
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal self-assembly mimosine peptides with enhanced antimicrobial activity: towards a new generation of multitasking chelating agents†
Joanna Izabela
Lachowicz
 *a,
Gabriele
Dalla Torre
bc,
Rosita
Cappai
d,
Enrico
Randaccio
e,
Valeria M.
Nurchi
d,
Remigiusz
Bachor
e,
Zbigniew
Szewczuk
e,
Lukasz
Jaremko
f,
Mariusz
Jaremko
f,
Maria Barbara
Pisano
g,
Sofia
Cosentino
g,
Germano
Orrù
h,
Antonella
Ibba
h,
Joni
Mujika
b and
Xabier
Lopez
*b
*a,
Gabriele
Dalla Torre
bc,
Rosita
Cappai
d,
Enrico
Randaccio
e,
Valeria M.
Nurchi
d,
Remigiusz
Bachor
e,
Zbigniew
Szewczuk
e,
Lukasz
Jaremko
f,
Mariusz
Jaremko
f,
Maria Barbara
Pisano
g,
Sofia
Cosentino
g,
Germano
Orrù
h,
Antonella
Ibba
h,
Joni
Mujika
b and
Xabier
Lopez
*b
aDepartment of Medical Sciences and Public Health, University of Cagliari, Cittadella Universitaria, 09042 Monserrato, Italy. E-mail: lachowicz@unica.it
bKimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, Donostia International Physics Center (DIPC), P.K. 1072, Donostia, Euskadi, 20080 San Sebastian, Spain. E-mail: xabier.lopez@ehu.es
cUCIBIO/REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre, s/n, 4169-007 Porto, Portugal
dDepartment of Life Sciences, University of Cagliari, Cittadella Universitaria, 09042 Monserrato, Italy
eFaculty of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland
fDivision of Biological and Environmental Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), 23955, Thuwal, Saudi Arabia
gDepartment of Medical Sciences and Public Health, University of Cagliari, Cittadella Universitaria, 09042 Monserrato, Italy
hDepartment of Surgical Sciences, University of Cagliari, Cittadella Universitaria, 09042 Monserrato, Italy
First published on 28th January 2020
Abstract
Mimosine is a non-protein amino acid with various properties, such as antibacterial, anti-inflammatory, anti-cancer and anti-virus among others. Due to its structural similarity with deferiprone (DFP), mimosine is a potential excellent metal chelator. In the present work, we combine experimental and theoretical (DFT) approaches in order to investigate the properties of mimosine peptides. Six different peptides were synthesized and their complex stoichiometry and stability were characterized by means of UV-Vis spectrophotometry. Then, the binding mode and self-assembly features of the peptides were evaluated using a DFT approach, taking into account different number of mimosine amino acids and varying the length of the spacer between the mimosine residues, and there was good agreement between experimental data and computational calculations. Further elucidations of the structural properties of these peptides allowed us to propose improvements in the structure of the mimosine moiety which can lead to enhanced affinity for high-valent metals. Moreover, we demonstrate that these peptides show an anti-microbial activity against Gram positive bacteria that is enhanced by the formation of a complex with iron(III) ions. The mimosine peptides could be an alternative to antimicrobial peptides (AMPs), which are expensive and susceptible to proteolytic degradation. In summary, in the present work, we propose a new generation of multipurpose mimosine-based peptides as new metal self-assembly chelators which could be a turning point in biomedical and nanotechnological applications.
1. Introduction
One out of three proteins use metal ions as cofactors. The binding of metal ions to proteins changes their structures and properties, and it is a key feature for their structural, regulatory, and/or enzymatic functions. For example, peptides that self-assemble through metal–ligand complexes form nanostructures that possess very different structures and functions from the original peptides. This particular property was recently used for the synthesis of metal-triggered, self-assembled peptides in nanotechnology,1 to synthesize nanofiber materials for cell culture and tissue engineering,2 to assemble peptide nanotubes3 and helical ribbons, etc. Thus, the formation of molecular building blocks that can spontaneously form regular and stable macroscopic structures, via covalent or noncovalent bonds is a very active and fast-growing process in nanotechnology.In addition to the 22 amino-acids used by eukaryotes, there are more than 140 non-proteinogenic amino-acids that are not naturally encoded or found in the genetic code of any organism. Some of them possess a biological function (e.g. components of bacterial cell walls, neurotransmitters and toxins) that may be used as natural or man-made pharmacological compounds.4
Antibiotics are among the most extensively used drugs. Their abuse in human medicine and in animal farming leads to bacteria resistance.5 Now, we are witnessing the end of a golden epoch of antibiotics6 and new solutions are critical for human health. Antimicrobial peptides (AMPs) have emerged as an alternative to antibiotics and in 2003 Daptomycin (MIC 32 μg ml−1 (ref. 7)) was approved by the FDA as an antibiotic against a variety of Gram-positive bacteria (methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE) and penicillin-resistant Streptococcus pneumoniae8 (PRSP)). The bactericidal activity of AMPs is triggered mainly by membrane disruption and reduction in bacterial resistance; however most AMPs are expensive, highly cytotoxic and susceptible to proteolytic degradation. AMPs can be improved by the design of peptide mimics (peptidomimetics), whose backbone is not based on the regular amino acid linked in a chain.9,10 One of the strategies in peptidomimetics is the use of unnatural amino acids.11
Mimosine [β-[N-(3-hydroxy-4-oxypyridyl)]-α-aminopropionic acid] (MIM, Scheme 1) is a non-protein amino-acid biosynthesized by the Mimosoideae family of plants. The chemistry,12 methods for estimation,13 biosynthesis,14 and the degradation of this secondary metabolite15 have been extensively described in the literature. Over the past few years, mimosine has been found to be involved in different biological processes such as antibacterial,16 anti-cancer,17 anti-inflammatory,18 anti-fibrosis,19 anti-influenza,20 anti-virus,21 herbicidal22 and insecticidal,23 among others.12 In the 90s, mimosine was investigated as a potential inhibitor of the G1/S phase,24 and, over the last twenty years, as a potential drug for cancer.25 The activity of mimosine-based peptides has been linked to its metal chelating properties. For instance, the coordination of the essential metal ions, in blood plasma, causes an inhibition of the growth of wool and defleecing in sheeps.26
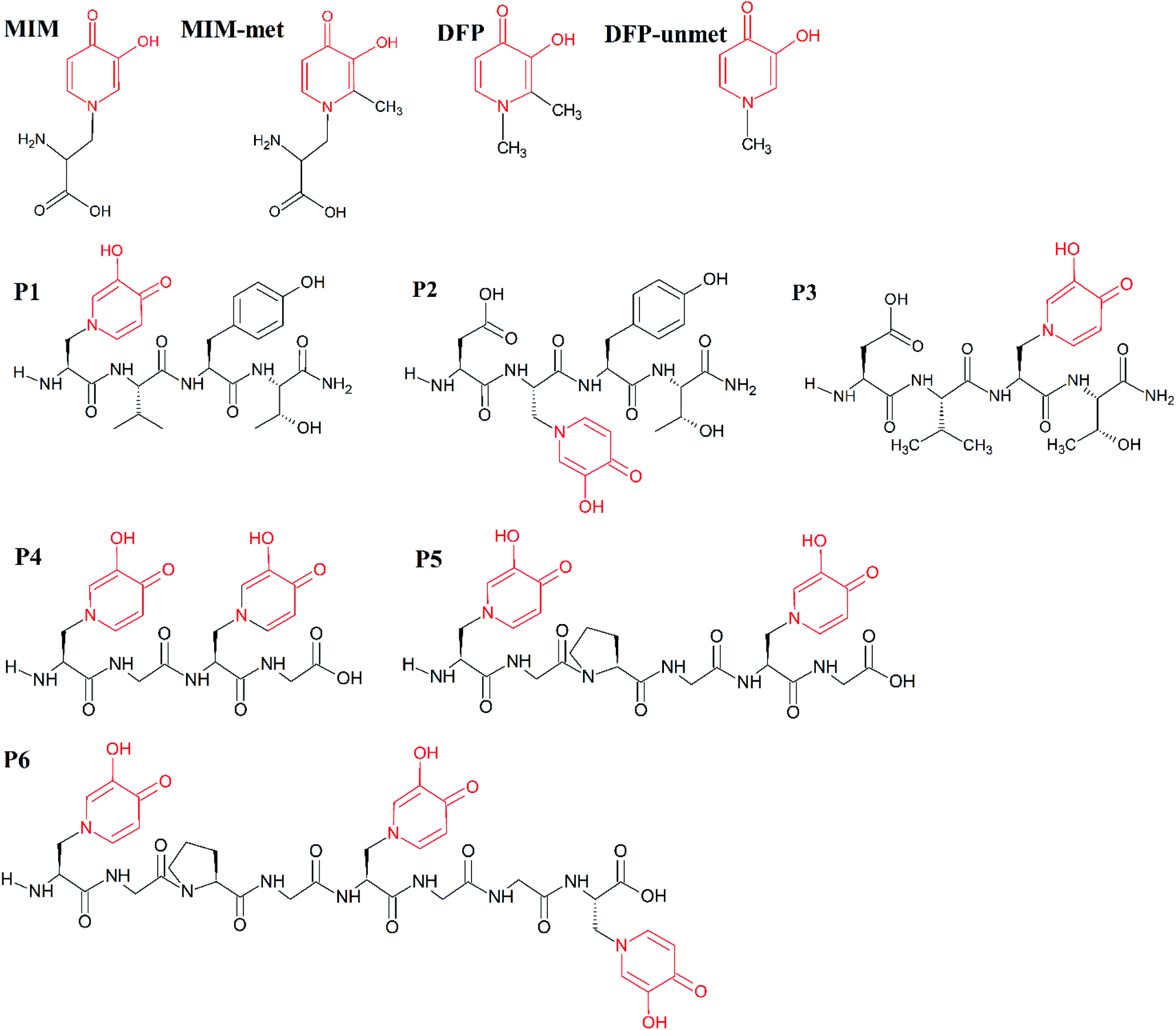 | ||
| Scheme 1 Molecular structures of the studied molecules (MIM, MIM-unmet, DFP, DFP-unmet, P1–P6). Structural similarities between these molecules are indicated in red. | ||
The mimosine sidechain is structurally similar to that of deferiprone (DFP, Ferriprox, 1,2-dimethyl-3-hydroxy-4-pyridinone, Scheme 1) and therefore, they show similar features in their metal-coordination ability. DFP is used as a drug to remove the excess iron in thalassaemia major.27 It has been used in the treatment of thalassaemia major in Europe and Asia since 1994, and was approved for its use in the US,28 in 2011. Its coordination of Cu(II),29,30 Cd(II),31 Al(III),32 Fe(III),33 Ga(III),34 Gd(III),29 and In(III)34 metal ions, as well as actinides35 and lanthanides,36 has been extensively described in the literature. DFP and its derivatives have been proposed as potential remedies for diseases related to metal disorders, e.g. anaemia, thalassemia, hemochromatosis, etc., for which the delivery, or removal, of metal ions is desirable. Recently, hydroxypyridinones have been used to improve the retention and tissue uptake of metal-based insulin-enhancing agents. DFP-based therapy protects against the toxicity of heavy metals (used in contrast agents), chemotherapeutics, exposition to harmful radioisotopes and the neurotoxicity of redox metals.37 Fe(III)–, VO(IV)–, Cu(II)–, Zn(II)– and Ni(II)–mimosine complexes have been investigated by various research groups.38,39 Mimosine dipeptides and tetrapeptides have been previously synthesized and investigated as potential neuraminidase,40 tyrosinase40,41 and cyclooxigenase41 inhibitors. However, to our knowledge, no other applications have been considered, nor any synthesis of mimosine hexapeptides has been performed, to this day.
Recently, we have investigated by means of QM/MM and DFT calculations the potential application of several mimosine-based peptides as improved metal chelators.42 We found that this class of ligands may reach a metal binding affinity comparable to or even higher than DFP.42 In this paper, we present synthetic, DFT, and experimental iron(III)- and copper(II)-complex formation studies of tetra- and hexapeptides containing one, two or three mimosine residues. The antimicrobial activities of the synthesized complexes against Gram positive, Gram negative bacteria and dermatophytes are also investigated and reported here (Section 3.2). We found very good agreement between experimental and theoretical studies, which point to these mimosine-based peptides as effective iron(III) and copper(II) chelators. The effect on the affinity of key structural features of these peptides, such as the number of mimosine residues and length and flexibility of the spacer, is also discussed and rationalized. The DFT model established here can be used for the design of the self-assembly of metal ions and mimosine peptides for medical and nanotechnological applications.
2. Experimental
2.1. Reagents
All solvents and reagents were used as supplied. Fmoc amino acid derivatives were purchased from Novabiochem. FeCl3, CuCl2 × 2H2O, NaOH, HCl, L-mimosine was obtained from Sigma Aldrich. (Benzotriazol-1-yloxy)tris(pyrrolidino)phosphonium hexafluorophosphate (PyBOP), N-[(dimethylamino)(1H-1,2,3-triazolo-[4,5-b]pyridin-1-yl)methylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU), and MBHA-Rink amide resin (0.69 mmol g−1) were obtained from Merck Millipore. Fmoc-Gly-Wang resin (0.73 mmol g−1), 2-chlorotrityl chloride resin (1.00 mmol g−1 and trifluoroacetic acid (TFA) were obtained from IrisBiotech. Solvents for peptide synthesis (N,N-dimethylformamide (DMF), dichloromethane (DCM), and (N-ethyldiisopropylamine (DIEA)) were obtained from Sigma Aldrich.2.2. Synthesis
The synthesis of the Fmoc-Mim derivative was performed using the method described by Upadhyay et al.40 Briefly, mimosine (200 mg) and sodium carbonate (Na2CO3) (220 mg) were dissolved in distilled water (3 mL). Fmoc-Osu (500 mg) dissolved in 3.6 mL of 1,4-dioxane was added dropwise to the solution and stirred for 2 h at room temperature. Afterwards, 12 mL of Na2CO3 (0.1 M) was added. The mixture was stirred for 7 h at 26 °C and was then filtered and washed with 20 mL of ethyl acetate to remove the excess of Fmoc-Osu and by-products. The water fraction was kept in an ice bath, the pH was adjusted to 4.0 (by adding 6 N HCl) and the mixture was incubated overnight at 4 °C. The resulting precipitate was filtered, washed with distilled water, and dried under reduced pressure to give Fmoc mimosine. The obtained product was analyzed by ESI-MS.The synthesis of peptides on the MBHA-Rink (peptides 1–3, Table S1†), Fmoc-Gly-Wang resin (peptide 4, Table S1†) and 2-chlorotrityl chloride resin was performed manually in polypropylene syringe reactors (intavis AG) equipped with polyethylene filters, according to a standard Fmoc (9-fluorenylmethoxycarbonyl) solid phase synthesis procedure.43
All products were purified using the analytical HPLC Thermo Separation system with UV detection (210 nm) having a YMAC-Pack RP C18 Column (4.6 × 250 mm, 5 μm), with a gradient elution of 0–40% B in A (A = 0.1% TFA in water; B = 0.1% TFA in acetonitrile/H2O, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) over 30 min (flow rate 1 mL min−1). The main fraction, corresponding to the peptide, was collected and lyophilized.
:1) over 30 min (flow rate 1 mL min−1). The main fraction, corresponding to the peptide, was collected and lyophilized.
All products were analyzed on a microTOF-Q mass spectrometer (Bruker Daltonics, Bremen, Germany) and on an FTICR (Fourier Transform Ion Cyclotron Resonance) PAex-Qe Ultra 7 T mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with a standard ESI source. The instruments were operated in the positive-ion mode and calibrated daily with a Tunemix™ mixture (Agilent Technologies, Palo Alto, CA, USA). The mass accuracy was better than 5 ppm. Analyte solutions (70 μl) were introduced at a flow rate of 3 μl min−1. The parameters of the instruments were as follows: for microTOF-Q MS: scan range: 50–1600 m/z; drying gas: nitrogen; flow rate: 4.0 L min−1, temperature: 200 °C; potential between the spray needle and the orifice: 4.2 kV; for FTICR MS: scan range: 100–1600 m/z; drying gas: nitrogen; flow rate: 1.5 L min−1, temperature: 200 °C; potential between the spray needle and the orifice: 4.2 kV. For MS spectra analysis (Table 1), a Bruker Compass Data Analysis 4.0 software was used.
| Species | DFP 32 | Mimosine58 | Species | DFP 59 | DFP 32 | Mimosine59 | Mimosine38 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| logK |
logK |
logK |
logK |
logK |
logK |
||||||
| LH | 9.82 | 8.76 | LH | 8.80 | 9.82 | 8.86 | 9.02 | ||||
| LH2 | 3.66 | 7.14 | LH2 | 3.35 | 3.66 | 7.00 | 7.18 | ||||
| LH3 | 2.48 | LH3 | — | — | 2.62 | 2.56 | |||||
| LH4 | — | — | 1.1 | <1 | |||||||
| Species | logK |
λ max | ε | logK |
Species | logK |
logβ |
logβ |
λ max | ε | logβ |
| CuLH2 | 18.1 | ||||||||||
| FeLH | 12.00 | CuLH | 1.60 | 16.36 | 755 | 34 ± 5 | 16.63 | ||||
| FeL | 15.01 | 540 | 1700 | CuL | 9.35 | 10.42 | 9.48 | ||||
| CuLH−2 | −9.47 | ||||||||||
| FeL2H2 | 9.50 | CuL2H2 | — | 31.26 | 693 ± 10 | 37 ± 10 | 32.04 | ||||
| CuL2H | 21.98 | 24.40 | 24.43 | ||||||||
| FeL2 | 12.02 | 491 | 3700 | CuL2 | 7.58 | 19.09 | 16.81 | 690 | 33 | 17.23 | |
| CuL2H−1 | 8.49 | 5.79 | |||||||||
| Cu2L | 15.70 | 735 ± 15 | 28 ± 5 | ||||||||
| Cu2L2 | 32.2 | 662 | 50 | 29.98 | |||||||
| Cu2L3 | 29.52 | ||||||||||
| FeL3H3 | 8.00 | ||||||||||
| FeL3H2 | 6.28 | ||||||||||
| FeL3H | 7.43 | ||||||||||
| FeL3 | 10.40 | 457 | 4400 | 7.42 | |||||||
2.3. NMR measurements
2.0 mg of each peptide was dissolved in 500 μL of a mixture of 10% D2O and 90% of H2O (v/v). After the peptide was dissolved, the pH of the solution was manually adjusted to 7.40, prior to each measurement. All NMR experiments were performed using either 700 MHz (for peptides 1 and 2) or 950 MHz (for peptides 3–5) spectrometers at 25 °C. All NMR data were processed by NMRPipe44 and analyzed by using Sparky45 software. Complete assignments of the 1H and 13C resonances, for all the peptides (Tables S3–S7†), were done by the application of a standard procedure46 based on the inspection of the 2D homonuclear TOCSY (with mixing times of 10 and 80 ms) and ROESY (with mixing times of 300) experiments. The NMR spectra were acquired after 2 months of synthesis and ESI-MS spectra acquisition. No peptide degradation was observed.2.4. UV-Vis measurements
UV-Vis spectra were recorded using a SPECTROstar Nano (BMG LABTECH). 5 μl total volume of the peptide and metal water solutions were placed in multi-well plates, and spectra were collected, using a range of wavelengths from 200 to 900 nm. The final pH was a result of mixing peptide- and water solutions with metal acidic solutions. The peptide and metal concentrations, for each experiment, are reported in the Results and Discussion section. The pHs of the solutions were measured using a METROHM Microelectrode 6.0224.100 daily calibrated with a Mettler TOLEDO InLab® Solutions buffer (pHs 4.01 and 9.21).2.5. ESI-MS measurements
The obtained mass spectra were recorded in the positive mode on a LCMS 9030 qTOF Shimadzu mass spectrometer (Shimadzu, Kyoto, Japan). The m/z range was between 100 and 1500, interface voltage (+) was 4.5 kV, interface temperature 300 °C, DL temperature 250 °C, heat block temperature 400 °C, nebulizing gas (nitrogen) flow 3 L min−1, and total flow 0.4 mL min−1 of the H2O/MeCN mixture (1:1, v:v). Equimolar ratios of the peptide and metal ions (Fe(III) ions (FeCl3), Cu(II) (CuSO4) were mixed in water, dissolved in methanol and analyzed by mass spectrometry.
2.6. DFT calculations
Optimization was carried out using the B3LYP functional47,48 with Grimme's D3 dispersion correction49 along with the Becke–Johnson (BJ) damping scheme and the 6-31+G(d) basis set. To confirm that the optimized structures were real minima on the potential energy surfaces, frequency calculations were carried out at the same level of theory. All structures showed positive force constants for all normal modes of vibration. The frequencies were then used to evaluate the zero-point vibrational energy (ZPVE) and thermal (T = 298°K) vibrational corrections to the Gibbs free energies within the harmonic oscillator approximation. To calculate the entropy, the different contributions to the partition function were evaluated using the standard statistical mechanics expressions in the canonical ensemble and the harmonic oscillator and rigid rotor approximation. In addition, energies were refined with single point calculations at the B3LYP-D3(BJ)/6-311++G(3df,2p) level of theory. All calculations were performed with the IEFPCM solvation model50 in order to properly investigate the thermodynamics of metal–ligand complexes in solution.All calculations were performed with the Gaussian16 Rev. A03 package.51
Binding energies and enthalpies were calculated according to the following substitution reaction:
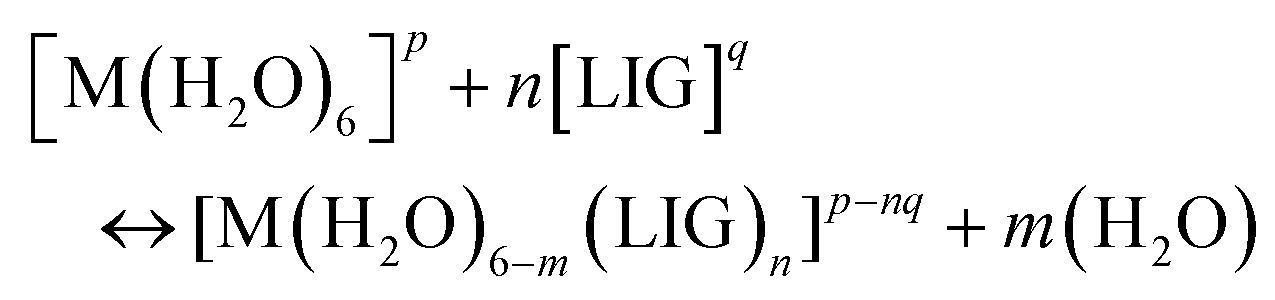 | (1) |
Ligands were considered in their unprotonated form since this is the way that experimental stability constants are calculated, and good agreement was found between DFT binding energies and experimental logβ values as in a previous work.52
The binding energies in solution associated with eqn (1) can be calculated as:
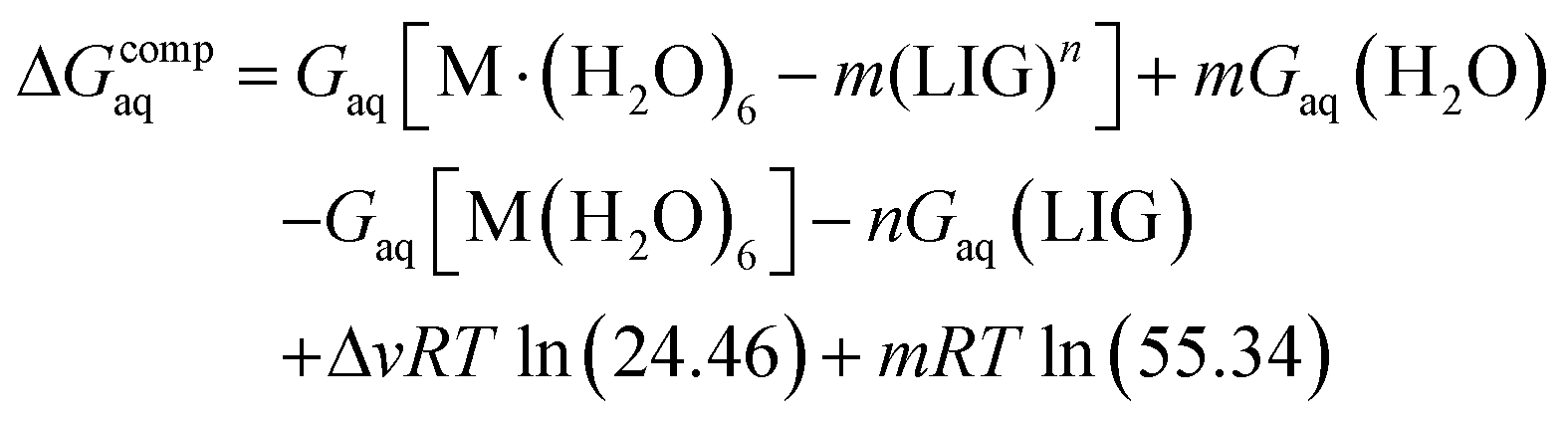 | (2) |
Since the energies were determined using an ideal gas at 1 atm as the standard state, the penultimate term in eqn (2) corresponds to the volume change due to the transformation from 1 atm to 1 M in solution, where Δv refers to the change in the number of species in the reaction. The last term is the entropic factor that accounts for the concentration of 55.34 M of water in liquid water.53 The ΔvRTln(24.46) and mRTln(55.34) corrections correspond to the changes in the standard state, accounting, on the one hand, for the change due to the transformation from 1 atm to 1 M in solution, and to an entropic factor that accounts, on the other hand, for the concentration of 55.34 M of water in liquid.53
2.7. Antimicrobial activity
The bacterial and fungal suspensions, adjusted to a 0.5 McFarland standard turbidity (equivalent to 1.5 × 108 CFU mL−1 or 1–5 × 106 spores per mL−1),54 were prepared in NB and in sterile phosphate-buffered saline (PBS) with 0.05% Tween 80 (PBS-Tween), respectively.
A sterilized filter paper disc (5 mm in diameter) containing either a 10 μL of peptide or a Cu(II)–peptide complex sample was placed on Muller Hinton (MHA) or Sabouraud Dextrose agar (SDA) plates previously seeded with the prepared bacterial and fungal suspensions. Different stock solutions of peptides were prepared, depending on the residual amount. Peptides 1, 3 and 5 were dissolved in 0.2 mL of sterile distilled water to obtain the final concentrations of 50, 340, and 810 μg per disc, respectively. Peptides 2 and 4 were dissolved in 0.5 mL of sterile distilled water to obtain 84 and 149 μg per disc concentrations, respectively. The stock solution of peptide 6 prepared in DMSO (100%) was further diluted in NB to obtain the final concentrations of 140 and 14 μg per disc. These concentrations were chosen according to each peptide maximum-water solubility. The Cu(II)–peptide complexes involving peptides 2, 4 and 6 were prepared in 1:3, 1:1 and 1:1 metal-to-peptide molar ratios, respectively. 25 μg of amoxicillin, 5 μg of ofloxacin or 15 μg of Ketoconazole (Oxoid) and blank disc impregnated with 10 μL of NB or PBS-Tween were used as positive and negative controls, respectively.
Serial doubling dilutions of the different peptides and their metal complexes were prepared in 100 μL of NB, in 96-well microtiter plates. The following concentrations were tested: P3, 3400–106.25 μg mL−1, P4, 7450–232.81 μg mL−1, P5, 8100–253.125 μg mL−1, P6, 6900–215.625 μg mL−1, for the following metal–peptide complexes: Fe(III)–P1 333.7–3.0 μg mL−1, Fe(III)–P2 (1:3 metal:peptide molar ratio) 560–17.5 μg mL−1, Fe(III)–P4 (1:1 metal:peptide molar ratio) 967.5–30.2 μg mL−1, Fe(III)–P6 (1:1 metal:peptide molar ratio) 896.1–28.0 μg mL−1. The bacterial suspensions, prepared as described above, were further diluted in the broth media, and 100 μL volume of this diluted inoculum was added to each well of the plate, resulting in the final inoculum of 5 × 105 CFU mL−1. Controls for the sterility of the NB and the peptides, and the culture (inoculum) were included; DMSO (for P6) was also monitored to check the effect of the solvent on the growth of microorganisms. Furthermore, ofloxacin (8 μg mL−1) was used as the positive control for Gram-positive and negative bacteria. MICs and MBCs were determined after 24 h of incubation of the plates, at 37 °C. Microbial growth was indicated by the presence of turbidity and a “pellet” on the well bottom. MICs were determined presumptively as the first well, in ascending order, which did not produce a pellet. To confirm MICs and to establish MBCs, 10 μL of broth was removed from each well and inoculated on Tryptic Soy Agar (Microbiol) plates. After incubation under the conditions described above, the number of surviving bacteria was determined. The MIC was the lowest concentration that resulted in a significant decrease of the inoculum's viability (>90%), and the MBC concentration was found to be the one for which 99.9% (or more) of the initial inoculum was killed. All tests were conducted in triplicate, and the modal MIC and MBC values were selected.
The MIC dilutions were prepared with daily prepared solutions and with 2 month-old solutions (stored at −5 °C). No differences were observed in the experimental results.
3. Results and discussion
3.1. Metal coordination
The number of mimosine residues, their positions in the peptide backbone, and the length of the spacer between neighboring mimosine residues determine the stoichiometry of metal–peptide complexes and their structures (Scheme 2). Some of the predicted stoichiometries of the M2+ and M3+ metal-ion complexes with mimosine peptides are shown in Scheme 2, together with their possible structures.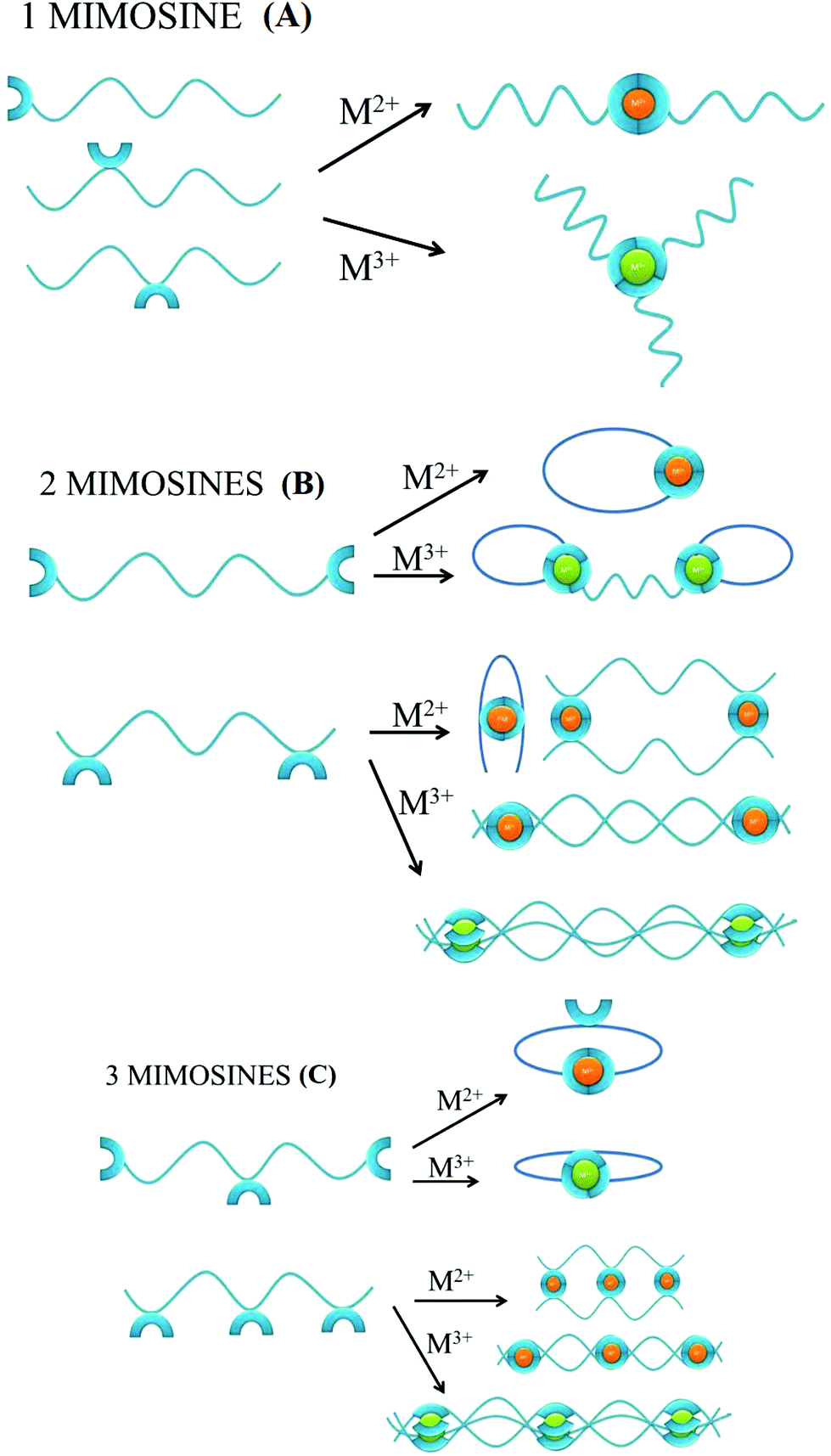 | ||
| Scheme 2 Possible types of the self-assembled metal–peptide aggregates, according to the number of mimosine residues, the position of the mimosine in the backbone of the peptide, and the oxidation state of the metal. | ||
In the peptide containing one mimosine residue (Scheme 2A), the most probable stoichiometry with M2+ is a 1:2 (metal:ligand) molar ratio, whereas in the trivalent metal ion, a 1:3 (metal:ligand) stoichiometry is expected. The position of the mimosine residue in the peptide backbone influences the ternary structure of the metal complex, and in some cases, where the conformational constraints occur, even the changes in the metal–ligand stoichiometry.
The addition of the second mimosine residue in the peptide backbone (Scheme 2B) gives more possible stoichiometries of the metal complex and makes the coordination system more complicated. The peptide that contains mimosine amino acids at the N- and C-ends of the peptide can form cyclic complexes with divalent metal ions, if the length of the linker between mimosine residues is long enough for obtaining appropriate geometry of the metal complex. In the case of the short spacer between neighboring mimosine residues (e.g. mimosine residues in the middle of the peptide backbone), it is likely that bend, dimeric or polymeric structures will be formed. For trivalent metal ions, the two mimosine peptides will form 2:3 (metal:ligand) complexes of different ternary structures (closed or linear) or polymers, depending on the length of the linker between mimosine units.
The presence of a third mimosine residue (Scheme 2C) makes the predictability of the complex stoichiometry harder. Two mimosine residues at the N- and C-ends of the peptide, and one in the middle, will form cyclic complexes with trivalent metal ions, but also with divalent ones. In the last case, one mimosine residue will have free binding sites, which could be the starting point to form higher stoichiometric complexes. Three mimosine residues in close proximity in the backbone will lead to the formation of 3:2, 3:3 (metal:ligand) linear complexes or polymeric structures with divalent and trivalent metal ions.
The possible formation of polymeric structures by mimosine peptides is intriguing. The two mimosine peptides (Scheme 3A) can form with M2+ metal ions in linear polymers, while three mimosine peptides can be assembled both in linear and in non-linear polymers (Scheme 3B).
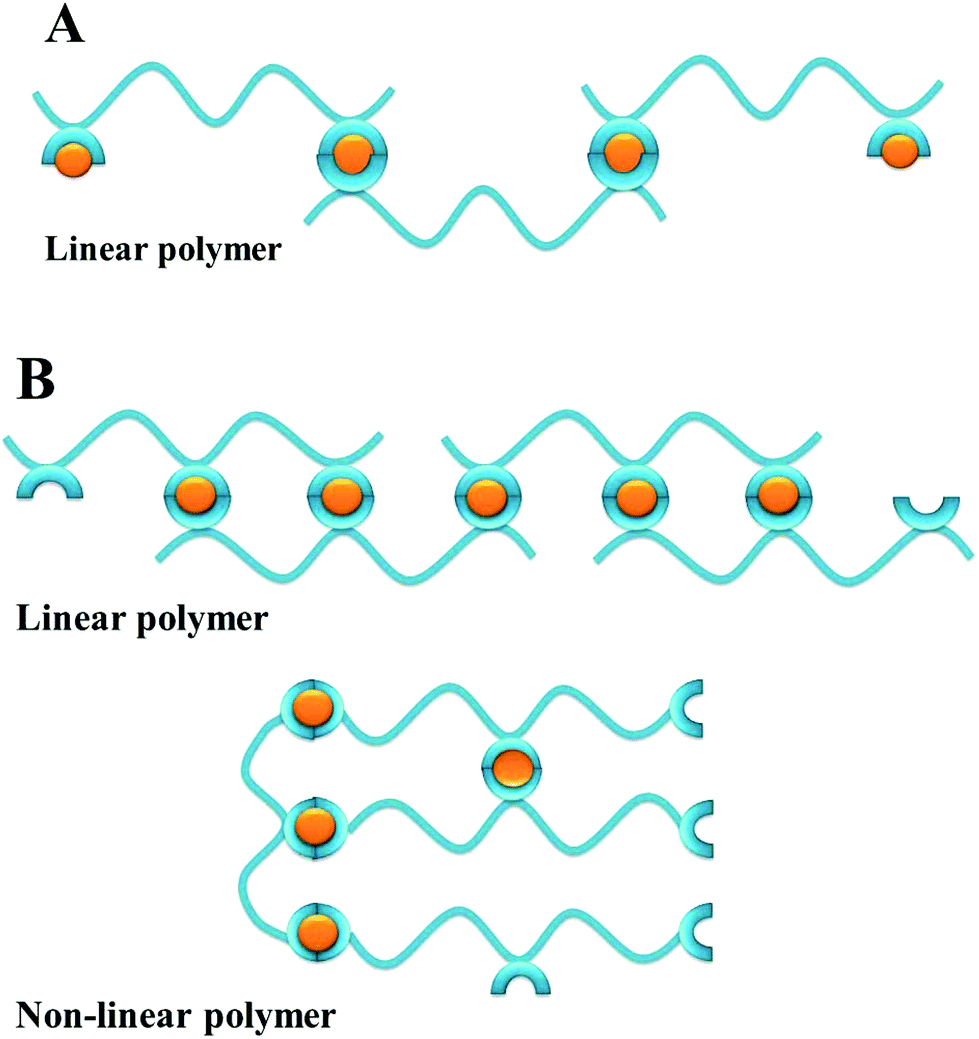 | ||
| Scheme 3 Possible types of the self-assembled metal–peptide polymers with M2+, according to the number of mimosine residues and the position of the mimosine in the backbone of the peptide. | ||
The formation of polymeric structures instead of dimeric complexes depends on the length of the linker between neighbouring mimosine residues, as well as linker rigidity (peptide bonds or proline residues) and steric incumbrances in the metal complex. Moreover, the metal and peptide concentrations in the solution, pH, ionic strength, and temperature determining polymerization/aggregation process56 influence the polymerization/aggregation process, but they are not the object of the present studies.
In order to predict accurately the stoichiometry and the structure of the investigated complexes, we establish a DFT calculation protocol and probe peptides with a variable number of mimosine residues and different lengths of the linker.
The geometries of the 1:3 Fe(III)–ligand complexes with MIM and DFP show a perfect octahedral coordination mode (Fig. S1†), in agreement with the X-ray structure of the 1:3 Fe(DFP)3 complex34 (Fig. 1C). The octahedral geometry of the metal coordination shell is characterized by O–Fe–O angles between 80–90 and ∼167 degrees, with the Fe–O distances 1.9–2.0 Å (Table S8†). Similarly, 1:2 and 1:1 Fe(III)–ligand complexes retain octahedral geometry with coordination sites filled by water molecules in the axial positions (Fig. S2 and S3†). On the other hand, in the 1:1 Cu(II)–ligand complex, one water molecule leaves the copper coordination sphere (Fig. S5†), and the system shows a pentacoordinate binding mode. In the 1:2 Cu(II)–ligand complex, two water molecules move away from their axial positions (Fig. S4†) and a stable tetrahedral complex is therefore formed, which is coherent with the X-ray structure for 1:2 Cu(II)–DFP (Fig. 1D).57 The calculations with our DFT model are in line with experimental data (Table S2 and Fig. S8†).
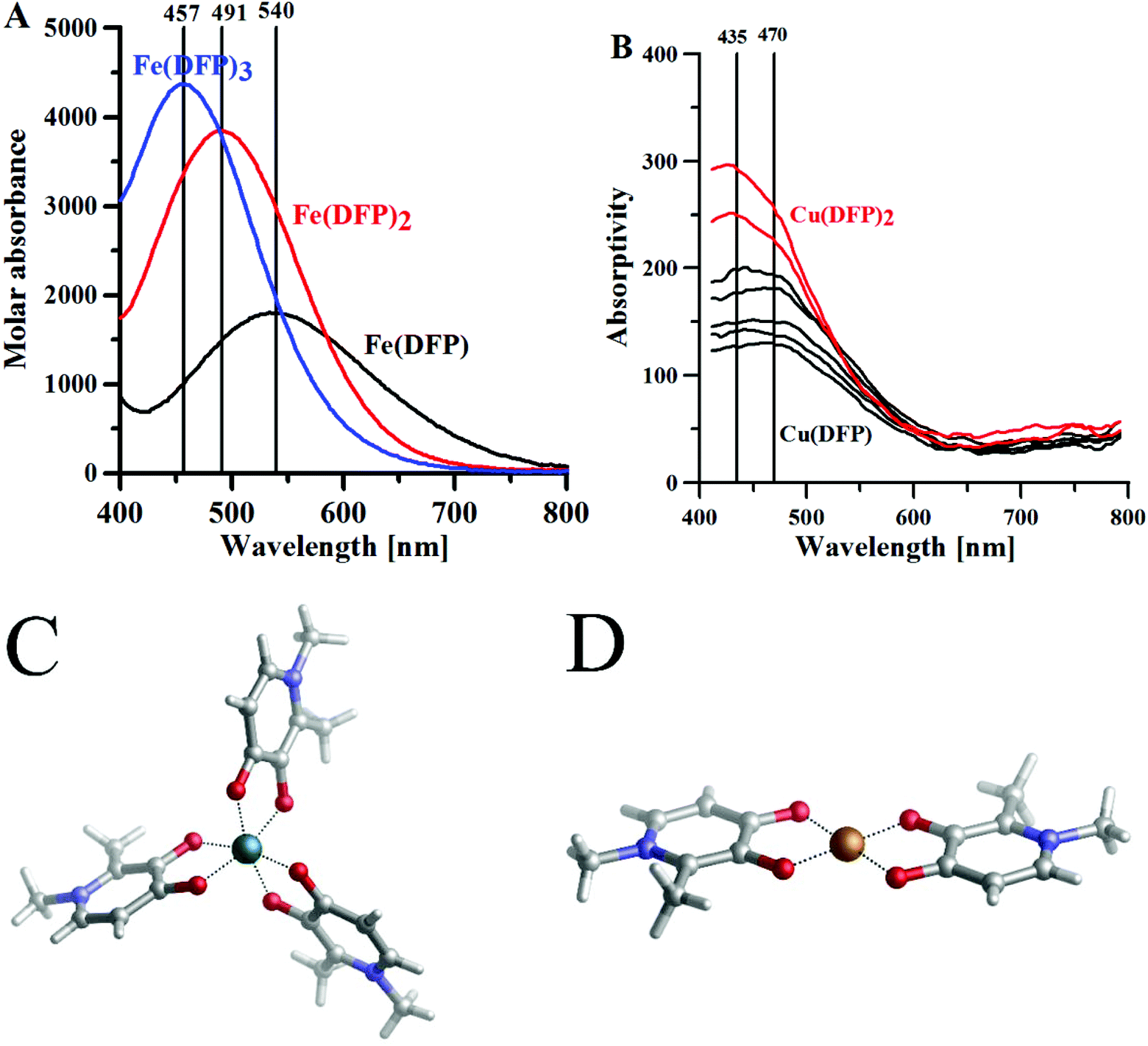 | ||
| Fig. 1 The maximum absorptivity spectra of (A) Fe3+–DFP complexes, [DFP] = 0.5 mM, l = 1 cm; (B) Cu(II)–DFP complexes, metal:ligand molar ratio 1:2, [DFP] = 0.5 mM, l = 10 cm. (C) Crystal structure CCDC (JAWSEF01, additional database identifier JUHXEP, 1183333) of Fe(DFP)3 complexes by E. T. Clarke et al.31 (D) The crystal structure (CCDC: 1291877, WELTEM) of the Cu(DFP)2 complex by A. El-Jammal et al.57 | ||
In aqueous solution, DFP forms complexes with a 1:1 metal:ligand stoichiometry and at a pH lower than 1 (Fig. S6†) these complexes are transformed into 1:2 stoichiometry complexes above pH 2 and 1:3 complexes above pH 3.32 The DFP–Fe(III) complexes are violet (at acidic pH) or red (neutral and basic pH), in water, and each complex is characterized by its specific absorptivity band (Fig. 1A). The crystal structure of DFP with Fe(III) ions (Fig. 1C) shows that the metal ion is coordinated in octahedral geometry by three pairs of carbonyl and dissociated hydroxyl groups.
With respect to L-mimosine, the protonation of the amino- and carboxylic groups makes the equilibrium of the iron complexation more complicated; nevertheless, electronic absorption spectra are similar to those obtained for DFP iron complexes, showing that amino- and carboxylic groups do not bind to the iron(III) ions.39 The constants of formation for the DFP and MIM complexes with iron(III) ions, shown in Table 1, are in good correlation with the computed binding enthalpies (ΔHcomp) and free energies (ΔGcomp) for the complexes of similar stoichiometry, and with those calculated with our DFT model (Table S2 and Fig. S8†).
The aqueous solution complexes of DFP with copper(II) ions are green colored and the Vis spectra (Fig. 1B) in the 400–600 nm range show the charge transfer bands (Amaxλ ∼ 480 nm) of O(phenol) to Cu(II) ions. Indeed, the crystal structure determined by A. El-Jammal et al.57 (Fig. 1D) confirms the formation of a Cu(DFP)2 complex and the coordination of copper ions by the pair of carbonyl and dissociated hydroxyl groups in octahedral geometry, with two axial water ligands. As previously shown in the DFT calculations, the copper(II) complexes with DFP are less stable than the iron(III) complexes. The [Cu(DFP)]+ forms at pH < 1 (Fig. S6†) and is replaced with the Cu(DFP)2 complex above pH 2.5. The constants of stability of the copper(II)–DFP and mimosine complexes, presented in Table 1, are in good agreement with those calculated using our DFT model (Table S2†).
The copper(II)–MIM and copper(II)–DFP absorption spectra were studied by Stunzi et al.59 According to the authors, the 1:1 (copper(II):MIM) stoichiometry complexes have almost the same wavelength maxima (Table 1) as the corresponding DFP complexes. This finding confirms that the metal binding sites are similar for MIM and DFP. The addition of the spectra of CuDFP+ and CuGly+ (λmax 715 nm)60 reproduces approximately the spectrum of Cu2(mimosine)2+, whereas the spectrum of the dimeric 2:2 mimosine is similar to the mixed 1:1:1 Cu–Gly–DFP spectrum and has λmax (660 nm) between those for the CuGly2 (617 nm) and Cu(mimosine)22− (690 nm) complexes. The small extinction coefficient Cu2(mimosine)2 complex may be explained by a slightly distorted structure, essentially strain-free.
The formation of a dinuclear copper(II)–mimosine complex was confirmed by EPR studies, performed by Chruscinska et al.38 A dinuclear species predominates in equimolar solution, for pHs ranging from pH 4 to 11 (Fig. S6†), and two sets of hyperfine components, each consisting of seven lines, at the approximate intensity ratio of 1:2:3:4:3:2:1, and in the parallel region of the ΔMS = ±1 resonances, are present in the EPR spectrum. The measured zero field splitting corresponds to an intermetallic distance of approximately 5.0 Å. The atoms from the donor sets are as follows: (CO, O−) and (NH2, COO−).
Our DFT calculations confirm the stability of the dinuclear copper(II)–MIM complex. Fig. 2 shows the characteristics of the structure in which both Cu(II) cations are pentacoordinated by interacting with the backbone NH2, the COO− groups of one MIM, and the carbonyl and hydroxyl oxygen atoms of the side chain of the other MIM. The metal coordination shell is completed by an explicit water molecule. The distances and angles computed (Table S10†) confirmed that the ligands are placed in a near-optimum arrangement. Compared to the Cu(DFP)2 structure, the Cu–O distances in the dinuclear mimosine complex are slightly longer (1.94–1.97 Å) and the Cu–metal binding site angles are slightly different (90° and 170°) than the corresponding distances (1.91–1.93 Å) and angles (90° and 180°) in the DFP copper complex (Table S10†). The DFT calculated distance between two copper ions in the dinuclear complex is 5.222 Å and stays in line with the EPR experimental data.38 The stability of the complex was determined, via the following reaction:
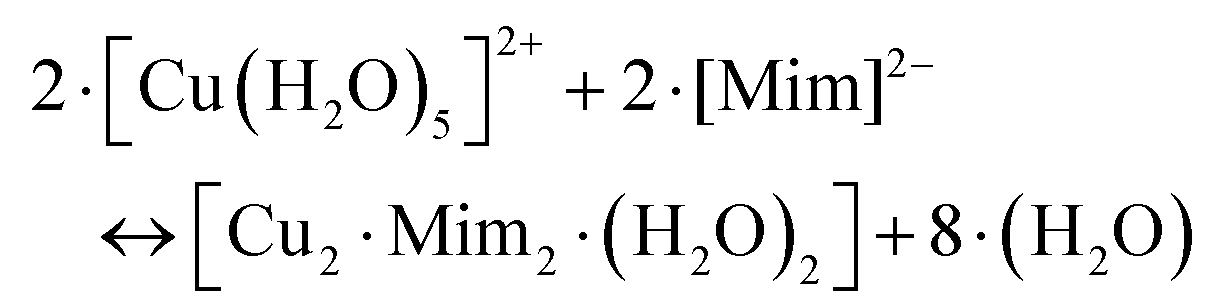 | (3) |
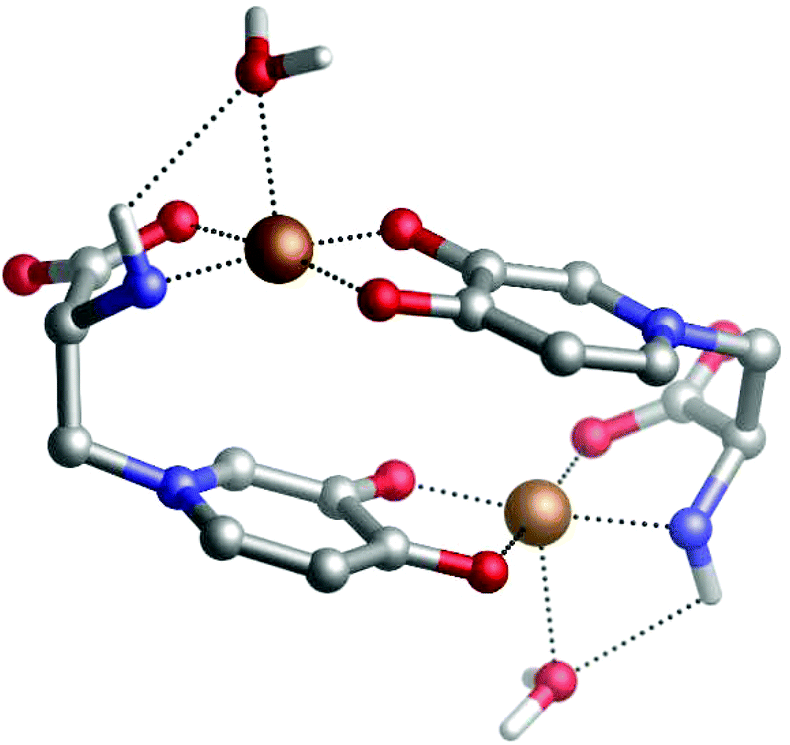 | ||
| Fig. 2 DFT optimized structure of a dinuclear copper(II)–mimosine complex. | ||
The formation of metal complexes was studied experimentally using UV-Vis spectrometry, and the structure and stability of the formed complexes were determined by DFT calculations.
Electronic absorption spectra of the Peptide 1 (P1) and P2 complexes with iron ions are shown in Fig. 3A. The P1 peptide is considered to be a [LH3]2+ ligand, where the amine, amide and hydroxyl groups are the dissociating groups. In the solution containing iron(III) and P1 at a molar ratio 1:1, and at pH 1.0, one band with a maximum absorptivity at 540 nm can be seen on the Vis spectrum, and it can be associated with the [Fe(P1)H2]4+ complex, as with the DFP complexes. The dissociated hydroxyl group is coordinating the iron ion, whereas both N-end and C-end (amide protected) of the peptide remain protonated, at pH 1.0. Compared to amines, amides are weaker bases, and therefore do not present noticeable acid–base properties in water;61 they are protonated only at a low pH (<4). In the solution of the metal and peptide at a 1:3 molar ratio, at pH 4.0, two additional bands appear, with maximum values at 491 and 457 nm; these bands can be attributed to the [Fe(P1)2H2]3+ and [Fe(P1)3H3]3+ complexes, respectively. The positive charge of the [Fe(P1)2H2]+ complex is associated with the metal ion and the protonated amine on the N-end of the peptides. The ESI-MS spectra (Fig. S9†) recorded in 50/50 H2O/MeCN solution showed the formation of the [Fe(P1)H−1]+ ([C26H34FeN6O8]+; Fig. S9a†) and [Fe(P1)2]+ ([C52H70FeN12O16]; Fig. S9b†) complexes.
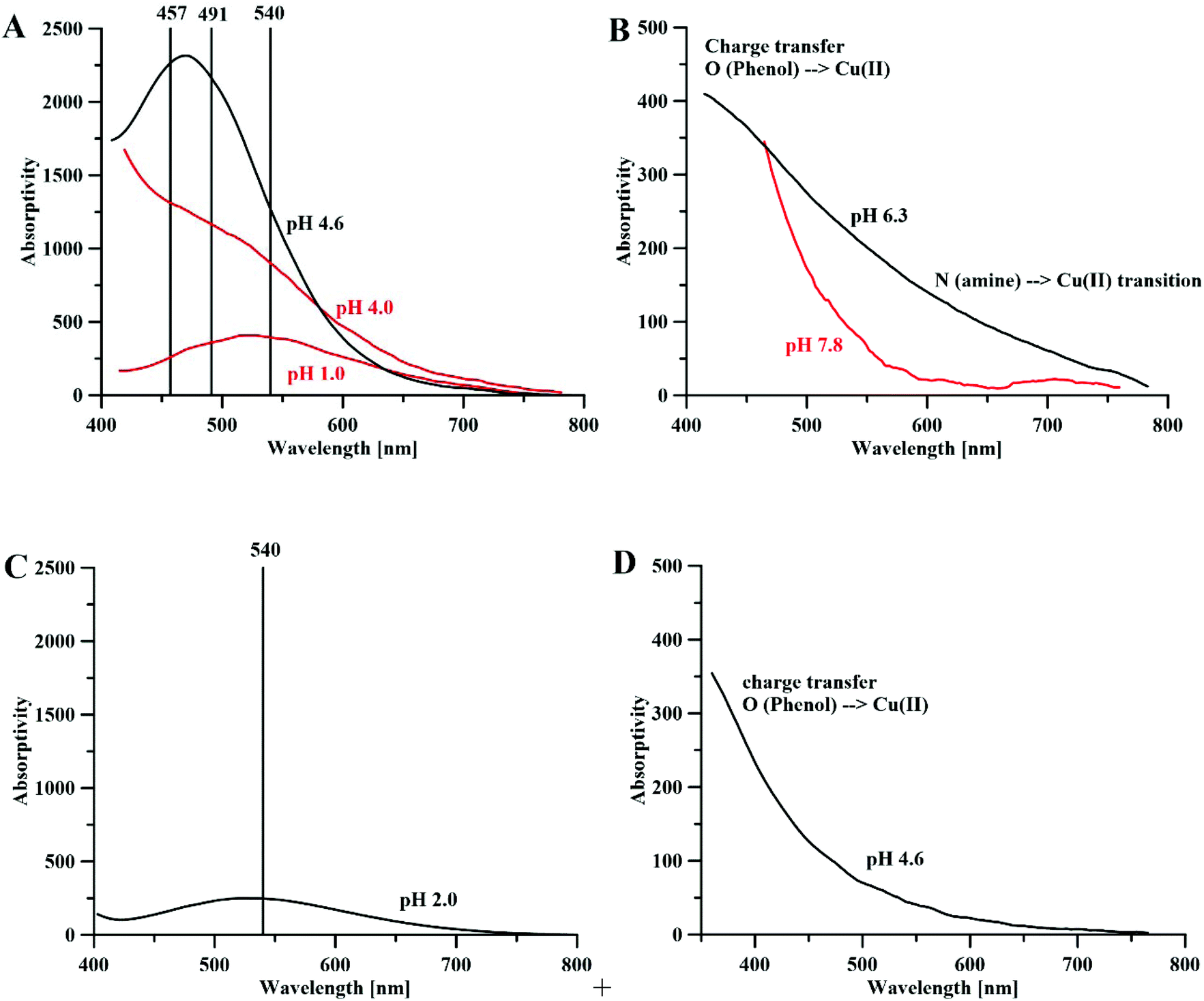 | ||
| Fig. 3 Absorptivity spectra of (A) Fe3+–P1 (red) and Fe3+–P2 complexes (black), metal:ligand molar ratios 1:1 and 1:3 (for P1) and the ratio 1:3 (for P2), [P1] = [P2] 10 mM, l = 0.13 cm; (B) the Cu2+–P1 (red) and Cu2+–P2 (black) complexes, the metal:ligand molar ratio 1:2, [P1]=[P2] = 10 mM, l = 0.13 cm. (C) Fe3+–P4 complexes, the metal:peptide molar ratio 1:1, [P4] = 30 mM, l = 0.13 cm and (D) Cu2+–P4 complexes, the metal:peptide molar ratio 1:1, and [P4] = 30 mM and l = 0.13 cm. | ||
The P2 peptide is considered a [LH4]2+ ligand with amine, amide, hydroxyl and carboxylic dissociating groups. Fig. 3A shows the absorptivity spectrum of the P2 complex with iron(III) ions, at pH 4.6. The large band centered at 475 nm is composed of two bands (457 and 491 nm) of almost the same intensity, and one band at 540 nm of a lower intensity that represents the formation of all three types of complexes: [Fe(P2)H]2+, [Fe(P2)2H2]+ and [Fe(P2)3].
The similarities between the UV-Vis spectra of the 1:3 (metal:ligand) stoichiometry, P1- and P2-peptide complexes (Fig. 3A) and that of DFP (Fig. 1A) suggest that only MIM residues are involved in the coordination of the iron, whereas carboxylic groups are not part of the metal coordination core. Indeed, the DFT optimized complexes in solution show that carboxylic acid groups are not involved in the coordination of the iron, and that the metal ions are bound by the oxygen atoms of MIM residues (Fig. 4). For each one-mimosine peptide, we optimized the most likely 1:1 metal–ligand geometry and calculated its binding energy (Table 2); even though in Table 2 we also reported the binding energies of 1:1, 1:2 and 1:3 metal–DFP complexes, it is important to bear in mind that a direct comparison of the stability of these compounds with mimosine-containing peptides is not reliable. Indeed, it is not correct to compare the stability of bidentate ligands (DPF complexes) with those of hexadentate ligands (MIM-peptides) because of different entropic contributions related to their respective chelate effects.
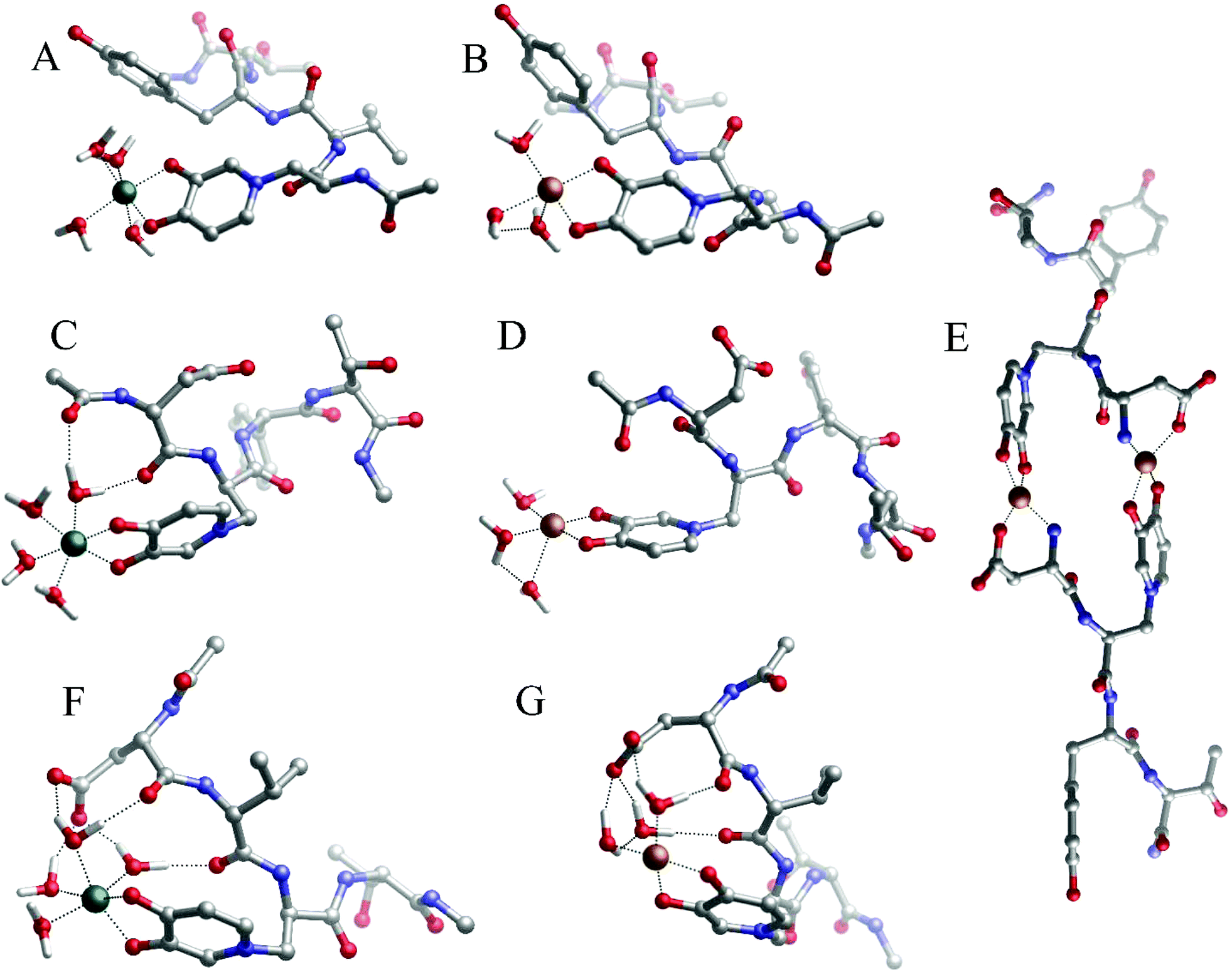 | ||
| Fig. 4 Geometries of (A) Fe(P1), (B) Cu(P1), (C) Fe(P2), (D) Cu(P2), (E) Cu2(P2)2, (F) Fe(P3) and (G) Cu(P3) complexes optimized with DFT in solution. | ||
| Ligand name | Metal:ligand stoichiometry |
Fe(III) | Cu(II) | ||
|---|---|---|---|---|---|
| ΔHaq | ΔGaq | ΔHaq | ΔGaq | ||
| DFP | 1:3 |
−159.7 | −165.3 | — | — |
| DFP | 1:2 |
−124.6 | −131.2 | −92.5 | −97.6 |
| DFP | 1:1 |
−78.3 | −81.5 | −53.5 | −56.1 |
| Pept1 | 1:1 |
−70.2 | −65.4 | −51.8 | −53.9 |
| Pept2 | 1:1 |
−79.2 | −76.4 | −47.7 | −50.9 |
| Pept2 | 2:2 |
−159.7 | −178.2 | ||
| Pept3 | 1:1 |
−100.2 | −91.4 | −76.3 | −69.5 |
| Pept4 | 1:1 |
−109.7 | −123.0 | −74.3 | −79.9 |
| Pept4 | 2:2 |
−170.8 | −187.0 | ||
| Pept5 | 1:1 |
−106.7 | −119.1 | −65.3 | −73.9 |
| Pept5 | 2:2 |
−197.1 | −205.9 | ||
| Pept6 | 1:1 |
−121.9 | −152.9 | −60.9 | −88.2 |
A comparison of the free energies of the 1:1 complexes (Table 2) shows that Peptide 3 (P3) forms the most stable complexes with either copper and iron when compared with Peptide 1 and Peptide 2.
The reason for this higher stability could be due to the intra-molecular H-bonds that are formed between the coordinating water molecules and backbone carbonyl and carboxylate residues of P3 (Fig. 4F and G); these interactions are supposed to stabilize the overall system when compared with P1 and P2, although a thorough conformational analysis of all peptides is beyond the scope of the present work.
Likewise, the 1:1 Fe(III)–P2 complex is more stable (ΔGaq = −76.4 kcal mol−1) than the complex formed with P1 (ΔGaq = −65.4 kcal mol−1), whereas the 1:1 Cu(II)–P2 and Cu(II)–P1 complexes have similar stability. Again, in the case of the Fe(II)–P2 complex (Fig. 4C), intra-molecular hydrogen bonds are additional forces that concur in the higher stabilization of the system.
For the same reasons outlined above, the 1:1 Fe(III)–P3 and Cu(II)–P3 complexes are remarkably more stable than the respective 1:1 metal–DFP complexes (Table 2).
The Vis spectra of the Cu2+–P1 complexes are shown in Fig. 3B. At a slightly basic pH, only the high intensity charge transfer O(phenol) → Cu2+ band appears to occur, which suggests the formation of a [Cu(P1)2H2]2+ complex. The absence of the bands in the 600–800 nm range implies that neither the amine group (N-end and C-end of the peptide) in the metal coordination shell nor the free metal ion in the solution is present. The ESI-MS spectra (in 50/50 H2O/MeCN solution; Fig. S10†) showed the presence of the [Cu(P1)H−1]+ ([C26H34CuN6O8]+) complex (Fig. S10a†).
The P2 complexes with copper(II) ions, at pH 6.3, are different from those of P1 (Fig. 3B). Next to the band of charge transfer O(Phenol) → Cu2+, we notice the presence of the d–d transition band (∼650 nm) N(amine) → Cu2+ transition, which confirms the implication of the nitrogen atoms in the metal coordination shell. The high intensity of the absorptivity spectrum (in agreement with the literature Cu(II)–mimosine complex data59 in Table 1) suggests the formation of the dinuclear complex [Cu2(P2)2] (Fig. 4E), where each copper(II) ion is coordinated by the mimosine (CO, O−) and aspartic acid (NH2, COO−) residues.
The copper(II) 1:1 stoichiometry complexes of P1, P2 and P3, characterized by DFT calculations (Fig. 4A–D, F and G), confirm that the Cu(II) ion is coordinated by the pair of hydroxyl and carbonyl atoms of the MIM residue and three water molecules.
Considering the stability of the Cu2(P2)2 complex, it is evident that unlike the dinuclear copper(II)–MIM complex where Cu(II) ions appear penta-coordinated, in the Cu2(P2)2 system the two Cu(II) ions are tetra-coordinated by interacting with the MIM moiety of one peptide and the terminal carboxylic and amino groups of the other peptide (Fig. 4E). However, the interaction of Asp1 amino acid through its side chain carboxylic group and terminal amine slightly distorts the planarity between the metal ligands (Table S10†). Nevertheless, the metal coordination shell of the P2 dinuclear complex is similar to that of the 2:2 copper(II)–MIM complex, with the Cu(II)–ligand binding site distances and angles being nearly the same (Table S10†). The stabilities of such complexes were computed according to eqn (3).
Cu(II) is tetra-coordinated both in solution and within the complex. The computed free energy value of the tetra-coordinated Cu2(P2)2 (−178.2 kcal mol−1) complex is considerably more stable than the penta-coordinated Cu2(MIM)2 (−163.1 kcal mol−1) one.
:1 metal-to-peptide molar ratio. Fig. 3C presents the absorptivity spectrum of the iron(III) ions with the P4 peptide, at pH 2.0. The single band with the maximum absorptivity at 540 nm suggests the formation of a monomeric [Fe(P4)H3]3+ species, positively charged due to its iron ion and amine group, whereas the second hydroxyl group and the carboxylic groups remain protonated at this low pH. It is unlikely that a dimeric complex is formed at such very low pH.
DFT calculations in water show the formation of a monomeric species Fe(III)/P4 (Fig. 5A), where MIM residues are close enough to form a cyclic complex, and where two MIM and two water molecules form a complex exhibiting octahedral geometry (Table S11† and Fig. 5A).
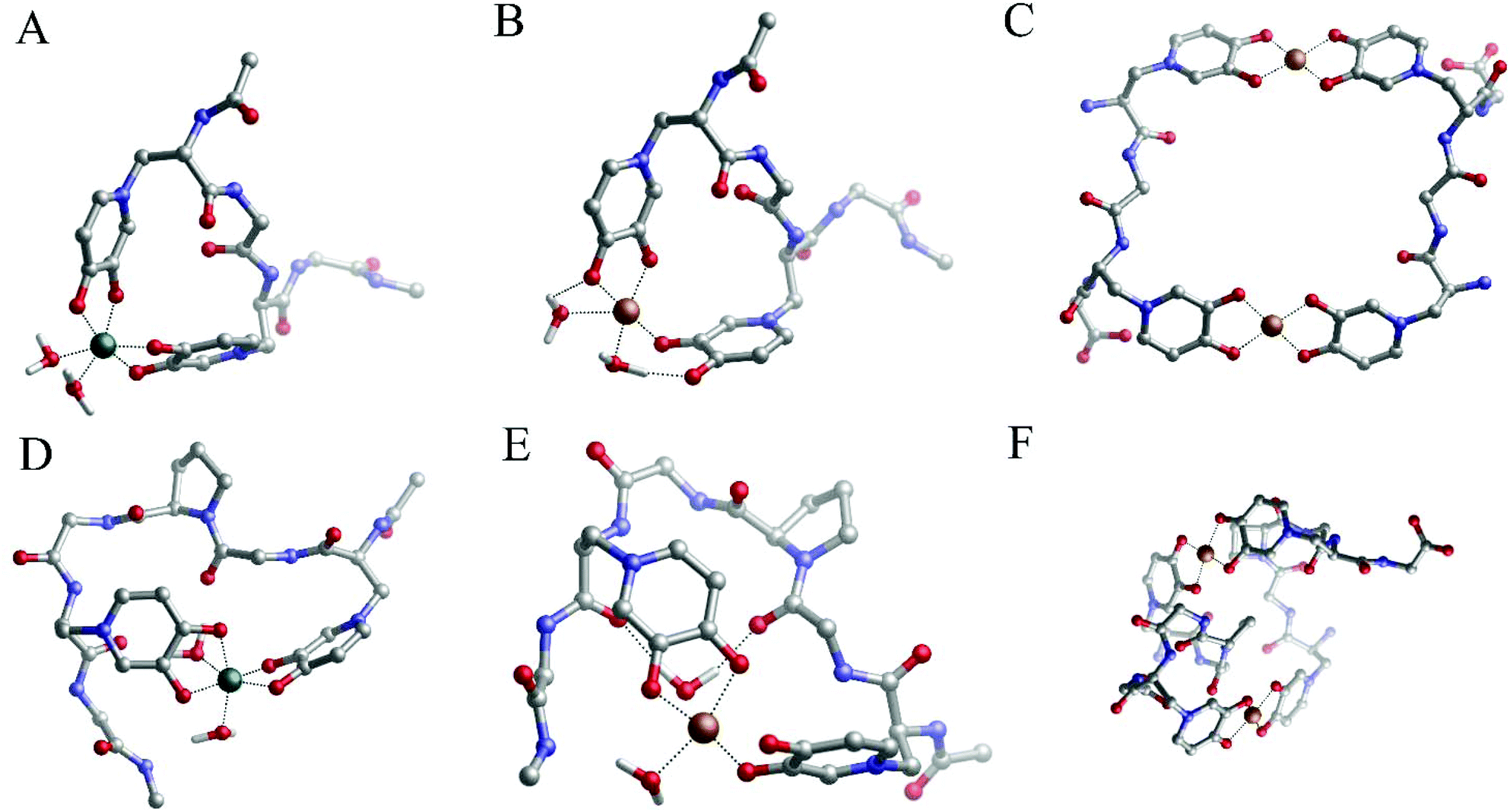 | ||
| Fig. 5 Geometries of (A) the Fe(III)/P4 1:1 complex, (B) the Cu(II)–P4 1:1 complex, (C) the Cu(II)–P4 2:2 complex, (D) the Fe(III)–P5 1:1 complex, (E) the Cu(II)–P5 1:1 complex and (F) the Cu(II)–P5 2:2 complex optimized with DFT in solution. | ||
Fig. 3D shows the absorptivity spectrum of the Cu(II)–P4 system, in which only the band of charge transfer O(Phenol) → Cu2+ is present. This implies the existence of a coordination of the copper(II) by 2 (CO, O–) pairs of oxygen atoms of two MIM residues (Fig. 5B). The spacer between the two MIM residues is too short to allow both MIM residues participate in the coordination of the copper ion (Fig. 5B) and, due to the steric encumbrance, it is more likely that a dinuclear complex [Cu2(P4)2] (Fig. 5C) is formed, instead of a monomeric species [Cu(P4)]. The formation of a dimeric complex, in which each metal ion is coordinated by two MIM residues (Fig. 5C), is consistent with the absorptivity spectrum of the Cu(II)/P4 system obtained in our study (Fig. 3D).
Peptide 5 contains two MIM residues spaced by three amino acid residues. It has the same dissociating groups as peptide 4, and is considered to be a [LH4]+ ligand, but it is longer than P4 due to the presence of additional glycine and proline residue. The proline residue was introduced in the middle of the peptide's backbone to support turn formation upon metal complexation.1
DFT calculations highlight that the 1:1 iron to P5 complex has two MIM residues and two water molecules in the metal-coordination core (Fig. 5D). The Fe(III) coordination shells are very similar, with both P4 and P5 systems satisfying the optimal octahedral coordination mode of iron (geometric data are given in Table S11†). Therefore the slightly lower stability of Fe(III)–P5 compared to the Fe(III)–P4 complex (Table 2) could be due to the rigidity of the peptide backbone that contains the proline residue; however, we reiterate that a careful conformational analysis (beyond the scope of this work) is pivotal in order to examine this point.
According to DFT calculations, P5 can form a monomeric Cu(II)–P5 complex in which two MIM residues and two water molecules coordinate copper ions in the distorted octahedral complex (Fig. 5E). Also in this case, the stability of the monomeric Cu(II)–P5 complex is lower than that of the monomeric Cu(II)–P4 system (Table 2) and, analogously to P4, it is highly possible that copper dimeric species (Fig. 5F) are formed. Finally, a comparison of the stabilities of the P5 and P4 dimeric complexes with the copper(II) ion reveals that the former is remarkably more stable than the latter.
Fig. 6A shows the Vis spectra of the iron(III)–P6 system titration with the growing quantity of iron ions. We can clearly see that the spectrum reaches its highest absorptivity for a 1:1 metal to peptide molar ratio, which is equivalent to the maximum absorptivity of the Fe(DFP)3 complex (Fig. 1A). Based on this result, we can hypothesize the formation of a monomeric Fe(P6) complex, where the metal ion is coordinated by six oxygen atoms of three mimosine residues (Fig. 7A).
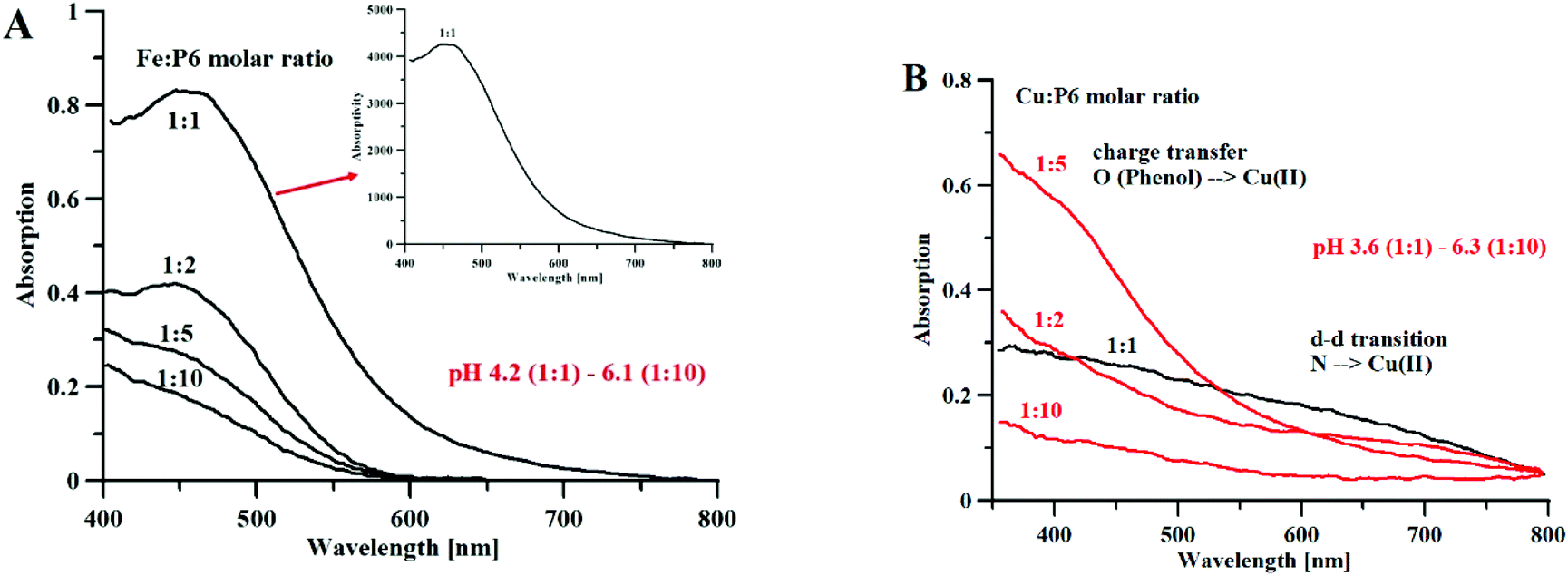 | ||
| Fig. 6 (A) Absorption and absorptivity spectra of Fe3+–P6 complexes, [P6] = 1.5 mM, l = 0.13 cm (B) Absorption spectra of Cu2+–P6 complexes, [P6] = 3.5 mM, l = 1.0 cm. | ||
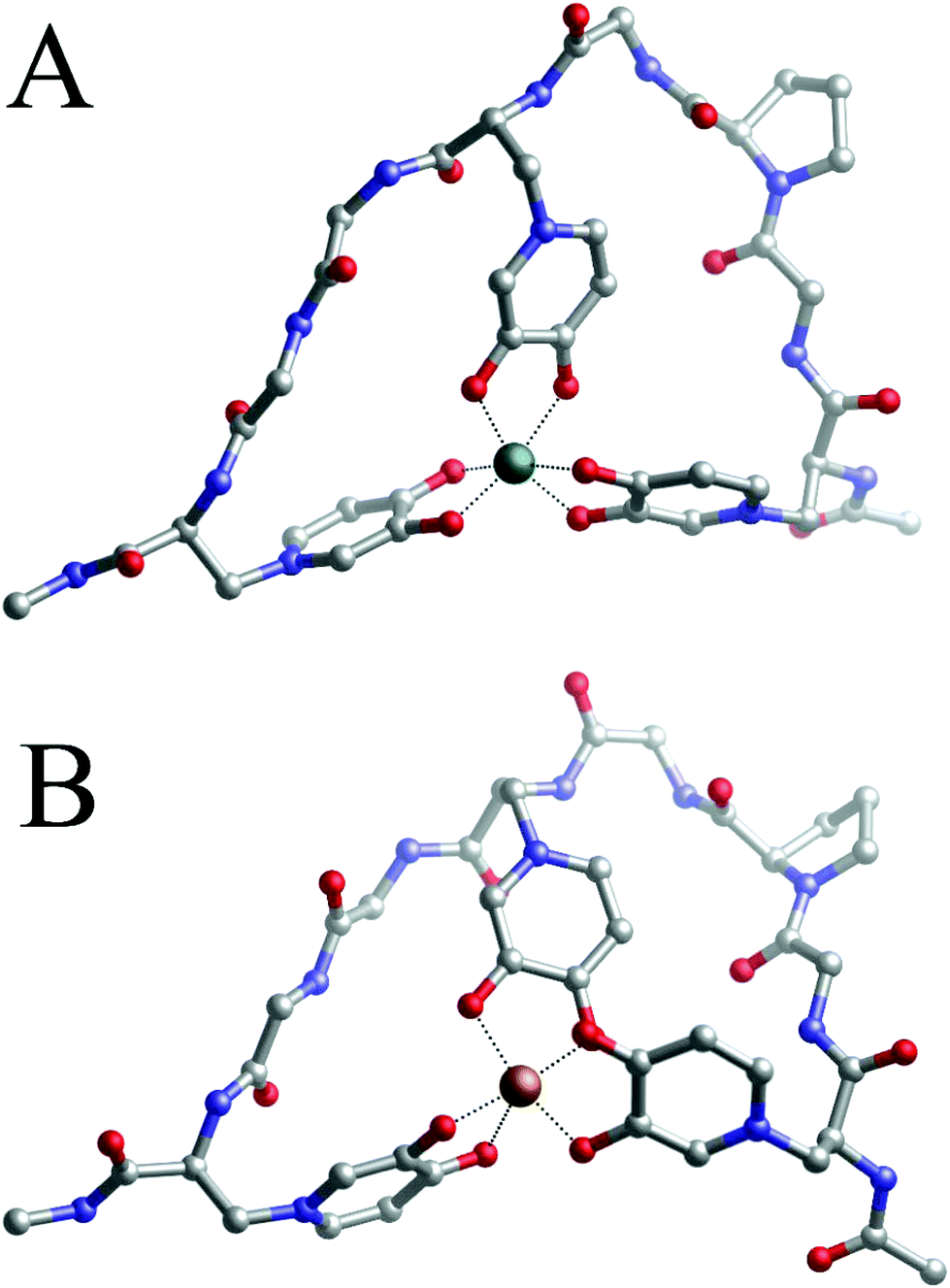 | ||
| Fig. 7 Geometries of Fe(III)–P6 (A) and the Cu(II) (B)–P6 1:1 complex optimized with DFT in solution. | ||
Fig. 6B shows the results of an analog experiment with copper(II) ions. At a high metal-to-ligand molar ratio (1:10), and a pH of 6.3, only the band of charge transfer O(Phenol) → Cu2+ can be observed. In the solutions of 1:5 and 1:2 metal to ligand molar ratios, the band of charge transfer is predominant (over the band of the N(amine) → Cu2+ d–d transition). In the solution containing equimolar concentrations of metal and peptide, at pH 3.6, the intensity of the charge transfer band is almost equal to the band of the d–d transition, which makes it likely that mixed coordination complexes are formed.
The 1:1 complexes formed by peptide 6 and Fe(III) or Cu(II) were characterized by DFT methods and their binding energies computed, according to eqn (2). For both complexes, the optimized structures are shown in Fig. 7A and B respectively, and their computed binding energies in solution are presented in Table 2.
The spacers between the mimosine residues are long enough to involve three mimosine residues in the metal-coordination process. In fact, the angles computed on the metal coordination shell of Fe(P6) are very similar to the ones computed on Fe(DFP)3 (see Table S11†), indicating that the peptide is long enough to allow the three mimosine side chain to arrange in a near-optimum disposition. Interestingly, the free energy (Table 2) of the Fe(III)–P6 complex (−152.9 kcal mol−1) is less negative compared to that of the 1:3 Fe(III)–DFP (−165.3 kcal mol−1) and very similar to that of the 1:3 Fe(III)–MIM complex (−151.8 kcal mol−1, Table S2†). Such a situation indicates that, in the case of P6, the entropic contribution related to the chelate effect is not a major driving force in the stabilization of 1:1 Fe(III)–P6, as highlighted by the very similar free energy of the 1:3 Fe(III)–MIM complex. A possible explanation could be that the introduction of one proline residue turns the backbone of the peptide, but makes it more rigid and decreases its overall complex stability. Clearly, the addition of a proline residue is not a good strategy for the design of novel mimosine-containing chelating agents with improved chelation performance. These findings are in agreement with the results obtained in our previous work involving proline-containing and proline-free mimosine peptides coordinating Al(III) ions.42
Regarding the Cu/P6 monomeric complex, apparently the metal adopts octahedral coordination mode because the angles between the oxygen ligands are appropriate for this coordination mode (see Table S12†). However, one of the mimosine residues interacts only though one oxygen atom (Cu–O distances of 1.98 and 2.80 Å), and a second one is in the mono-/bidentate borderline (Cu–O distances of 1.97 and 2.41 Å). Even if two MIM residues are involved in the coordination of copper, the geometry of the complex is distorted, and two of the MIM residues are not in planar position, with respect to the central metal ion. It is evident that the length of the linker between the neighboring metal binding MIM residues is too short to allow the formation of a complex of octahedral geometry, similar to that of Cu(DFP)2, where two ligand molecules are in planar and equatorial positions. It is more likely that stoichiometries different than 1:1 are formed with the co-participation of the amine group in the complex core, as it can be deduced from Vis spectra with the evident N(amine) → Cu2+ d–d transition band (Fig. 6B).
Theoretical TDDFT spectra of metal complexes with DFP and all studied peptides confirmed our analysis of the UV/Vis experimental spectra and showed good agreement with the experiments. For a complete analysis of the theoretical spectra, we refer to the ESI (Fig. S11–S15†).
As seen in Fig. S8,† the binding energies computed in solution (ΔGcomp, see Table S2†) are in very good agreement with the available logβ values with a correlation coefficient of 0.9873, confirming the suitability of our methodology for the characterization of the complexes investigated in this study. Interestingly, the DFP ligand interacts more tightly with the two metals than it does with the MIM residue; with Fe(III) and the 1:1, 1:2 and 1:3 complexes containing DFP being, respectively, 9.2, 15.4 and 15.6 kcal mol−1 more stable than the MIM-containing ligands. In the case of Cu(II), the 1:1 and 1:2 ligands that contain DFP are, respectively, 6.1 and 13.2 kcal mol−1 more stable than their MIM-containing counterparts.
Considering both families of ligands (deferiprone-based and mimosine-based), the introduction of a methyl group significantly increases the stability of the formed complexes, especially the stability of the iron complexes (Table S2†). For Fe(III), for example, the Fe–DFP 1:1, 1:2 and 1:3 complexes are 4.3, 8.0 and 7.0 kcal mol−1 more stable than the corresponding Fe–DFP-unmet (Table S2†). Similarly, in the case of Cu(II), the presence of methyl in the Cu(II)–DFP complexes also leads to more stable complexes than the Cu(II)–DFP-unmet complexes (2.7 and 3.5 kcal mol−1 for the 1:1 and 1:2 complexes). However, the increase in stability, upon addition of the methyl group, is lower than that obtained with the Fe(III)–ligand complexes. Interestingly, a similar trend is also observed for the MIM-based ligands, for which the inclusion of the –CH3 substituent stabilizes all complexes in a similar fashion. The addition of a methyl in position 2 in the mimosine ligand (MIM-met) stabilizes the Fe(III) 1:1, 1:2 and 1:3 complexes by 4.5, 7.5 and 4.9 kcal mol−1, respectively; the Cu(II) 1:1 and 1:2 complexes are stabilized by 2.4 and 6.5 kcal mol−1, respectively.
In general, we can conclude that the addition of a 2-methyl group is important for augmenting the affinity of DFP to the metal, but the introduction of the 3-methyl substituent in mimosine would lead to a lower increase in affinity. This is probably due to the positively charged amino group of the MIM residue, which acts as an electron-withdrawing group (EW), thus lowering the electron density from the coordination site of the ligands. As a result, the EW amino group (positively charged) partially compensates the ED effect of the methyl group. Nevertheless, the addition of this 2-methyl group could help in the improvement of the chelation performance of these compounds.
3.2. Antimicrobial activity
Antimicrobial peptides (AMPs) (also known as host defence peptides, HDPs) are part of the innate immune response, in all classes of life, produced to kill bacteria, viruses, fungi and even cancer cells.62–65 Most AMPs have a positive charge (+2 to +9) and hydrophobic amino acids (more than 30%), while the length varies between 10 and 50 amino acid residues. The mechanism of AMP action against microbials is complicated and the same AMP may act on different targets.63 One of the best known mechanisms is through membrane permeabilization, which consequently leads to a loss of cellular components and cell death.64 The non-specific membrane interactions between AMPs and bacteria differ from those of antibiotics, and they target specific molecular receptors of pathogens and make it difficult for bacteria to acquire resistance to AMPs.64Different approaches have been used to ameliorate AMP bactericidal activity with a short half-time and at the same time reduce hemolytic activity, e.g. changes in the amino acids’ sequence, cyclization, and synthesis of multivalent constructs. It was observed that the proline-rich peptides have a higher membrane internalization, while Gly-rich peptides are more selective for Gram-negative bacteria, fungi and cancer cells.65,66 Also many short-chain pro-rich peptides showed a high activity against Gram-negative bacteria.67 The long peptides with a high ratio of Pro and Gly amino acids usually exhibit linear, rather than secondary, structures.68–70 AMPs have mostly a positive charge (from +2 to +9), which allows AMPs to interact with negatively charged lipid head groups.71 The increase of the positively charged amino acids in the peptides’ sequence increases the affinity for the microbial membrane and improves the overall antimicrobial activity. However, also anionic AMPs (containing Gln and Asp) participate in the eukaryotic innate immune system.72 Their negative charge ranges from −1 to −2, and they generally need cations (e.g. Zn2+) as cofactors for antibiotic activity.73
AMPs with high hydrophobicity can damage the membrane structure, which results in cell lysis or the formation of transient pores and the transport of peptides inside the cell; this property enables them to interact with intracellular targets.74
The preliminary screening carried out on the six peptides by the disk diffusion method (Table S14†) reveals that the peptides tested at higher concentrations possess an antibacterial activity. In particular, P5 (810 μg per disk equivalent to 81 mg mL−1) exhibits an inhibitory effect against S. aureus and B. cereus, and P3 (340 μg per disk) against S. aureus. The other four peptides tested with lower concentrations show no inhibition activity against the microbial strains used as indicators.
The physicochemical properties of the peptide, with respect to charge, amphipathic nature, hydrophobicity capacity, are key factors impacting on their mode of action and selectivity towards microbial cells.75 The binding of the metal may induce changes to net charge and the conformational plasticity of the peptides that may favor the interaction with the bacterial cell wall and the phospholipid bilayer component of the membrane, resulting in the potentiation of antibacterial activity; however, additional experiments need to be performed to confirm such hypothesis.
In order to better investigate the antibacterial activity of the mimosine-derived peptides and their complexes, the MIC and MBC values were determined following the broth microdilution method. The results are reported in Tables S15 and S16.† The copper complexes with the peptides investigated in our study showed no antibacterial activity, whereas the iron(III) complexes with the P4 and P6 peptides gave a positive antibacterial response. The free P4 shows an MIC of 7450 μg mL−1, and the iron(III) complex of P4 shows up to fifteen times lower MIC values against S. aureus (484 μg mL−1), B. cereus (484 μg mL−1), and E. coli (967 μg mL−1). A good inhibition activity was recorded against the strains S. aureus and B. cereus by the P6 Fe complex, with MIC values of 448 μg mL−1, which are much lower than the MIC activity of the free P6 peptide (>6900 μg ml−1).
The enhanced antagonistic activity of the iron complexes against Bacillus could be due to the siderophore mimicking property of the mimosine peptides, these being structurally similar to the siderophores produced by Bacillus strains (e.g. petrobactin), but more in-depth studies aimed at understanding the mechanism of action are needed for supporting this hypothesis. The siderophore mimicking property of mimosine peptides could be applied for diagnostic and theragnostic treatment of bacterial infections,76 while mimosine is structurally similar to that of DFP and could form stable complexes with Gd(III)77 and other lanthanide metals.37
Mimosine based peptides are structurally and functionally different from classical antibiotics and their MIC activity should be compared with AMPs rather than antibiotics. The MIC comparative studies of different AMPs made by Ebbensgaard et al.5 showed that unstructured AMPs (Myxinidin-NH2, Pyrrhocoricin, Apidaecin IA, Metalnikowin I) are less effective than α-helical and β-sheet structured peptides and their MIC activity requires more than 256 μg mL−1 concentration. Such a value is comparable with the results reported for the Fe(III)–P6 complexes.
Even if mimosine peptides require high doses to reach MIC activity, their month-long stability, both in the solid state and in solution, makes them good candidates for being an alternative to antibiotics (or mixed) therapies. Recent studies point out that the contemporary use of different antibiotic agents can reduce the dose of each drug in the combination. Such mixed therapies may lower the development of bacterial resistance in vitro (compared to monotherapy). AMPs/peptidomimetics are well suited for synergic combinations with conventional antibiotics,64 while disrupting bacterial membranes and facilitating antibiotics to reach their cytosolic targets.78
Natural and synthetic antimicrobial peptides have been shown to prevent biofilm formation, kill bacteria or disrupt the biofilm structure.88 AMPs prevent bacteria adhesion on surfaces89 and damage mature biofilms by detachment or killing bacteria.90 The exact mechanism of biofilm degradation is poorly understood, but the fast destruction of biofilm embedded cells90 may indicate that they act by disrupting the membranes of the bacteria.
In this study the antibiofilm activity of the most effective inhibitor (peptide 6) and its Fe complex is evaluated. Three different strains of bacterial pathogens (S. aureus, S. intermedius, and P. aeruginosa), described in the literature as linked to biofilm-associated diseases in humans and animals,91,92 are used to examine the antibiofilm activity of peptide 6 and its iron complex. A preliminary evaluation of P6 against bacteria in Planktonic status shows (Table S17†) an antimicrobial activity (MIC > 1 mM L−1; MBC > 1 mM L−1), whereas P6 in sessile form shows a slightly different behavior. Peptide 6 is found unable to completely inhibit the formation of biofilms, but a consistent reduction (up to 70%) related to the peptide's concentration is observed for S. aureus (from 1 to 0.5 mM peptide concentration range). The antibiofilm data show that the P6 Biofilm–interaction is not iron-dependent (P > 0.05 between two conditions +Fe/−Fe), as seen in Fig. S7A,† suggesting that P6 activity against biofilm formation is rather not related to siderophore mimicking. With respect to the other bacterial strains, S. intermedius and P. aeruginosa, the obtained values, at different peptide concentrations, with and without iron, are not significant; nevertheless, the addition of iron ions to the peptide solution slightly reduces the formation of a biofilm (Fig. S7B and C†). The antibiofilm results reported here are preliminary, and should be more thoroughly examined by the genic expression of biofilm-related genes, for example those involved in the bacteria quorum-sensing network.92
The anti-biofilm activity of AMPs can be enhanced by mixed therapy with antibiotics.93–95 Combine use of AMPs with known antibiotics is useful since they can target different strains of bacteria with different metabolisms cells in low pH, hypoxic or low nutritious environments.96
4. Conclusions
We have presented a series of mimosine-based peptides, which according to our experimental and theoretical data, are effective iron(III) and copper(II) chelators. These peptides bind Fe(III) more strongly than Cu(II), as non-peptidic complexes based on deferiprone. Quite interestingly, the binding energies of these peptide complexes can be modulated by a number of key structural factors, such as the number of mimosine residues in the peptide, and length and flexibility of the spacer between mimosine residues. In this sense, peptide 6, with three mimosine residues, forms the most stable complexes with both Fe(III) and Cu(II), followed by peptides 4 and 5, with two mimosine residues.For each peptide, the basic metal:peptide stoichiometry was investigated. The formation of oligomeric and polymeric structures depends on environmental factors (peptide and metal concentration, pH, ionic strength and temperature) and needs further investigation in diffusion NMR studies. The present results and the combination of theoretical and experimental methodologies can be helpful for the design of self-assembly between metal ions and mimosine peptides, which could lead to various medical and nanotechnological applications. In fact, we have also proved that Fe–mimosine complexes can display a significant antimicrobial activity, which highlights the potential interest of the compounds presented in this work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant no. UMO-2015/17/D/ST5/01329 from the National Science Centre, Poland. This work was also supported by Grants PGC2018-099321-B-I00 from the Ministry of Science and Universities through the Office of Science Research (MINECO/FEDER), and Grant IT588-13 from the Basque Government.References
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200–5219 RSC.
- D. E. Przybyla and J. Chmielewski, Biochemistry, 2010, 49, 4411–4419 CrossRef CAS PubMed.
- M. Panciera, M. Amorín and J. R. Granja, Chem. – Eur. J., 2014, 20, 10260–10265 CrossRef CAS PubMed.
- A. Ambrogelly, S. Palioura and D. Söll, Nat. Chem. Biol., 2007, 3, 29–35 CrossRef CAS PubMed.
- A. Ebbensgaard, H. Mordhorst, M. T. Overgaard, C. G. Nielsen, F. M. Aarestrup and E. B. Hansen, PLoS One, 2015, 10, e0144611 CrossRef PubMed.
- A. Lewies, L. H. Du Plessis and J. F. Wentzel, Probiotics Antimicrob. Proteins, 2019, 11, 370–381 CrossRef CAS PubMed.
- K. Sabol, J. E. Patterson, J. S. Lewis, A. Owens, J. Cadena and J. H. Jorgensen, Antimicrob. Agents Chemother., 2005, 49, 1664–1665 CrossRef CAS PubMed.
- J. N. Steenbergen, J. Alder, G. M. Thorne and F. P. Tally, J. Antimicrob. Chemother., 2005, 55, 283–288 CrossRef CAS PubMed.
- S. Rotem and A. Mor, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1582–1592 CrossRef CAS PubMed.
- B. Findlay, G. G. Zhanel and F. Schweizer, Antimicrob. Agents Chemother., 2010, 54, 4049–4058 CrossRef CAS PubMed.
- N. Molchanova, P. Hansen and H. Franzyk, Molecules, 2017, 22, 1430 CrossRef PubMed.
- B. C. Q. Nguyen and S. Tawata, Phytother. Res., 2016, 30, 1230–1242 CrossRef CAS PubMed.
- S. Tawata, in Pesticide and Alternatives, ed. J.E Casida, Elsevier Science Publishers, Amsterdam, Netherlands, 1990, pp. 541–544 Search PubMed.
- J. W. Hylin, Phytochemistry, 1964, 3, 161–164 CrossRef CAS.
- B. Tangendjaja, J. Hogan and R. Wills, Aust. J. Agric. Res., 1983, 34, 289–293 CrossRef CAS.
- R. Anitha, S. Jayavelu and K. Murugesan, Phytother. Res., 2005, 19, 992–993 CrossRef CAS PubMed.
- A. Zalatnai and J. Bocsi, Anticancer Res., 2003, 23, 4007–4009 CAS.
- S. Frydas, N. Papaioannou, M. Papazahariadou, M. Hatzistilianou, E. Karagouni, M. Trakatelli, G. Brellou, C. Petrarca, M. Castellani and P. Conti, Int. J. Immunopathol. Pharmacol., 2005, 18, 85–93 CrossRef CAS PubMed.
- C. Baltazar, R. Mun, H. Tajmir-Riahi and J. Bariyanga, J. Mol. Struct., 2018, 1161, 273–278 CrossRef CAS.
- Y. Shan, Y. Ma, M. Wang and Y. Dong, Curr. Med. Chem., 2012, 19, 5885–5894 CrossRef CAS PubMed.
- Y. Dai, B. Gold, J. K. Vishwanatha and S. L. Rhode, Virology, 1994, 205, 210–216 CrossRef CAS PubMed.
- T. Xuan, A. Elzaawely, F. Deba, M. Fukuta and S. Tawata, Agron. Sustainable Dev., 2006, 26, 89–97 CrossRef CAS.
- R. Williams and R. Hoagland, Allelopathy J., 2007, 19, 423–430 Search PubMed.
- M. Lalande, Exp. Cell Res., 1990, 186, 332–339 CrossRef CAS PubMed.
- A. Restivo, L. Brard, C. Granai and N. Swamy, J. Clin. Oncol., 2005, 23, 3200–3200 Search PubMed.
- K. Ward and R. N. Harris, Aust. J. Biol. Sci., 1976, 29, 189–196 CrossRef CAS PubMed.
- J. Savulescu, Br. Med. J., 2004, 328, 358 CrossRef PubMed.
- FDA NEWS RELEASE: FDA Approves Ferripox (deferiprone) to Treat Patients with Excess Iron in the Body, Oct. 14, 2011, http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm275814.htm.
- R. Ma, J. J. Reibenspies and A. E. Martell, Inorg. Chim. Acta, 1994, 223, 21–29 CrossRef CAS.
- R. J. Motekaitis and A. E. Martell, Inorg. Chim. Acta, 1991, 183, 71–80 CrossRef CAS.
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1992, 191, 57–63 CrossRef.
- V. M. Nurchi, G. Crisponi, T. Pivetta, M. Donatoni and M. Remelli, J. Inorg. Biochem., 2008, 102, 684–692 CrossRef CAS PubMed.
- J. Lachowicz, V. Nurchi, G. Crisponi, M. Jaraquemada-Pelaez, M. Arca, A. Pintus, M. Santos, C. Quintanova, L. Gano and Z. Szewczuk, Dalton Trans., 2016, 45, 6517–6528 RSC.
- E. T. Clarke, A. E. Martell and J. Reibenspies, Inorg. Chim. Acta, 1992, 196, 177–183 CrossRef CAS.
- A. E. Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed.
- D. M. Doble, M. Melchior, B. O'Sullivan, C. Siering, J. Xu, V. C. Pierre and K. N. Raymond, Inorg. Chem., 2003, 42, 4930–4937 CrossRef CAS PubMed.
- K. H. Thompson, C. A. Barta and C. Orvig, Chem. Soc. Rev., 2006, 35, 545–556 RSC.
- E. Chruscinska, E. Garribba, G. Micera and A. Panzanelli, J. Inorg. Biochem., 1999, 75, 225–232 CrossRef CAS.
- R. C. Scarrow, P. E. Riley, K. Abu-Dari, D. L. White and K. N. Raymond, Inorg. Chem., 1985, 24, 954–967 CrossRef CAS.
- A. Upadhyay, J. Chompoo, N. Taira, M. Fukuta, S. Gima and S. Tawata, J. Agric. Food Chem., 2011, 59, 12858–12863 CrossRef CAS PubMed.
- B. C. Q. Nguyen and S. Tawata, Molecules, 2015, 20, 14334–14347 CrossRef CAS PubMed.
- J. Mujika, G. Dalla Torre, J. Lachowicz and X. Lopez, RSC Adv., 2019, 9, 7688–7697 RSC.
- W. C. Chan and P. D. White, Fmoc solid phase peptide synthesis, Oxford University Press, 2000 Search PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- T. Goddard and D. Kneller, SPARKY 3.114, University of California, San Francisco, USA, 2007 Search PubMed.
- K. Wüthrich, Europhys. News, 1986, 17, 11–13 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. Stephens, F. Devlin, C. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Revision A, 2016, vol. 3 Search PubMed.
- G. Dalla Torre, J. I. Mujika, E. Formoso, E. Matito, M. J. Ramos and X. Lopez, Dalton Trans., 2018, 47, 9592–9607 RSC.
- J. Alí-Torres, L. Rodríguez-Santiago and M. Sodupe, Phys. Chem. Chem. Phys., 2011, 13, 7852–7861 RSC.
- B. Gandhi, R. Summerbell and T. Mazzulli, J. Clin. Microbiol., 2018, 56, e01481–e01417 CAS.
- G. Orru, C. Demontis, A. Mameli, E. Tuveri, P. Coni, G. Pichiri, F. Coghe, A. Rosa, P. Rossi and G. D'Hallewin, Front. Microbiol., 2017, 8, 2067–2078 CrossRef PubMed.
- W. Wang, S. Nema and D. Teagarden, Int. J. Pharm., 2010, 390, 89–99 CrossRef CAS PubMed.
- A. El-Jammal, P. L. Howell, M. A. Turner, N. Li and D. M. Templeton, J. Med. Chem., 1994, 37, 461–466 CrossRef CAS PubMed.
- W. C. Tsai and K. H. Ling, J. Chin. Biochem. Soc., 1973, 2, 70–86 CAS.
- H. Stunzi, D. Perrin, T. Teitei and R. Harris, Aust. J. Chem., 1979, 32, 21–30 CrossRef.
- R. P. Martin, L. Mosoni and B. Sarkar, J. Biol. Chem., 1971, 246, 5944–5951 CAS.
- J. I. Lachowicz, M. Crespo-Alonso, F. Secci, C. Caltagirone, G. Alberti, R. Biesuz, J. O. Orton and V. M. Nurchi, J. Trace Elem. Med. Biol., 2018, 50, 580–588 CrossRef CAS PubMed.
- K. Reddy, R. Yedery and C. Aranha, Int. J. Antimicrob. Agents, 2004, 24, 536–547 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- J. Wang, X. Dou, J. Song, Y. Lyu, X. Zhu, L. Xu, W. Li and A. Shan, Med. Res. Rev., 2019, 39, 831–859 CrossRef CAS PubMed.
- E. de Souza Cândido, D. A. Sousa, J. C. Viana, N. G. de Oliveira-Júnior, V. Miranda and O. L. Franco, Peptides, 2014, 55, 65–78 CrossRef PubMed.
- N. Ilić, M. Novković, F. Guida, D. Xhindoli, M. Benincasa, A. Tossi and D. Juretić, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 1004–1012 CrossRef PubMed.
- W. Li, J. Tailhades, N. M. O'Brien-Simpson, F. Separovic, L. Otvos, M. A. Hossain and J. D. Wade, Amino Acids, 2014, 46, 2287–2294 CrossRef CAS PubMed.
- E. Sikorska and E. Kamysz, J. Pept. Sci., 2014, 20, 952–957 CrossRef CAS PubMed.
- T. Niidome, H. Mihara, M. Oka, T. Hayashi, T. S. Kazutoshiyoshida and H. Aoyagi, J. Pept. Res., 1998, 51, 337–345 CrossRef CAS PubMed.
- E. J. Veldhuizen, V. A. Schneider, H. Agustiandari, A. Van Dijk, J. L. Tjeerdsma-van Bokhoven, F. J. Bikker and H. P. Haagsman, PLoS One, 2014, 9, e95939 CrossRef PubMed.
- X. Wang, J. C. B. Junior, B. Mishra, T. Lushnikova, R. M. Epand and G. Wang, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1350–1361 CrossRef CAS PubMed.
- N. B. Leite, L. C. Da Costa, D. dos Santos Alvares, M. P. dos Santos Cabrera, B. M. de Souza, M. S. Palma and J. R. Neto, Amino Acids, 2011, 40, 91–100 CrossRef CAS PubMed.
- S. G. Dashper, N. M. O'Brien-Simpson, K. J. Cross, R. A. Paolini, B. Hoffmann, D. V. Catmull, M. Malkoski and E. C. Reynolds, Antimicrob. Agents Chemother., 2005, 49, 2322–2328 CrossRef CAS PubMed.
- A. Schmidtchen, M. Pasupuleti and M. Malmsten, Adv. Colloid Interface Sci., 2014, 205, 265–274 CrossRef CAS PubMed.
- N. Malanovic and K. Lohner, Pharmaceuticals, 2016, 9, 59–92 CrossRef PubMed.
- H. Kornreich-Leshem, C. Ziv, E. Gumienna-Kontecka, R. Arad-Yellin, Y. Chen, M. Elhabiri, A.-M. Albrecht-Gary, Y. Hadar and A. Shanzer, J. Am. Chem. Soc., 2005, 127, 1137–1145 CrossRef CAS PubMed.
- M. Rogosnitzky and S. Branch, BioMetals, 2016, 29, 365–376 CrossRef CAS PubMed.
- R. Domalaon, T. Idowu, G. G. Zhanel and F. Schweizer, Clin. Microbiol. Rev., 2018, 31, e00077–e00017 CrossRef CAS PubMed.
- R. M. Donlan, Emerging Infect. Dis., 2002, 8, 881–890 CrossRef PubMed.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed.
- F. Reffuveille, C. de la Fuente-Núñez, S. Mansour and R. E. Hancock, Antimicrob. Agents Chemother., 2014, 58, 5363–5371 CrossRef PubMed.
- N. Høiby, T. Bjarnsholt, C. Moser, G. Bassi, T. Coenye, G. Donelli, L. Hall-Stoodley, V. Hola, C. Imbert and K. Kirketerp-Møller, Clin. Microbiol. Infect., 2015, 21, S1–S25 CrossRef PubMed.
- U. Römling, S. Kjelleberg, S. Normark, L. Nyman, B. E. Uhlin and B. Åkerlund, J. Intern. Med., 2014, 276, 98–110 CrossRef PubMed.
- J. D. Bryers, Biotechnol. Bioeng., 2008, 100, 1–18 CrossRef CAS PubMed.
- J. C. Carmen, B. L. Roeder, J. L. Nelson, R. L. R. Ogilvie, R. A. Robison, G. B. Schaalje and W. G. Pitt, Am. J. Infect. Control, 2005, 33, 78–82 CrossRef PubMed.
- I. K. Paterson, A. Hoyle, G. Ochoa, C. Baker-Austin and N. G. Taylor, Sci. Rep., 2016, 6, 37853–37863 CrossRef CAS PubMed.
- M. Yasir, M. Willcox and D. Dutta, Materials, 2018, 11, 2468–2483 CrossRef PubMed.
- G. Batoni, G. Maisetta and S. Esin, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1044–1060 CrossRef CAS PubMed.
- L. A. Segev-Zarko, R. Saar-Dover, V. Brumfeld, M. L. Mangoni and Y. Shai, Biochem. J., 2015, 468, 259–270 CrossRef CAS PubMed.
- L. Montanaro, A. Poggi, L. Visai, S. Ravaioli, D. Campoccia, P. Speziale and C. R. Arciola, Int. J. Artif. Organs, 2011, 34, 824–831 CrossRef CAS PubMed.
- M. Erriu, C. Blus, S. Szmukler-Moncler, S. Buogo, R. Levi, G. Barbato, D. Madonnaripa, G. Denotti, V. Piras and G. Orrù, Ultrason. Sonochem., 2014, 21, 15–22 CrossRef CAS PubMed.
- N. M. Mishra, Y. Briers, C. Lamberigts, H. Steenackers, S. Robijns, B. Landuyt, J. Vanderleyden, L. Schoofs, R. Lavigne and W. Luyten, Org. Biomol. Chem., 2015, 13, 7477–7486 RSC.
- H. Rudilla, E. Fusté, Y. Cajal, F. Rabanal, T. Vinuesa and M. Viñas, Molecules, 2016, 21, 1223–1235 CrossRef PubMed.
- S. M. Ribeiro, C. de la Fuente-Núñez, B. Baquir, C. Faria-Junior, O. L. Franco and R. E. Hancock, Antimicrob. Agents Chemother., 2015, 59, 3906–3912 CrossRef CAS PubMed.
- L. Grassi, G. Maisetta, S. Esin and G. Batoni, Front. Microbiol., 2017, 8, 2409–2417 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C9DT04545G |
| This journal is © The Royal Society of Chemistry 2020 |