
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic/molecular layer deposition and electrochemical performance of dilithium 2-aminoterephthalate†
Juho
Heiska
 ,
Mikko
Nisula
,
Eeva-Leena
Rautama
,
Antti J.
Karttunen
and
Maarit
Karppinen
*
,
Mikko
Nisula
,
Eeva-Leena
Rautama
,
Antti J.
Karttunen
and
Maarit
Karppinen
*
Department of Chemistry and Materials Science, Aalto University, FI-00076 Espoo, Finland. E-mail: maarit.karppinen@aalto.fi
First published on 8th January 2020
Abstract
Control of the redox potential of lithium terephthalate Li2TP anode material is demonstrated by functionalizing its terephthalate backbone with an electron-donating amino group; this lowers – as intended – the redox potential of Li2TP by 0.14 V. The two Li-organic electrode materials, Li2TP and Li2TP-NH2, are fabricated as crystalline thin films from gaseous precursors using the atomic/molecular layer deposition (ALD/MLD) technique. The amino-functionalized material possesses a previously unknown crystal structure, addressed here by applying the USPEX evolutionary algorithm for the structure prediction and then LeBail fitting of the experimental XRD pattern based on the predicted structure model. The ALD/MLD fabrication yields in situ lithiated active electrode materials without any conductive additivies or binders and thus allows a straightforward evaluation of their intrinsic electrochemical properties. Comparison between Li2TP and its amino-functionalized derivative reveals inferior capacity retention and rate capability characteristics for the latter, which somewhat counterveils the pros-and-cons balance between the two Li-organic electrode materials. From galvanostatic cycling experiments and post-mortem XRD and SEM analysis, the issue with Li2TP-NH2 is revealed to be in the morphology changes occurring during the discharge/charge cycling.
Introduction
Lithium-ion batteries have found their way to a diverse range of portable applications owing to their superior performance, in particular regarding the specific energy and efficiency. However, it is well known that the present battery chemistries possess some safety issues; there are also concerns related to the availability of their raw materials and the notable CO2 emissions during their manufacturing.1 Alternative next-generation battery chemistries are thus continuously searched for; they should be based on cheap and abundant elements, and exhibit competitive electrochemical performance while being easy to recycle. Organic electrode materials tick many of these requirements and they have been gaining increasing attention in recent years.2,3 There are however several issues to be addressed, related e.g. to the fact that small organic molecules suffer from drastic dissolution in the conventional liquid electrolytes, which results in a capacity decay upon cycling. Another challenge is their intrinsically low electronic conductivity. Different ways have been adopted to fight these challenges such as coating, grafting, chemical modification, and polymerization.Organic molecules are remarkably flexible for different chemical modifications, which opens up enormous possibilities to tune the properties of organic electrode materials.4 A typical organic electrode material comprises one or more redox active functional groups connected to a larger conjugated backbone. The type of the functional group is the determining factor of the potential where the redox reactions occur. The resultant loss or gain of electron(s) is then stabilized by the conjugated backbone. Here we focus on a carbonyl-type material for which upon reduction, the electron is added to the lowest unoccupied molecular orbital (LUMO).
When an additional electron-donating functional group is attached to the carbon skeleton it causes the LUMO energy to become more positive, thereby lowering the redox potential.5 A somewhat linear relationship between the redox potential and LUMO energy has been reported.6 The challenge in the evaluation of the effects of different functional groups is to find simple model systems with a minimum number of other variables.7 The present study was inspired by our recent success in depositing high-quality lithium terephthalate (Li2TP) thin films using the atomic/molecular layer deposition (ALD/MLD) technique, and testing these films as a negative electrode material in a pristine form, e.g. without any conductive additives or binder material.8,9 The ALD/MLD technique for metal-organics is a strongly emerging branch of the conventional atomic layer deposition (ALD) technology for inorganic thin films, which has been in industrial use already for decades.10,11 Like ALD, the combined ALD/MLD technique for metal–organic thin films is based on sequential, self-saturating surface reactions of gaseous precursors and has the capacity to yield thin films with incontestable thickness control and uniformity.12 The recent developments in the field of ALD/MLD have opened new avenues towards new interesting metal–organic materials.8,13–18 Here we extend the ALD/MLD material repertoire to dilithium 2-aminoterephthalate (Li2TP-NH2), which then allows us to evaluate the role of the electron-donating amino group on the redox potential of the terephthalate skeleton, with the anticipation that the somewhat high redox potential of Li2TP could be decreased by introducing the additional amino group (Fig. 1). Prior to the present work, Li2TP-NH2 has been synthesized in bulk form and found to be electroactive but not significantly different from Li2TP regarding the redox potential.19 On the other hand, these carboxylates behave similarly in sodium ion batteries,20–23 and the sodium equivalent Na2TP-NH2 has shown the expected decrease in redox potential in bulk samples.24
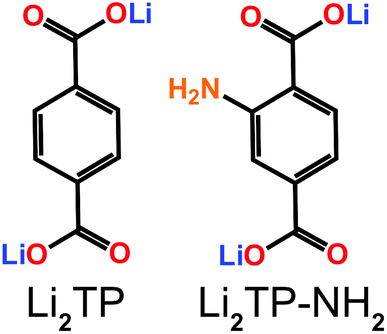 | ||
| Fig. 1 Molecular structures of Li2TP and Li2TP-NH2. | ||
Results and discussion
ALD/MLD process development
We have previously developed a facile ALD/MLD process for Li2TP films based on Li(thd) and terephthalic acid (TPA) precursors.8 Here we adopted the same lithium precursor for the Li2TP-NH2 process and replaced the organic precursor to its amino-substituted version, i.e. 2-aminoterephthalic acid (TPA-NH2), to make the comparison straightforward. First, we confirmed the most important characteristics of an ideal ALD/MLD process, that is, the saturation of the so-called growth-per-cycle (GPC) value (calculated from the XRR-determined film thickness value) with increasing precursor pulse lengths; these experiments were carried out at the deposition temperature 200 °C, and the number of ALD/MLD cycles was fixed to 100 (Fig. 2). In all our depositions the N2 purging time was kept constant, at 4 s after the Li(thd) pulse and at 30 s after the organic precursor pulse. It was revealed that for both the Li2TP-NH2 and Li2TP processes the film growth saturated with a relatively short (4 s) Li(thd) pulse time, while the organic precursors required a little longer pulsing time, i.e. 6 s for TPA-NH2 and 10 s for TPA. The resultant GPC values were appreciably high, and also quite similar for the two processes, i.e. 3.6 Å per cycle for Li2TP-NH2 and 3.0 Å per cycle for Li2TP. Also, in both cases the growth rate decreased significantly with increasing deposition temperature (Fig. 2b); this is a trend seen for most of the reported ALD/MLD processes.8,14,15,17,18,25,26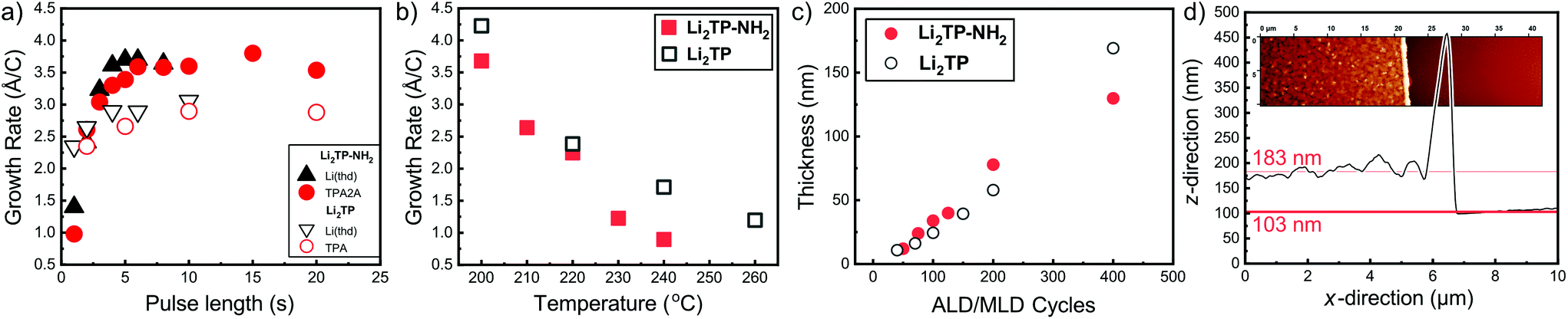 | ||
| Fig. 2 Growth-per-cycle (GPC) values for the Li(thd) + TPA-NH2 and Li(thd) + TPA8 processes as a function of (a) precursor pulse lengths (at 200 °C with 100 ALD/MLD cycles), and (b) deposition temperature with the following precursor pulse lengths: 4 s of Li(thd) and 10 s of organic in both cases. In (c) thicknesses of Li2TP-NH2 and Li2TP films with increasing number of ALD/MLD cycles. (d) Film thickness determination using AFM (for a Li2TP-NH2 film grown with 200 ALD/MLD cycles). | ||
We also confirmed the linearity of the film growth with an increasing number of ALD/MLD cycles after a small incubation period in the beginning (Fig. 2c). The precise determination of the film thickness with XRR was possible for the thinner films only; for the thicker films the surface roughness increased such that the XRR method became less reliable.27 From the AFM data the mean surface roughness of the film with 100 cycles was determined to be ∼3 nm, while for the thicker films with 200 and 400 ALD/MLD cycles the roughness was ∼10 and ∼14 nm, respectively. For the thickest films, we employed spectroscopic ellipsometry for the thickness determination. Additionally, an estimation of the film thickness (∼80 nm) for a film deposited with 200 ALD/MLD cycles was obtained using AFM; a part of the film was scratched (with a surgical knife) and then the surface was scanned with an AFM tip to probe the depth difference between the film surface and the silicon substrate surface (Fig. 2d).
The Li2TP-NH2 films were all crystalline after 200 ALD/MLD cycles. In Fig. 3a we show a part of the GIXRD pattern around the main peak for representative films to demonstrate that the FWHM (full width at half maximum) decreases with an increasing number of deposition cycles, indicating an increase of the crystallite size as the films get thicker. The same observation can be made from the AFM images shown in Fig. 3b. The AFM data suggest an island-type growth mode (similar to that previously seen for the Li2TP films);8 it seems that initially the Li2TP-NH2 films grow as amorphous, but soon islands of crystallites start to form which then serve as the preferred sites for the further film growth. This is in line with the fact that the film density (calculated from the critical angle value obtained from the XRR pattern) was initially somewhat higher, i.e. 1.59 g cm−3 for the film deposited with 100 cycles, then 1.40 and 1.44 g cm−3 for films deposited with 200 and 400 cycles, respectively.
 | ||
| Fig. 3 (a) GIXRD patterns with calculated FWHM values, and (b) AFM images for Li2TP-NH2 films deposited at 200 °C with increasing number of ALD/MLD cycles. The z-scale in AFM images is 0–50 nm for the sample with 100 cycles and 0–250 nm in samples with 200 and 400 cycles. | ||
Structure of Li2TP-NH2 films
The Li2TP-NH2 films were crystalline, but the diffraction pattern could not be indexed according to any relevant crystal structure. It should be noted that even though the Li2TP-NH2 phase has been previously synthesized in bulk form (plus XRD pattern is given),28 its crystal structure has not been reported. Moreover, unlike the case with e.g. the zirconium analogues, ZrTP and ZrTP-NH2,29 the Li2TP-NH2 structure is apparently not identical with the Li2TP structure (ESI Fig. 2†). We used the USPEX evolutionary algorithm30 to predict the most probable crystal structure candidates for Li2TP-NH2. The lowest-energy structure candidate revealed through USPEX is a layered monoclinic structure with two Li2TP-NH2 formula units in the unit cell (Fig. 4a). Interestingly, there is a direct subgroup-group relation between this Li2TP-NH2 structure candidate (Pc) and the layered structure seen for Li2TP (P21/c) in both bulk and in our ALD/MLD samples.8,31 The USPEX prediction also yielded a few other low-energy monoclinic structures for Li2TP-NH2 with energies not much higher than in the case of the Pc structure (see ESI†).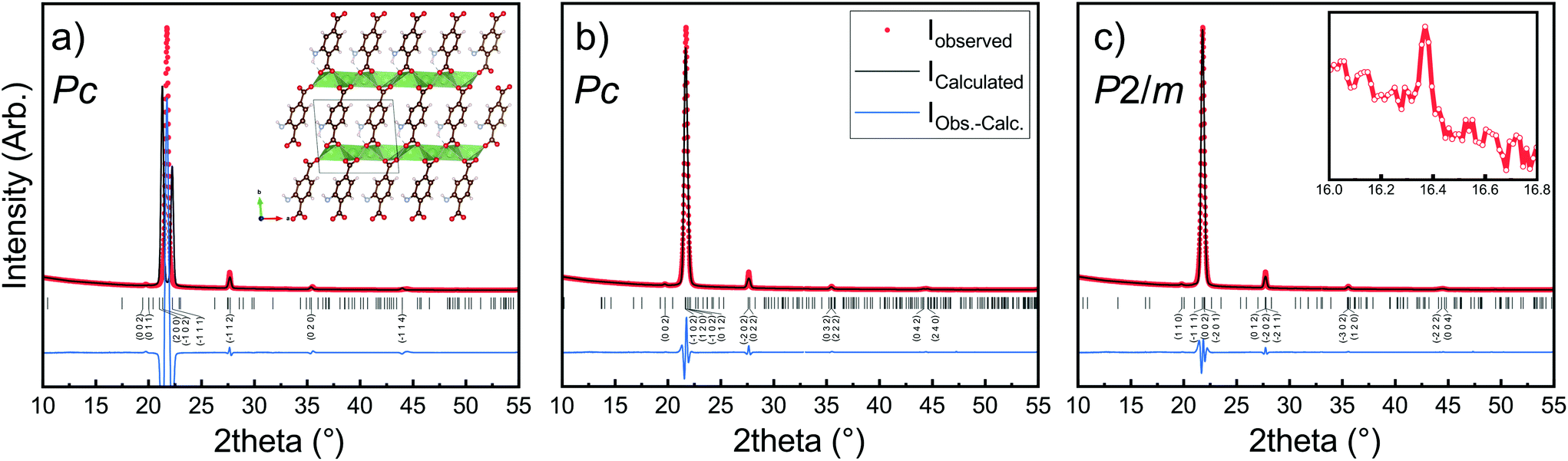 | ||
| Fig. 4 XRD pattern and LeBail fits of Li2TP-NH2 with a thickness of around 1000 nm. In (a) the simulated profile fit using lattice parameters predicted by USPEX evolutionary algorithm, in (b) LeBail fits with tiny reflection at 16° omitted, in (c) the tiny peak is included (inset). Visualization of the structure can be found in ESI.† Substrate peaks are removed for clarity. | ||
In Fig. 4b, we have indexed and LeBail fitted an experimental XRD pattern for our Li2TP-NH2 thin-film sample; the fitting confirms that the structure must be close to the USPEX-predicted monoclinic Pc structure (with lattice parameters a = 8.68 Å, b = 5.23 Å, c = 8.99 Å, and β = 93.76°), the monoclinic distortion being clearly seen as the splitting of the main reflection around 2θ ≈ 22°. However, a closer look up indicates an additional tiny reflection around 2θ ≈ 16° (Fig. 4c), which is not explained with the Pc model. Considering this reflection, the best match was found for a cell with P2/m symmetry (a = 8.57 Å, b = 5.28 Å, c = 8.23 Å, β = 99.85°). For both the aforementioned structure models, the lattice parameters are rather close to the USPEX-predicted lattice, with a = 8.48 Å, b = 5.07 Å, c = 9.16 Å, and β = 94.77°. It should be noted that neither the experimental nor the simulated diffraction patterns match with the previously reported pattern for a bulk Li2TP-NH2 sample.28 Steric hindrance of the amino group most likely plays a role here, in such a way that the different synthesis conditions could lead to different amino group arrangements.32,33
The chemical bonding scheme in our Li2TP-NH2 thin films was studied with FTIR spectroscopy. A comparison of the thin-film spectrum to that of its TPA-NH2 precursor confirms the disappearance of the characteristic carbonyl group (only seen for the acid precursor) and the appearance of the carboxyl feature upon the metal carboxylate bond formation (ESI Fig. 3†). The detailed interpretation of spectral features is given in ESI Fig. 4,† where we compare our thin-film Li2TP-NH2 spectrum with the previously reported FTIR spectrum for a bulk Li2TP-NH2 sample28 and also with a spectrum calculated based on the USPEX predicted monoclinic Pc structure for Li2TP-NH2; for the peak interpretation (ESI Table 2†),34,35 we also considered the spectra reported for Li2TP,36 Na2TP-NH2,33 and TPA-NH2.37
Most interesting observations were made by comparing the spectra of our present Li2TP-NH2 and Li2TP thin films, see Fig. 5. The dominant peaks in terephthalate systems are caused by the asymmetric (υas) and symmetric (υs) stretches of carboxylate around 1600 cm−1 and 1400 cm−1, respectively.14 The position of these peaks is often strongly affected by the nature of the substituents. In particular, electron donating groups attached to aromatic carbon skeleton are expected to shift υas to the higher energies (blue shift), while electron withdrawing groups such as –NH2 should result in a shift to the lower energies (red shift). Electron withdrawing substituents also shift υs to the lower energies but this behavior is less predictable depending also on the position of the substituent (relative to carboxylate).38 Indeed, from Fig. 5 it is seen that υas red shifts from 1572 cm−1 (Li2TP) to 1568 cm−1 (Li2TP-NH2) upon the addition of the –NH2 group, and υs from 1393 cm−1 (Li2TP) to 1372 cm−1 (Li2TP-NH2). The separation (Δ) between υas and υs is indicative of the binding mode of the carboxylate unit.14,38 For our Li2TP-NH2 thin-film sample, the obtained Δ value of 196 cm−1 suggests a bridging-type connection, which is in accordance with the USPEX-predicted structure model.
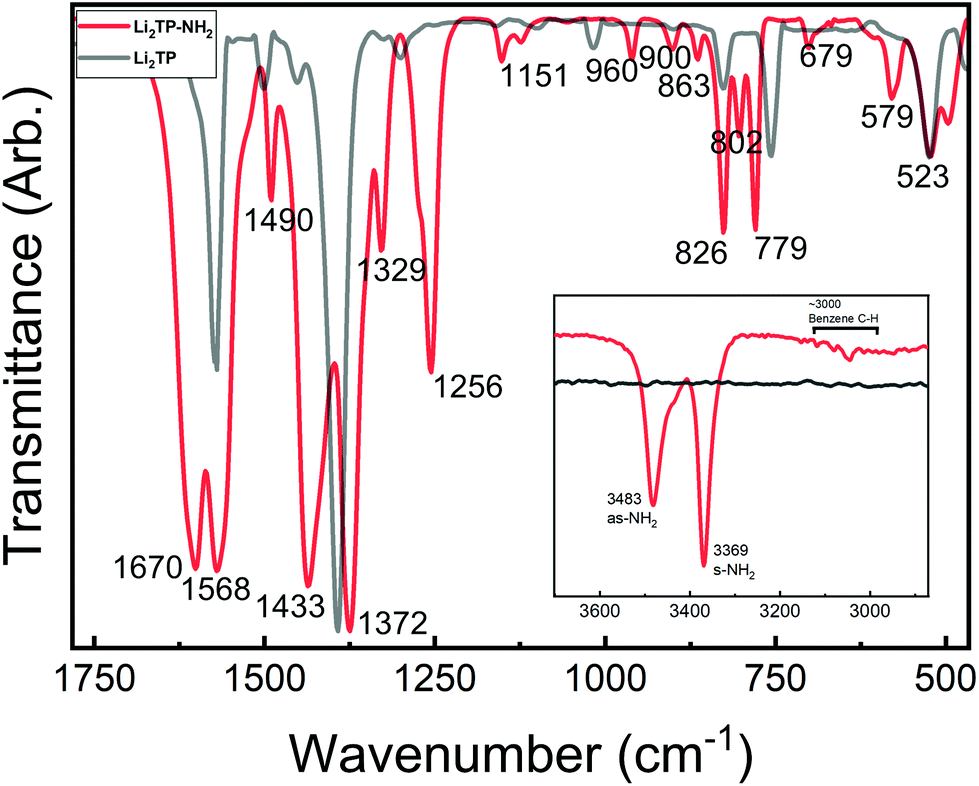 | ||
| Fig. 5 FTIR spectra for Li2TP-NH2 and Li2TP; inset shows the antisymmetric (as) and symmetric (s) vibrations of the amino group at higher wavenumbers. | ||
Another important observation is related to the stretching modes of the benzene ring (19a and 19b; ESI Table 2†), being composed of partly stretching and partly bending character of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. In para disubstituted benzene (like Li2TP) the 19a mode lies at higher wavenumbers,39 while in asymmetric trisubstituted benzene the 19b mode is at a higher frequency, according to the normal coordinate analysis.35 This is important since with monosubstituted benzenes the vibration 19a has been found to become more intense with donor substituents, while 19b gets stronger with acceptor substituents. A similar correlation could explain the strong peak observed at 1433 cm−1 for our Li2TP-NH2 thin film. Another observation is that the amino group is apparently not reacting with the Li(thd), as the characteristic antisymmetric and symmetric peaks due to the amino group remain intact. However, this does not mean that the amino group would not show any coordination effect on the lithium oxide layer; indeed a small shoulder is seen in the asymmetric stretch of the amino peaks.
C bonds. In para disubstituted benzene (like Li2TP) the 19a mode lies at higher wavenumbers,39 while in asymmetric trisubstituted benzene the 19b mode is at a higher frequency, according to the normal coordinate analysis.35 This is important since with monosubstituted benzenes the vibration 19a has been found to become more intense with donor substituents, while 19b gets stronger with acceptor substituents. A similar correlation could explain the strong peak observed at 1433 cm−1 for our Li2TP-NH2 thin film. Another observation is that the amino group is apparently not reacting with the Li(thd), as the characteristic antisymmetric and symmetric peaks due to the amino group remain intact. However, this does not mean that the amino group would not show any coordination effect on the lithium oxide layer; indeed a small shoulder is seen in the asymmetric stretch of the amino peaks.
Electrochemical characteristics
Cyclic voltammetry in coin-cell configuration was used to investigate the redox behaviors of the Li2TP-NH2 and Li2TP films. The voltammograms were recorded between 0.25–3.0 V for Li2TP-NH2 and 0.01–3 V for Li2TP; in the first case scanning to too low potentials was found to be detrimental and was hence avoided. The 1st and 2nd cycles were measured with a scan rate of 0.1 mV s−1, followed by cycles with higher rates of 0.5 and 1 mV s−1 (Fig. 6). For both materials, the 1st cycle was found drastically different from the following cycles, presumably due to solid electrolyte interphase (SEI) formation.40 After the 1st cycle, Li2TP-NH2 shows wide reduction and oxidation peaks centered around 0.62 and 0.82 V, respectively, while Li2TP exhibits relatively sharp peaks at 0.76 V (reduction) and 1.0 V (oxidation). Hence the electron-donating amino group indeed lowers the redox peak potential of Li2TP by 0.14 V. The magnitude of the decrease in the potential is not enormous but it is in line with the results previously reported for the Na-based counterparts (0.19 V; quasi-open circuit voltages).24 According to DFT calculations by Renault et al.28 lithiation of Li2TP-NH2 occurs preferably on the carbonyl next to the amino group. This preferred process can be observed as a shoulder of the main reduction peak; this shoulder is however seen with the slowest scan rate of 0.1 mV s−1 only, indicating slow diffusion or kinetics of the electrode (Fig. 6a; red curve). Finally we note that the positive impact of the amino group in lowering the redox potential loses some of its significance due to the poorer electrode kinetics and/or slower diffusion into the electrode, the reason of which will be discussed in more detail later on.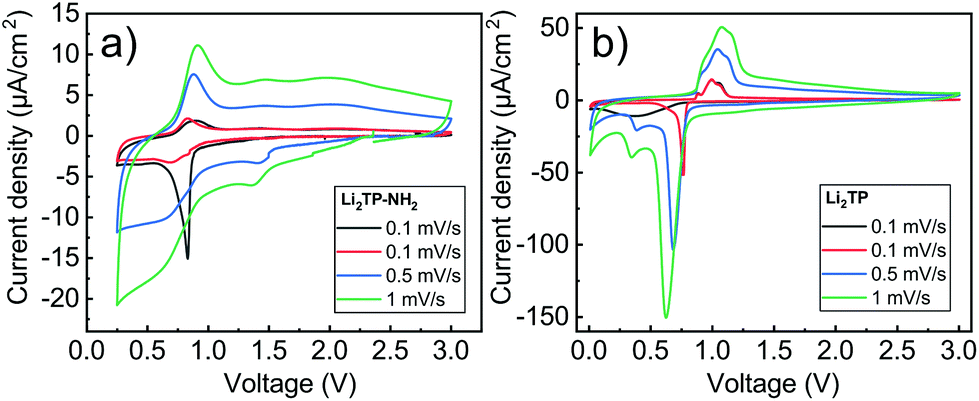 | ||
| Fig. 6 Cyclic voltammograms for (a) Li2TP-NH2 (0.25–3.0 V), and (b) Li2TP (0.01–3.0 V) with various scan rates. The dark grey line represents the 1st cycle, and the red, blue, and green lines represent the subsequent cycles. Each cycle starts at the original open circuit voltage, being ca. 2 V for both the samples. | ||
Galvanostatic charge/discharge cyclings were performed with various current rates to investigate the cycling performance (Fig. 7); in these experiments, the voltage range was 0.4–3.0 V for both materials. In line with the cyclic voltammetry results, the performance of Li2TP-NH2 was inferior to that of Li2TP in the rate capability tests. The capacity retention between 1.1 and 53.0 μA cm−2 was 67% for Li2TP but only 24% for Li2TP-NH2. After the initial cycles, the coulombic efficiency on average was 99% for Li2TP and 97% for Li2TP-NH2. The capacity values were ∼9 μA h cm−2 for Li2TP and ∼8 μA h cm−2 for Li2TP-NH2.
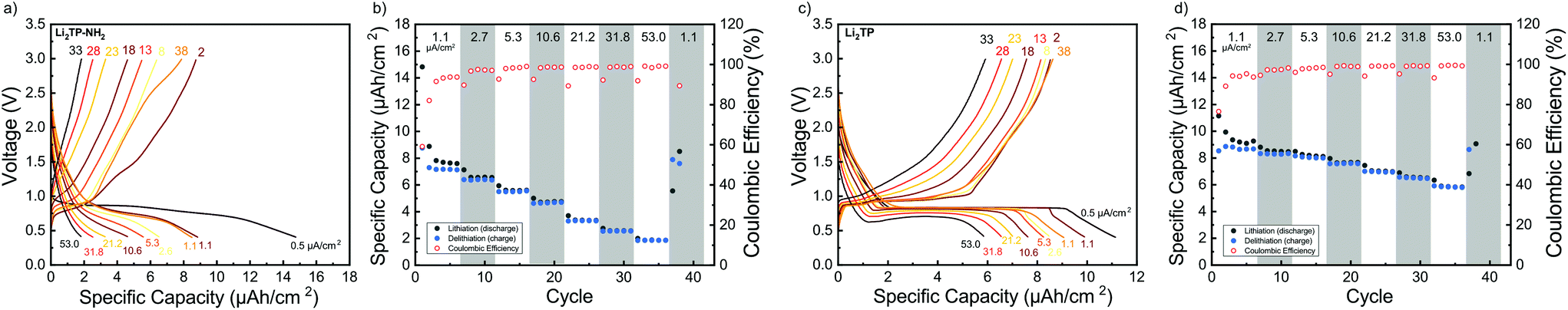 | ||
| Fig. 7 Rate capability measurements for Li2TP-NH2 (a and b) and Li2TP (c and d): cycling starts with a format cycle with a small current of 0.5 μA cm−2, thereafter the current is increased after every 5 cycles before returning to the original value (1.1, 2.7, 5.3, 10.6, 21.2, 31.8, 53.0 μA cm−2). In (a) and (c) the shape of the charge–discharge curve at different cycles and in (b) and (d) capacity plotted with coulombic efficiency. | ||
While the targeted role of the additional functional groups is to decrease or increase the redox potential,41 they may also have unintended consequences. It has been shown that electron donor functional groups may disturb the π–π orbital stacking of the benzene rings and actually decrease the conductivity of the material.42 According to the calculations by Zhang et al.,43 Li+ hopping is fast in Li2TP and occurs mostly within the stacked π-orbitals of the benzene rings; this is also the path of the least resistance for the electrons in Li2TP.19,44 Upon replacing one of the hydrogen atoms in the benzene ring with an additional functional group a shift occurs for the HOMO and LUMO orbitals and thereby also for the π-orbital stacking. This might be one of the reasons for the poorer electrochemical performance. Functional groups may also introduce a steric hindrance for the Li+ diffusion.
The flat (dis)charge curve extending even up to high current densities for Li2TP is not seen for Li2TP-NH2 (Fig. 7). Such sloping potentials as observed for the present Li2TP-NH2 thin film and also previously reported for bulk Li2TP-NH2.28 Sloping voltage profiles are rather common in organic polymer batteries, where the redox active groups are interconnected and the potential strongly depends on the lithiation degree.45 Park et al.24 explained the difference as a change of the reaction type, from two-phase reaction to one-phase reaction. Electron donors are prone to cause charge reorganizations and formation of occupied states in the aromatic core; these changes may have an effect on the voltage profile.46 Therefore, there might be multiple reasons behind the observed change in the voltage profile.
From the rate capability measurement data shown in Fig. 7 it can be seen that the delithiation is never complete in the cycle succeeding the increase in current density, i.e., in these first discharge/charge cycles the capacity from lithiation is larger than the capacity from delithiation indicating that with the same current density, delithiation is the slower reaction. Afterwards, the capacity loss can be recovered with lower current rates, as seen in the increased delithiation capacity. A larger cut-off voltage for delithiation might solve the issue, but 3.0 V is already a very high voltage for a negative electrode material.
Its has been observed that the electronic conductivity of organic electrodes increases with the amount of intercalated lithium.44 Therefore, during lithiation, the material probably becomes more and more conducting such that it becomes easier to intercalate lithium to the lattice. This is actually what can be seen with Li2TP with the larger current densities during lithiation where the potential momentarily drops lower than the plateau. Hence, after a certain lithiation threshold, it becomes easier to continue the lithiation, presumably due to the decrease in resistivity. Clearly, this same behavior is not seen with Li2TP-NH2, presumably due to its intrinsically higher resistivity.
The performance of the films was also tested over 200 charge/discharge cycles. Cycling started again with a slow format cycle (0.5 μA cm−2) followed by cycling with 2.7 μA cm−2 (Fig. 8). It was seen that Li2TP retains 95% of its capacity and shows very stable performance over the whole cycling period, while Li2TP-NH2 retains 90% of its capacity, but also shows an interesting behavior, where the capacity first increases followed by a steady decrease (Fig. 8a). In previous works, similar abnormal excess capacity has been ascribed to the lithiation of the aromatic skeleton. This could have an effect on the reversibility of the redox reaction as some of the lithium inserted in low potentials would require very high potentials to leave the electrode.47 Amino groups have been reported to suppress the dissolution of organic electrodes,48 so the decrease in capacity is most likely not due to dissolution, and as can be seen in the Fig. 9b, the thickness of the film has only increased due to the formation of SEI. The coulombic efficiency of Li2TP is very high (>99%) after the initial cycles compared to rather poor efficiency of 95% of Li2TP-NH2. The lower coulombic efficiency is a direct sign that the redox reaction with Li2TP-NH2 is not as reversible as with Li2TP within this potential range. The coulombic efficiency of Li2TP-NH2 is noticeably low during the cycles related to the lithiation of the aromatic skeleton. Similar low coulombic efficiency with Li2TP was observed by Lee et al.47 when the low potential cut-off voltage was decreased to 0.0 V, where the aromatic skeleton of Li2TP was purposely lithiated. Therefore, both the gradual increase in capacity and the lower coulombic efficiency are possibly caused by the lithiation of the aromatic skeleton.
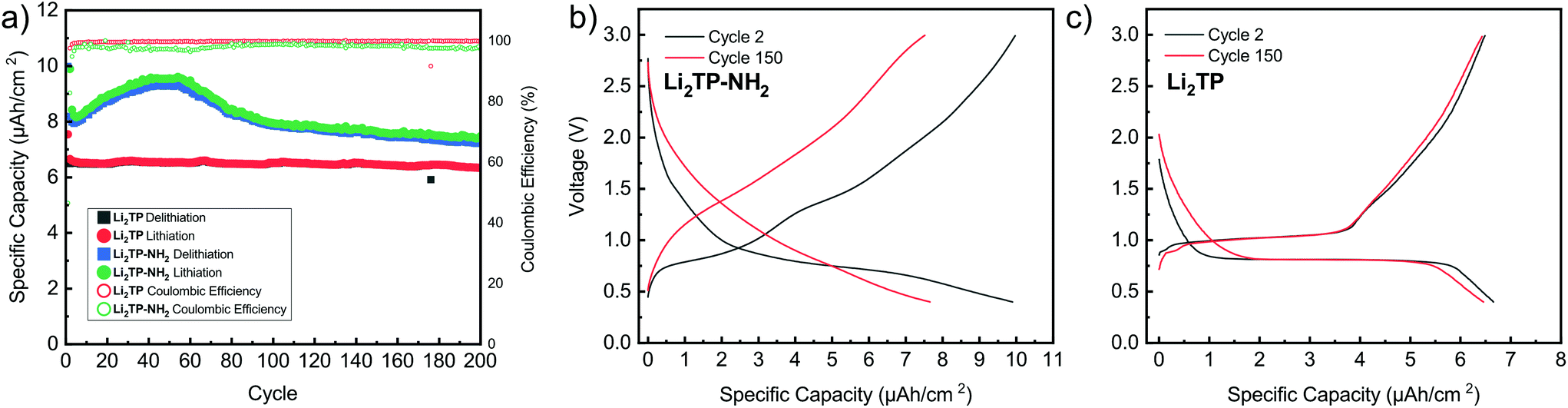 | ||
| Fig. 8 Performance of Li2TP-NH2 and Li2TP during extended cycling in a half-cell. In (a) capacity retention and coulombic efficiency over 200 cycles and in (b) and (c) the observed change in the charge–discharge curve of Li2TP-NH2 and Li2TP, respectively. | ||
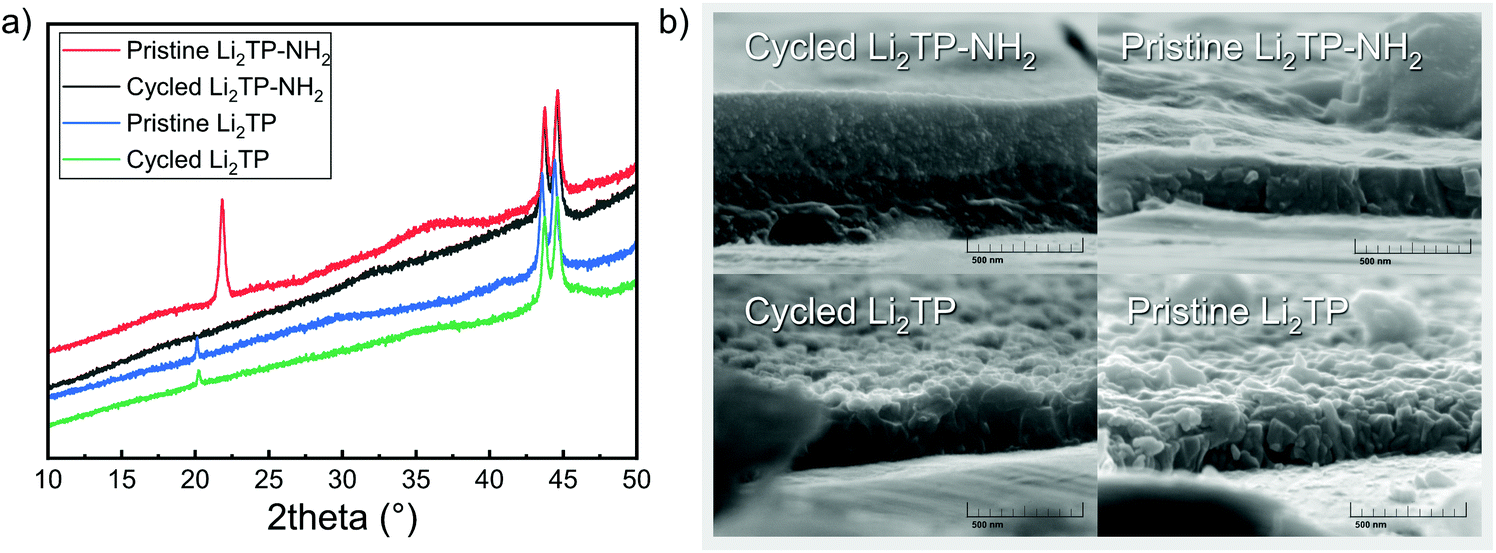 | ||
| Fig. 9 (a) Diffraction patterns of Li2TP-NH2 (the shift in baseline and peaks at 44° are due to steel substrate), and (b) SEM images for a pristine and cycled Li2TP-NH2 and Li2TP electrodes. | ||
The shape of the charge–discharge curve of Li2TP-NH2 changes drastically over the cycling while that of Li2TP remains essentially unchanged (Fig. 8b and c). The steady slope of Li2TP-NH2 disappears completely and the over-potential increases. The drastic changes in the shape of the charge–discharge curve are a direct indication of changes occurring on the material during cycling, which might be due to the amorphization of the material caused by the lithiation of the aromatic skeleton47 or other significant morphological changes of the electrode. In Fig. 9 we display post-mortem XRD and cross section SEM image for a pristine and cycled electrodes; both clearly show that the Li2TP-NH2 turns amorphous upon cycling. From the SEM images it is moreover seen that the cycled electrode is considerably thicker (formation of SEI) and it consists of a sea of small particles, while in the pristine electrode the crystallites are larger. The Li2TP on the otherhand shows only minor changes on the surface and the crystallites remain intact.
Material properties such as the electrode morphology49 and shape of crystallites50,51 of the active material may have a considerable effect on mass and electron transport of the electrode and thereby its electrochemical performance. In addition, it has been shown that particle pulverization can take place and is detrimental to the electrochemical performance of organic electrode materials.52 The observed morphological changes occurring during cycling for our Li2TP-NH2 electrode are very likely the cause of the capacity decay. We tentatively believe that the breakage of the crystal structure originates from the lithiation of the aromatic skeleton.47
Conclusions
We have widened the repertoire of lithium-bearing ALD/MLD processes to the amino-functionalized lithium terephthalate, i.e. dilithium 2-aminoterephthalate or Li2TP-NH2. The Li2TP-NH2 films were found to initially grow smooth and amorphous but after a certain threshold number of ALD/MLD cycles crystallites started to form. The Li2TP-NH2 crystals were not of any previously known crystal structure; we thus determined the structure with a combined prediction (USPEX), modelling (DFT) and fitting (LeBail) approach. Additional evidence for the resulting monoclinic structure was revealed from the FTIR data.Electrochemical measurements verified the validity of our initial design concept, that is, the possibility to use additional electron-donating functional groups to lower the redox potential of the lithium terephthalate Li2TP anode material. The decrease in the potential achieved with the amino group attached to the terephthalate backbone was 0.14 V. Unfortunately, this benefit was gained at the expense of the lower coulombic efficiency and poorer rate capability. We observed effects such as morphology, crystallinity changes, and delamination but no dissolution in the post-mortem analysis of the electrodes.
As these thin film electrodes are free from any conductive carbon or other additives and their preparation route is the same their performance should be indicative of the actual material. This especially makes the post-mortem analysis simpler as the interactions in the cells are minimized. We believe that manufacturing the organic electrode materials in the thin film form is the key to better understand their electrochemical performance.
Experimental
The Li2TP-NH2 and Li2TP (for reference) thin films were deposited from Li(thd) (thd = 2,2,6,6-tetramethyl-3,5-heptanedione), 2-aminoterephthalic acid (98%, Tokyo Chemical Industry Co. Ltd) and terephthalic acid (98%, Tokyo Chemical Industry Co. Ltd) precursors. The Li(thd) precursor powder was synthetized by mixing LiOH (98%, Alfa Aesar) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O and EtOH mixture with thd (Tokyo Chemical Industry Co. Ltd) in 1:1 H2O and EtOH mixture. The resulting white mixture was further dried in vacuum and purified by sublimation. The films were deposited in an F-120 flow-type hot-wall ALD reactor (ASM Microchemistry Ltd) at low pressure (∼5 mbar). The carrier and purging gas was nitrogen (produced from the air with nitrogen generator Parker HPN2-5000, with less than 10 ppm of oxygen). For the Li2TP films, the deposition process parameters were adopted from our previous work,8 while for the Li2TP-NH2 films the parameters were optimized in this work. The sublimation temperature for the TPA-NH2 was 185 °C and 175 °C for Li(thd). For the structure characterizations, Si(100) substrates were used, while for the electrochemical characterizations the films were grown on stainless steel disks (∅ 15.5 mm).
:1 H2O and EtOH mixture with thd (Tokyo Chemical Industry Co. Ltd) in 1:1 H2O and EtOH mixture. The resulting white mixture was further dried in vacuum and purified by sublimation. The films were deposited in an F-120 flow-type hot-wall ALD reactor (ASM Microchemistry Ltd) at low pressure (∼5 mbar). The carrier and purging gas was nitrogen (produced from the air with nitrogen generator Parker HPN2-5000, with less than 10 ppm of oxygen). For the Li2TP films, the deposition process parameters were adopted from our previous work,8 while for the Li2TP-NH2 films the parameters were optimized in this work. The sublimation temperature for the TPA-NH2 was 185 °C and 175 °C for Li(thd). For the structure characterizations, Si(100) substrates were used, while for the electrochemical characterizations the films were grown on stainless steel disks (∅ 15.5 mm).
X-ray diffraction patterns were collected for the films with PANanalytical X'Pert Pro diffractometer (Cu Kα1; λ = 1.540598 Å) both in the conventional (XRD) and the grazing-incidence (GIXRD; incident angle 0.5°) modes. The XRD patterns measured in Bragg-Brentano mode were indexed and fitted with the LeBail method using the options integrated into the FullProf software package (Treor90 and CheckCell).53 The film thickness and density were estimated for the films from the X-ray reflection (XRR) patterns measured with the same apparatus. It should be noted that the density calculation (see ESI†) is based on an approximated chemical composition of the film, which naturally courses some uncertainty; nevertheless, within a series of similar samples, the possible trends should be meaningful.8,54 For the thickest and roughest films, the film thickness was determined with spectroscopic ellipsometer measurements (Woollam Spectroscopic Ellipsometer; CompleteEASE software).
The bonding structure and the presence of intended functional groups were investigated with Fourier transform infrared spectroscopy (FTIR; Nicolet Magna 750). The FTIR measurements were performed in a range of 400 to 4000 cm−1 with a resolution of 4 cm−1. The atomic force microscope images were conducted in tapping mode with a Veeco Dimension 5000. Tips used were Mikromasch HQ: NSC14/AlBS tips with a typical radius of 8 nm and 5 N m−1 force constant.
For the electrochemical evaluation, the thin films were deposited on steel substrates. The thickness of the thin film could not be measured from the steel substrate, and therefore the exact mass of the electrode cannot be reliably determined. However, the mass of the electrode can be estimated from the growth rate on the silicon substrate. This will cause some systematic error in the data because the nucleation model on different substrates can differ.55 However, as the film's goal thickness was fixed to 170 nm, it makes much more sense just to compare the capacity per area and it makes the electrode comparison more practical. The theoretical capacity of Li2TP-NH2 is 277.8 mA h g−1. Assuming the film's thickness of 170 nm and calculated density, the areal capacity should be around 8 μA h cm−2 for both of the materials. This is relatively close value to the measured ones when taking into account all of the possible errors in calculated density, film thickness, and the exact area of the current collector as films grow in every direction and underneath the substrate. The samples were first dried in a vacuum oven (110 °C) for 24 hours and moved into argon filled glovebox with oxygen levels under 1 ppm. Afterwards, the films were directly applied as the working electrode in a CR2016 coin cell with a lithium metal counter electrode to evaluate their electrochemical performance. The electrolyte was 1 M LiPF6 in a 1:1 EC/DMC solution. Cyclic voltammetry measurements were carried out with Autolab PGSTA302N potentiostat, and galvanostatic cyclings in a Neware battery testing unit with various current densities. The galvanostatic cycling voltage range was chosen to limit the reactions occurring below 0.4 V. In cyclic voltammetry each cycle starts at the original open circuit voltage, being ca. 2 V for both the samples.
We also carried out crystal structure predictions for Li2TP-NH2 with the evolutionary algorithms implemented in the USPEX 9.4.4 code.30 All quantum chemical calculations within the USPEX simulations were performed using Quantum Espresso program package (version 6.0).56 We used density functional theory (DFT) with the PBE exchange–correlation functional and GBRV ultrasoft pseudopotentials.57,58 Kinetic energy cut-offs of 40 Ry and 200 Ry were applied for wavefunctions and charge densities, respectively. In the USPEX simulations, we applied a molecular crystal structure prediction algorithm.59 Each simulation included two Li+ cations per each 2-aminoterephtalate dianion and all structures where the dianion did not stay intact were discarded. The USPEX simulations were run with different numbers of formula units in the unit cell (Z = 2, 3, and 4). All simulations produced a similar type of layered structures, but the lowest-energy structures were obtained from two simulations carried out for Z = 2. The full USPEX and Quantum Espresso input files for one Z = 2 simulation are included in ESI.†
The low-energy structures produced by USPEX were re-optimized using the CRYSTAL17 program package, hybrid PBE0 density functional method, and triple-zeta-valence + polarization (TZVP) level of basis set. The Gaussian Type Orbital basis sets have been derived from Karlsruhe basis sets60 and adapted for solid-state calculations.13,61 For all low-energy structures, we applied a k-mesh of 3 × 5 × 3 for the reciprocal space sampling. The structures were optimized with the default optimization convergence criteria in CRYSTAL17 and the lowest-energy structures were confirmed to be true local minima by means of harmonic frequency calculations. The harmonic vibrational frequencies and IR intensities were obtained with the computational scheme implemented in CRYSTAL.62 For the simulated IR spectra, we applied a Lorentzian lineshape and an FWHM of 16 cm−1. The band assignments were carried out by visual inspection of the normal modes using the Jmol program package.63
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Advanced Grant Agreement (339478) and Academy of Finland (296299), and the use of the RawMatTERS Finland Infrastructure (RAMI) and OtaNano – Nanomicroscopy Center (Aalto-NMC) at Aalto University. Computational resources were provided by CSC-the Finnish IT Center for Science.Notes and references
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. Heiska, M. Nisula and M. Karppinen, J. Mater. Chem. A, 2019, 7, 18735–18758 RSC.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- Y. Liang and Y. Yao, Joule, 2018, 2, 1690–1706 CrossRef CAS.
- Q. Zhao, Y. Lu and J. Chen, Adv. Energy Mater., 2017, 7, 1601792 CrossRef.
- Z. Zhu and J. Chen, J. Electrochem. Soc., 2015, 162, 2393–2405 CrossRef.
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600–605 CrossRef CAS.
- M. Nisula and M. Karppinen, Nano Lett., 2016, 16, 1276–1281 CrossRef CAS PubMed.
- M. Nisula and M. Karppinen, J. Mater. Chem. A, 2018, 6, 7027–7033 RSC.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- A. S. Asundi, J. A. Raiford and S. F. Bent, ACS Energy Lett., 2019, 4, 908–925 CrossRef CAS.
- O. Nilsen, K. Klepper, H. Nielsen and H. Fjellvåg, ECS Trans., 2008, 16, 3–14 CAS.
- M. Nisula, J. Linnera, A. J. Karttunen and M. Karppinen, Chem. – Eur. J., 2017, 23, 2988–2992 CrossRef CAS PubMed.
- J. Penttinen, M. Nisula and M. Karppinen, Chem. – Eur. J., 2017, 23, 18225–18231 CrossRef CAS PubMed.
- E. Ahvenniemi and M. Karppinen, Chem. Commun., 2016, 52, 1139–1142 RSC.
- P. Sundberg and M. Karppinen, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- J. Penttinen, M. Nisula and M. Karppinen, Chem. – Eur. J., 2019, 25, 11466–11473 CrossRef CAS PubMed.
- A. Khayyami and M. Karppinen, Chem. Mater., 2018, 30, 5904–5911 CrossRef CAS PubMed.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribière, P. Poizot and J.-M. Tarascon, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed.
- L. Zhao, J. Zhao, Y. S. Hu, H. Li, Z. Zhou, M. Armand and L. Chen, Adv. Energy Mater., 2012, 2, 962–965 CrossRef CAS.
- X. Yan, C.-Y. Fan, X. Yang, Y.-Y. Wang, B.-H. Hou, W.-L. Pang and X.-L. Wu, Mater. Today Energy, 2019, 13, 302–307 CrossRef.
- F. Wan, X.-L. Wu, J.-Z. Guo, J.-Y. Li, J.-P. Zhang, L. Niu and R.-S. Wang, Nano Energy, 2015, 13, 450–457 CrossRef CAS.
- X. Yan, H. Ye, X.-L. Wu, Y.-P. Zheng, F. Wan, M. Liu, X.-H. Zhang, J.-P. Zhang and Y.-G. Guo, J. Mater. Chem. A, 2017, 5, 16622–16629 RSC.
- Y. Park, D.-S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24, 3562–3567 CrossRef CAS PubMed.
- X. Meng, J. Mater. Chem. A, 2017, 5, 18326–18378 RSC.
- A. Khayyami, A. Philip and M. Karppinen, Angew. Chem., Int. Ed., 2019, 58, 13400–13404 CrossRef CAS PubMed.
- O. Filies, O. Böling, K. Grewer, J. Lekki, M. Lekka, Z. Stachura and B. Cleff, Appl. Surf. Sci., 1999, 141, 357–365 CrossRef CAS.
- S. Renault, V. A. Oltean, M. Ebadi, K. Edström and D. Brandell, Solid State Ionics, 2017, 307, 1–5 CrossRef CAS.
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, Dalton Trans., 2013, 42, 13649 RSC.
- C. W. Glass, A. R. Oganov and N. Hansen, Comput. Phys. Commun., 2006, 175, 713–720 CrossRef CAS.
- J. a. Kaduk, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 474–485 CrossRef PubMed.
- K. B. Lausund, V. Petrovic and O. Nilsen, Dalton Trans., 2017, 46, 16983–16992 RSC.
- J. Sienkiewicz-Gromiuk, L. Mazur, A. Bartyzel and Z. Rzączyńska, J. Inorg. Organomet. Polym. Mater., 2012, 22, 1325–1331 CrossRef CAS.
- P. Larkin, Infrared and Raman Spectroscopy, Elsevier, 2011, vol. 9 Search PubMed.
- G. Varsányi, in Vibrational Spectra of Benzene Derivatives, Elsevier, 1969, pp. 141–393 Search PubMed.
- M. Fathima Beegum, L. Usha Kumari, B. Harikumar, H. T. Varghese and C. Yohannan Panicker, Rasayan J. Chem., 2008, 1, 117–124 Search PubMed.
- M. Karabacak, M. Cinar, Z. Unal and M. Kurt, J. Mol. Struct., 2010, 982, 22–27 CrossRef CAS.
- M. B. Hay and S. C. B. Myneni, Geochim. Cosmochim. Acta, 2007, 71, 3518–3532 CrossRef CAS.
- J. F. Arenas and J. I. Marcos, Spectrochim. Acta, Part A, 1979, 35, 355–363 CrossRef.
- V. A. Oltean, B. Philippe, S. Renault, R. Félix Duarte, H. Rensmo and D. Brandell, Chem. Mater., 2016, 28, 8742–8751 CrossRef CAS.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS.
- S. E. Wheeler, J. Am. Chem. Soc., 2011, 133, 10262–10274 CrossRef CAS PubMed.
- Y. Y. Zhang, Y. Y. Sun, S. X. Du, H.-J. Gao and S. B. Zhang, Appl. Phys. Lett., 2012, 100, 1–4 Search PubMed.
- N. Ogihara, N. Ohba and Y. Kishida, Sci. Adv., 2017, 3, e1603103 CrossRef PubMed.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- J. Lüder, F. Legrain, Y. Chen and S. Manzhos, MRS Commun., 2017, 7, 523–540 CrossRef.
- H. H. Lee, Y. Park, K. H. Shin, K. T. Lee and S. Y. Hong, ACS Appl. Mater. Interfaces, 2014, 6, 19118–19126 CrossRef CAS PubMed.
- B. Tian, Z. Ding, G.-H. Ning, W. Tang, C. Peng, B. Liu, J. Su, C. Su and K. P. Loh, Chem. Commun., 2017, 53, 2914–2917 RSC.
- S. Wang, L. Wang, K. Zhang, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS PubMed.
- N. Ogihara and Y. Kishida, Commun. Electrochem., 2015, 83, 861–863 CrossRef CAS.
- L. Fédèle, F. Sauvage, J. Bois, J. M. Tarascon and M. Bécuwe, J. Electrochem. Soc., 2014, 161, 46–52 CrossRef.
- C. Luo, Y. Zhu, Y. Xu, Y. Liu, T. Gao, J. Wang and C. Wang, J. Power Sources, 2014, 250, 372–378 CrossRef CAS.
- J. Rodríguez-Carvajal, Physica B: Condens. Matter, 1993, 192, 55–69 CrossRef.
- V. Holý, P. Ullrich and B. Tilo, High Resolution X-Ray Scattering from Thin Films and Multilayers, Springer-Verlag, Berlin, 1999 Search PubMed.
- R. L. Puurunen and W. Vandervorst, J. Appl. Phys., 2004, 96, 7686–7695 CrossRef CAS.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, DOI:10.1016/j.commatsci.2013.08.053.
- Q. Zhu, A. R. Oganov, C. W. Glass and H. T. Stokes, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 215–226 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. J. Karttunen, T. Tynell and M. Karppinen, J. Phys. Chem. C, 2015, 119, 13105–13114 CrossRef CAS.
- F. Pascale, C. M. Zicovich-Wilson, F. López Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt04572d |
| This journal is © The Royal Society of Chemistry 2020 |