g [P66614]Cl in an Ar-filled glovebox. The mixture was transferred into a 25 mL Teflon lined stainless steel autoclave in the glovebox. The autoclave was sealed, taken out of the glovebox, and then placed in an oven at 250 °C for 48 h. After the autoclave was cooled down to room temperature, a clear, light yellow Te solution was obtained as the unsaturated Te solution. If more Te powder (e.g. 20 or 24 mg) was used in the dissolution test, some undissolved Te powder was observed. The saturated Te solution was obtained by removal of the undissolved Te powder.2.3. General synthesis of Te nano- and microstructures using protic solvents
Taking ethanol as an example, typically, 0.5 mL Te solution in [P66614]Cl was placed in a 5 mL screw-cap glass bottle. Then 2 mL ethanol was added to the bottle immediately. The mixture was shaken strongly until a permanent black precipitate formed. Normally, this process needs less than one minute. The precipitate was separated by centrifugation, washed with acetone or toluene first and then with ethanol for several times. For water and ethylene glycol, a longer time is required to achieve Te precipitation.2.4. Materials characterization
Powder X-ray diffraction (PXRD) patterns were typically recorded using a PANalytical X'Pert Pro diffractometer with Cu-Kα1 radiation (λ = 154.0562 pm). The scanning electron microscopy (SEM) images were obtained at 3 kV using a Hitachi SU8020 field emission SEM coupled with energy-dispersive X-ray spectroscopy (EDX; Oxford X-MaxN). NMR spectra were measured on a Bruker AVANCE III HDX, 500 MHz Ascend (31P (202.45 MHz); 125Te (157.78 MHz)) equipped with a Prodigy-Cryo Probe. Chemical shifts were referenced to δH3PO4(85%) = 0.00 ppm (31P) and δMe2Te = 0.00 ppm (125Te). Chemical shifts (δ) are reported in ppm. Coupling constants (J) are reported in Hz.3. Results and discussion
The phosphonium IL [P66614]Cl was dried at 110 °C overnight under dynamic vacuum prior to use. The trialkylphosphane tellurides were prepared by dissolving Te in [P66614]Cl at high temperatures according to our previous report.14 For convenience, the trialkylphosphane tellurides obtained by dissolving Te in [P66614]Cl are defined here as “Te solution in [P66614]Cl”. In order to obtain Te nano- and microstructures, various solvents were tested (Table S1;† see Experimental section for more details). Both non-polar solvents (e.g. n-hexane, toluene, chloroform, or diethyl ether) and polar aprotic solvents (e.g. tetrahydrofuran, ethyl acetate, acetone, dimethylformamide, acetonitrile, or dimethyl sulfoxide) are miscible with trialkylphosphane tellurides in [P66614]Cl without Te precipitation, whereas polar protic solvents (e.g. water, alcohols, amides, or carboxylic acids) cause Te precipitation with a variety of sizes and morphologies of the obtained Te particles.3.1. Synthesis of Te nano- and microstructures using alcohols
The precipitation of Te powder from Te solutions in [P66614]Cl depends slightly on the alkyl chain length of the applied monohydric alcohol (see Experimental section for more details). Ethanol can cause precipitation within a few seconds by mixing it with Te solution in [P66614]Cl. With increasing alkyl chain length of the alcohols (n-butanol, n-hexanol, n-octanol, and n-decanol), the precipitation of Te powder took a verifiably longer time but not more than one minute. The precipitates were identified as crystalline Te by powder X-ray diffraction (PXRD; Fig. 1) and energy-dispersive X-ray spectroscopy (EDX; Fig. S1†). All reflections can be indexed with the trigonal unit cell of Te in space group P3121 (ICSD no. 76150).28 The SEM images (Fig. 2a–e) display that all obtained Te particles have quite homogeneous fusiform morphologies with lengths of 300–400 nm. With increasing alkyl chain length of the alcohols, these Te fusiforms maintain similar sizes and shapes but their edges become slightly sharper. The high-magnification SEM image (Fig. 2f) clearly reveals that a single Te fusiform is composed of even smaller Te nanofusiforms with lengths of 50–400 nm, i.e. Te nanofusiform assemblies form a Te microfusiform.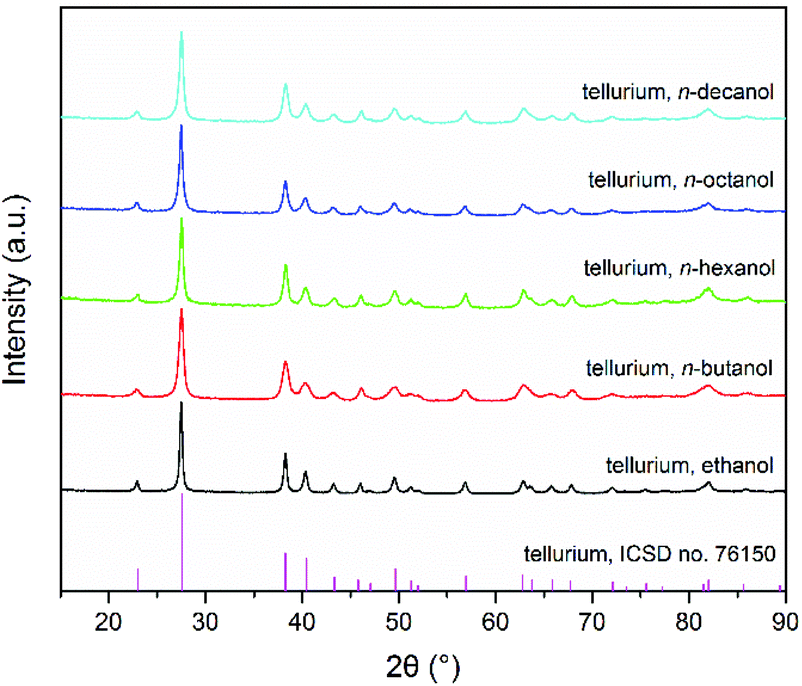 | ||
| Fig. 1 PXRD patterns (Cu-Kα1) of Te powders obtained by mixing monohydric alcohols with Te solution in [P66614]Cl. | ||
 | ||
| Fig. 2 SEM images of Te particles obtained by mixing Te solution in [P66614]Cl with ethanol (a), n-butanol (b), n-hexanol (c), n-octanol (d), or n-decanol (e) (scale bar, a–e, 300 nm). SEM image of a single 3D microfusiform particle obtained in n-butanol (f). | ||
The mixture of 2 mL ethylene glycol and 0.5 mL Te solution in [P66614]Cl turned out to be immiscible. Nonetheless, when these two phases were placed statically under air for several hours, a dark precipitate was observed in the upper IL phase (Fig. S2a†). The formation of crystalline Te was confirmed by PXRD. The SEM images are shown in Fig. 3. The low-magnification SEM image (Fig. 3a) demonstrates the overall morphology of aloe-like Te microstructures with a diameter of approximately 1.3 μm. The high-magnification SEM image in Fig. 3b shows that these microstructures are assemblies of lancets with typical lengths of about 600 nm in various directions. The rough surfaces of these lancets are clearly visible in Fig. 3b.
 | ||
| Fig. 3 SEM images of obtained Te particles using ethylene glycol as the protic solvent. | ||
As shown in Fig. 4, polyhedral nanoparticles of crystalline Te (as evidenced by PXRD) with diameters of about 50 nm were synthesized in the presence of allyl alcohol or propargyl alcohol as examples of unsaturated alcohols.
 | ||
| Fig. 4 SEM images of obtained Te nanoparticles using allyl alcohol (a) or propargyl alcohol (b) as the protic solvent. | ||
3.2. Synthesis of Te microstructures using amides
Protic amides, such as formamide and N-methylformamide, also lead to the formation of crystalline Te powders according to PXRD. Fig. 5a and b show the corresponding SEM images obtained from formamide, displaying also the formation of fusiform assemblies. However, the sizes with lengths of about 100 nm are much smaller than those obtained from monohydric alcohols. | ||
| Fig. 5 SEM images of obtained Te particles using formamide (a, b) or N-methylformamide (c, d) as the protic solvent. | ||
In contrast, Te particles obtained from N-methylformamide (Fig. 5c and d) show aggregates of intergrown elongated particles with diameters of approximately 400–600 nm (Fig. 5c). The magnified image reveals that each of these Te particles consists of much smaller, intergrown nanoparticles (Fig. 5d).
3.3. Synthesis of Te nanostructures using carboxylic acids
Carboxylic acids, such as formic acid, acetic acid, and propionic acid, lead quite rapidly to the precipitation of Te particles when mixed with Te solution in [P66614]Cl. SEM images reveal the formation of Te nanoparticles with a size of 30–50 nm, without creating complex 3D Te architectures (Fig. S3†).3.4. Synthesis of Te microstructures using water
Water, as one of the most widely used solvents, also leads to the formation of Te microstructures. Similar to ethylene glycol, 2 mL water added to a 0.5 mL Te solution in [P66614]Cl shows immiscibility. A dark Te precipitate formed in the upper IL phase (Fig. S2b†) when the water and Te solution phases were placed statically overnight. The formed Te precipitate consists of fusiform assemblies with typical lengths of about 4 μm, which are much larger than those obtained from monohydric alcohols (SEM image in Fig. S4†).3.5. Possible formation mechanism
As previously reported by us, relatively high amounts of Te powder can be dissolved in certain phosphonium-based ILs, such as trihexyltetradecylphosphonium dicyanamide ([P66614][N(CN)2]), trihexyltetradecylphosphonium decanoate ([P66614][decanoate]), and tetrabutylphosphonium decanoate ([P4444][decanoate]).14 NMR investigations show that the quaternary phosphonium cations undergo an SN2-type reaction to the respective trialkylphosphane telluride via the elimination of one of the alkyl chains. This process takes place at relatively high temperatures in the presence of Te and leads to its dissolution in the phosphonium-based ILs. In the case of [P66614]Cl, however, a very limited Te solubility was observed for both saturated and unsaturated Te solutions (for details see Experimental section) indicated by the absence of the respective resonances in the 125Te NMR spectra due to the very low concentration of dissolved Te-based species. In order to increase the concentration and to obtain meaningful 125Te NMR spectra, for both cases, the excess of [P66614]Cl was removed via flash chromatography and the resulted solutions are accordingly denoted as Tesat and Teunsat.The 31P NMR spectrum in Fig. 6b for Tesat indicates an upfield shifted resonance at δ(31P) = −13.6 ppm compared to that of the pure [P66614]Cl (δ(31P) = 33.2 ppm).29 The large 1JPTe coupling constant of 1665 Hz is in the range of typical phosphane tellurides.30 The corresponding 125Te NMR spectrum (Fig. 6d) shows one doublet resonance at δ(125Te) = −778.8 ppm with the same coupling constant 1JTeP = 1665 Hz. Both 31P and 125Te NMR spectra evidence a P–Te bond formation and the formation of trialkylphosphane tellurides. For Teunsat, however, the corresponding 31P and 125Te NMR spectra (Fig. 6a and c) show only one single resonance (δ(31P) = −22.7 ppm, and δ(125Te) = −781.7 ppm), respectively. The spectra variations between Tesat and Teunsat indicate that a rapid exchange between the free phosphane and phosphane tellurides happens, reaching a dynamic equilibrium. Based on the above investigations, we can conclude that trialkylphosphane tellurides form according to the reaction of [P66614]Cl with elemental Te via nucleophilic attack of Cl− at a relatively high temperature, which are well comparable with those of Te solution in [P66614][N(CN)2] and [P66614][decanoate].14
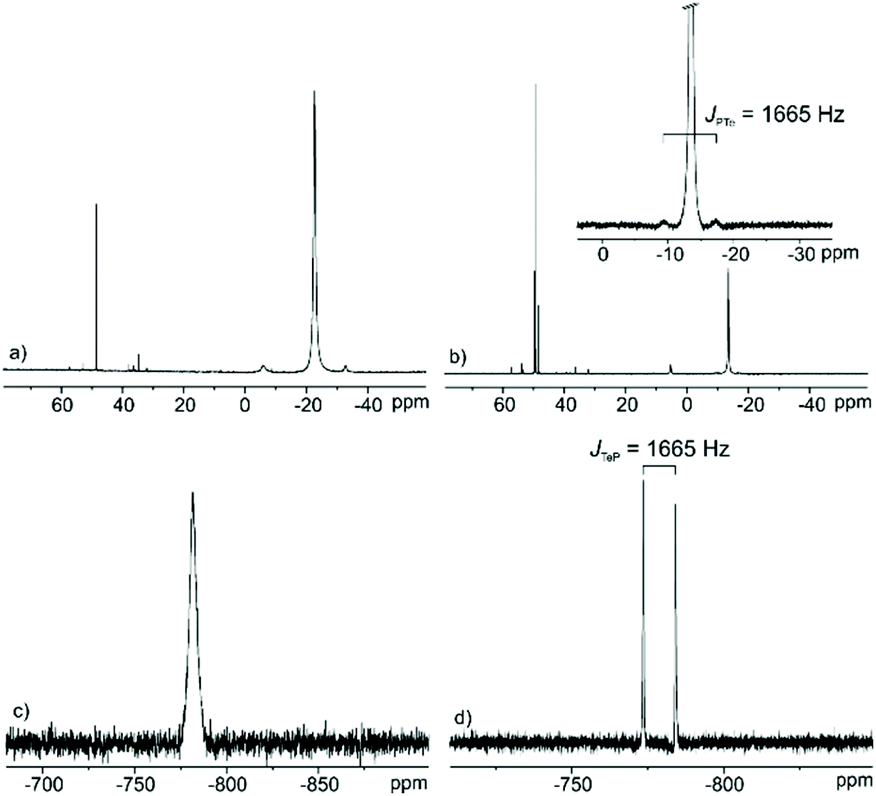 | ||
| Fig. 6 31P NMR spectra of Teunsat (a) and Tesat (b) as well as 125Te NMR spectra of Teunsat (c) and Tesat (d) solution in [P66614]Cl after removal of excess of ILs via flash chromatography. | ||
The Te solution in [P66614][N(CN)2] or [P66614][decanoate] was also tested for the Te precipitation reactions with an alcohol, but no precipitates were observed (Fig. S5†). This finding indicates that the Te precipitation reaction seems to only happen in the chloride system (e.g. [P66614]Cl). It is well known that commercially available [P66614]Cl usually contains around 0.1–0.5% HCl as a result of its preparation,17,31 and this impurity is difficult to remove using standard Schlenk line technique. From our findings, we can conclude that the presence of HCl impurities in [P66614]Cl decreased the solubility of Te (Fig. S6†). Although the presence of HCl impurity seems to reduce the solubility of Te in [P66614]Cl, it plays at the same time an essential role in the formation of Te microstructures after the addition of polar protic solvents. We verified this hypothesis by the neutralization of commercially available [P66614]Cl with sodium hydroxide solution to remove all HCl impurities and subsequently extensive drying on the Schlenk line. The neutralized and dried [P66614]Cl failed to form Te precipitation when contacted with alcohols (e.g. ethanol). However, when alcohols were replaced by carboxylic acids (e.g. acetic acid) to mix with the purified Te solution in [P66614]Cl, a very fast precipitation occurred and was identified to be crystalline Te by PXRD.
Based on our findings, we believe that (A) the trialkylphosphane tellurides formation with Te dissolving in [P66614]Cl and (B) the appropriate acidity combination from both HCl impurity and polar protic solvents are crucial for the formation of Te particles. The acidity of the added carboxylic acids is strong enough for a rapid P–Te bond cleavage, either in the presence or absence of HCl impurity in [P66614]Cl, resulting in the Te nanoparticles precipitation. In contrast, if protic solvents (e.g. water, alcohols, or amides) that possess a much lower pKa value compared to our tested acids, the HCl in [P66614]Cl turns out to be vital for 3D complex Te microstructures formation.
Morris and co-workers pointed out the so-called water deactivation effect in ILs, which at low concentrations the water is significantly deactivated through strong interaction with the anions of ILs, and exists as isolated single molecules or as small cluster assemblies.32,33 Similarly, trace amounts of HCl in [P66614]Cl are coordinated with chloride ions to form HCl2− anions as previously reported (eqn (1)).34
| Cl− + HCl ⇌ HCl2− | (1) |
The addition of protic solvents to the mixture of [P66614]Cl/[P66614][HCl2] liberates HCl, which leads to an increase in acidity and an accelerated cleavage of P–Te bonds. The morphologies and sizes of obtained Te particles are reproducible independent of the batches of commercial [P66614]Cl with slightly varying HCl concentration.
4. Conclusions
In summary, a variety of Te nano- and microstructures including 3D fusiform assemblies, 3D aloe-like architectures, and nanoparticles are obtained based on a convenient synthesis via the reaction of HCl containing Te solution in [P66614]Cl with polar protic solvents. Studies demonstrate that trace amount of HCl impurities in [P66614]Cl affects the formation of Te nano- and microstructures. Low concentration of HCl in [P66614]Cl tends to be deactivated through strong interaction with chloride ions to form HCl2− anions. The addition of protic solvents liberates HCl from the HCl2− anion which prompts P–Te bond cleavage and formation of Te particles.Currently, most reports of nanoparticles preparation in ILs do not include a careful analysis of their impurities. Considering that the impurities in ILs may affect the quality of nanoparticles, they should not be underestimated. The present investigation shows that trace impurities in ILs can be utilized as a tool to establish new strategies for inorganic material synthesis. However, the impact of IL impurities on the particles formation needs to be well understood.
Conflicts of interest
There are no conflicts to declareAcknowledgements
We would like to thank Dr Tian-Yi Li (IAPP, TU Dresden) for the help of removing excess [P66614]Cl in Teunsat and Tesat solution using flash chromatography. T. Z. acknowledges the support from China Scholarship Council (CSC No. 201404910507). This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG) within the Priority Program SPP 1708.Notes and references
- P. Walden, Bull. Acad. Imp. Sci. St.-Petersbourg, 1914, 8, 405–422 Search PubMed.
- V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS.
- S. P. M. Ventura, F. A. e Silva, M. V. Quental, D. Mondal, M. G. Freire and J. A. P. Coutinho, Chem. Rev., 2017, 117, 6984–7052 CrossRef CAS.
- M. Smiglak, J. M. Pringle, X. Lu, L. Han, S. Zhang, H. Gao, D. R. MacFarlane and R. D. Rogers, Chem. Commun., 2014, 50, 9228–9250 RSC.
- Q. Zhang and J. N. M. Shreeve, Chem. Rev., 2014, 114, 10527–10574 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- M.-R. Gao, J. Yuan and M. Antonietti, Chem. – Eur. J., 2017, 23, 5391–5403 CrossRef CAS PubMed.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- G. Gebresilassie Eshetu, M. Armand, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2014, 53, 13342–13359 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- P. Zhang, T. Wu and B. Han, Adv. Mater., 2014, 26, 6810–6827 CrossRef CAS PubMed.
- B. Kudłak, K. Owczarek and J. Namieśnik, Environ. Sci. Pollut. Res., 2015, 22, 11975–11992 CrossRef PubMed.
- T. Zhang, K. Schwedtmann, J. J. Weigand, T. Doert and M. Ruck, Chem. – Eur. J., 2018, 24, 9325–9332 CrossRef CAS PubMed.
- L. L. Lazarus, C. T. Riche, N. Malmstadt and R. L. Brutchey, Langmuir, 2012, 28, 15987–15993 CrossRef CAS PubMed.
- V. Gallo, P. Mastrorilli, C. F. Nobile, G. Romanazzi and G. P. Suranna, J. Chem. Soc., Dalton Trans., 2002, 4339–4342 RSC.
- M. Le Anh, A. Wolff, M. Kaiser, S. Yogendra, J. J. Weigand, J. Pallmann, E. Brunner, M. Ruck and T. Doert, Dalton Trans., 2017, 46, 15004–15011 RSC.
- H. Zhao, H. M. Brown, J. E. Holladay and Z. C. Zhang, Top. Catal., 2012, 55, 33–37 CrossRef CAS.
- Y. Su, H. M. Brown, X. Huang, X.-D. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117–122 CrossRef CAS.
- D. Tsiulyanu, S. Marian and H. D. Liess, Sens. Actuators, B, 2002, 85, 232–238 CrossRef CAS.
- Y. Wang, Z. Tang, P. Podsiadlo, Y. Elkasabi, J. Lahann and N. A. Kotov, Adv. Mater., 2006, 18, 518–522 CrossRef CAS.
- T.-Y. Wei, H.-Y. Chang and C.-C. Huang, RSC Adv., 2013, 3, 13983–13989 RSC.
- J. He, Y. Chen, W. Lv, K. Wen, Z. Wang, W. Zhang, Y. Li, W. Qin and W. He, ACS Nano, 2016, 10, 8837–8842 CrossRef CAS PubMed.
- Z. He, Y. Yang, J.-W. Liu and S.-H. Yu, Chem. Soc. Rev., 2017, 46, 2732–2753 RSC.
- J.-W. Liu, J. Xu, W. Hu, J.-L. Yang and S.-H. Yu, ChemNanoMat, 2016, 2, 167–170 CrossRef CAS.
- H. Zhu, H. Zhang, J. Liang, G. Rao, J. Li, G. Liu, Z. Du, H. Fan and J. Luo, J. Phys. Chem. C, 2011, 115, 6375–6380 CrossRef CAS.
- X. Wu, Y. Wang, S. Zhou, X. Y. Yuan, T. Gao, K. Wang, S. Lou, Y. Liu and X. Shi, Cryst. Growth Des., 2013, 13, 136–142 CrossRef CAS.
- Inorganic Crystal Structure Database, ICSD, Version 2019/1, FIZ Karlsruhe, Germany and NIST, Gaithersburg, USA, 2019 Search PubMed.
- T. Zhang, K. Schwedtmann, J. J. Weigand, T. Doert and M. Ruck, Chem. Commun., 2017, 53, 7588–7591 RSC.
- A. Nordheider, J. D. Woollins and T. Chivers, Chem. Rev., 2015, 115, 10378–10406 CrossRef CAS PubMed.
- C. J. Bradaric, A. Downard, C. Kennedy, A. J. Robertson and Y. Zhou, Green Chem., 2003, 5, 143–152 RSC.
- R. E. Morris, Chem. Commun., 2009, 2990–2998 RSC.
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS PubMed.
- R. Kohle, W. Kuchen and W. Peters, Z. Anorg. Allg. Chem., 1987, 551, 179–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt04604f |
| This journal is © The Royal Society of Chemistry 2020 |