
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved photon absorption in dye-functionalized silicon nanocrystals synthesized via microwave-assisted hydrosilylation†
Deski
Beri
ab,
Marius
Jakoby
a,
Ian A.
Howard
 ac,
Dmitry
Busko
a,
Bryce S.
Richards
*ac and
Andrey
Turshatov
*a
ac,
Dmitry
Busko
a,
Bryce S.
Richards
*ac and
Andrey
Turshatov
*a
aInstitute of Microstructure Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344, Eggenstein-Leopoldshafen, Germany. E-mail: andrey.turshatov@kit.edu; bryce.richards@kit.edu
bChemistry Department, Universitas Negeri Padang, Jl. Hamka, Air Tawar, 25131, Indonesia
cLight Technology Institute, Karlsruhe Institute of Technology, Engesserstrasse 13, 76131 Karlsruhe, Germany
First published on 28th January 2020
Abstract
Herein, we report a method to produce luminescent silicon nanocrystals (SiNc) that strongly absorb ultraviolet–visible light (300–550 nm) and emit in the near-infrared range (700–1000 nm) with a high photoluminescence quantum yield (PLQY). Using microwave-assisted hydrosilylation and employing reactive chromophores – such as ethenyl perylene, ethynyl perylene and ethylene-m-phenyl BODIPY – we are able to achieve a 10- and 3-fold enhancement of the absorption in the blue and green spectral range, respectively. The investigated dyes function both as passivating agents and highly efficient antenna, which absorb visible light and transfer the energy to SiNc with an efficiency of >95%. This enhanced absorption leads to a significant photoluminescence enhancement, up to ∼270% and ∼140% under excitation with blue and green light, respectively. Despite the gain in absolute brightness of the emission, we demonstrate that back energy transfer from the SiNc to the dyes leads to a decrease in the PLQY for dye-modified SiNc, as compared to unmodified SiNc. The synthesis of the SiNc-dye conjugates opens up new possibilities for applications of this abundant and non-toxic material in the field of solar energy harvesting, optical sensing and bioimaging via achieving strong NIR PL excited with visible light.
Introduction
The report by Canham1 on the luminescence properties of undoped porous silicon opened up new prospects for silicon-based materials. Today, applications include optoelectronics,2,3 photonics,4 light harvesting in solar cells5 and luminescent solar concentrators,6 sensors,7,8 biological fluorescence imaging,9 as well as medical applications10 (including photodynamic therapy of cancers).11 In addition to its excellent abundance and non-toxic properties,12 silicon is also regarded as a green and environmentally-friendly material when compared with other semiconductor nanocrystals.13 The photoluminescence (PL) properties of silicon nanocrystals (SiNc) can be tuned either by controlling the particle size or by modifying the SiNc surface with a layer of surface ligands.14 Several studies have focused on size modification to produce tunable emission from orange to near-infrared (NIR) region (∼600–1100 nm) with a high photoluminescence quantum yield (PLQY) of up to ∼60%.15–17 Meanwhile, other researchers used different passivation molecules to tune the emission while maintaining a fixed core size.18,19 A recent study found that amine-functionalized silicon can be used for the synthesis of SiNc resulting in blue emission with PLQY values as high as 90%.20 In general, the explanation of the tunable emission caused by size modification is based on quantum confinement,21 whereas the effect of surface passivation is still under debate.22Although SiNc exhibit strong PL when excited with UV light, excitation with visible light results in weak PL emission due to their weak absorption at such wavelengths. Applying bare SiNc as luminophores in the aforementioned applications is less effective if only UV-excitation is exploited. One of the most promising method to enhance visible absorption of SiNc is via the use of organic dyes as a light harvesting antenna. The dyes can strongly absorb visible light and then transfer this energy to the SiNc.23 In order to realize efficient energy transfer, the dyes should be physically confined near the surface of SiNc, such as in a micellar dispersion24–26 or chemically bonded to the SiNc surface.27 For most applications, chemical attachment would be more preferable, for example, to prevent dye migration when SiNc are embedded into a solid polymer matrix. Rosso-Vasic et al.28 have exploited chemical attachment of a Ru-bipyridine complex to amine-functionalized SiNc. An efficient energy transfer of 55% from Ru-pyridine ligands to the SiNc core was observed. Locritani et al. demonstrated that bonded pyrene molecules can increase brightness of SiNc emission by almost 300% at 378 nm excitation.29–33 In addition, Fermi et al. indicated that a porphyrin chromophore can be also used as antenna for SiNc. However, a significant decrease in PLQY from 40% (for bare SiNc) to 8% (for the conjugates with the porphyrin) was observed upon excitation at 427 nm.33 Recently, SiNc with attached diphenyl anthracene were used to improve optical properties of luminescent solar concentrators utilizing SiNc PL.34
In our previous work, we have demonstrated that microwave-assisted hydrosilylation (MAH) method can produce SiNc exhibiting a PLQY of up to 40% and excellent long-term stability (6 months without decrease of PLQY).35 In this article, we describe an approach to attach functional dyes to the core of SiNc via hydrosilylation reaction using a microwave (MW) reactor. As a result, we observed significant enhancement of SiNc NIR PL upon excitation with blue and green light. We investigated the photophysical properties of the obtained conjugates and estimated efficiency of the direct energy transfer (from dye chromophore to SiNc core) and back energy transfer (from SiNc core to the dye) using absolute PLQY and PL lifetime measurements.
Results and discussion
The synthesis of hydrogen-terminated silicon nanocrystals (H-SiNc) was performed using the method described by Sun et al.36 Briefly, SiNc were produced via thermal annealing of the sub-stoichiometric SiOx(x<2) precursors at 900 °C under a forming gas (5% hydrogen in argon) ambient. Partial phase separation, triggered by thermal annealing, resulted in SiNc encapsulated in a SiO2 matrix. The SiO2 matrix was then chemically etched using concentrated hydrofluoric acid (HF) acid (48% v/v) mixed with ethanol. A nonpolar solvent, in our case toluene, was used to extract H-SiNc from the polar etching solution. Approximately fifty successful batches of H-SiNc were synthesized, always using the same conditions for thermal annealing and etching. The functionalization with 1-hexene (C6), 3-ethenyl perylene (1), 3-ethynyl perylene (2) and ethylene-m-phenyl BODIPY (3) (Fig. 1) was performed using the MAH method. The product of the hydrosilylation reaction was organic-capped SiNc that can be easily dispersed in non-polar organic solvents (toluene or hexane, for instance). Fig. S1 (ESI†) summarizes effect of temperature and reaction time on PLQY of (C6)-SiNc synthesized in the MW reactor. The results of Fig. S1† suggest an optimal reaction temperature of 230 °C – the temperature that resuled in SiNc with the highest PLQY. Longer reaction times also resulted in higher PLQY values, with a maximum value of 33 ± 6% being achieved after 120 min reaction time. Thus, these reaction conditions (temperature of 230 °C and 120 min reaction time) were used in the reaction with functional dyes (1), (2) and (3). | ||
| Fig. 1 Molecular structure of the reactive dyes used in the surface modification of SiNc: (1) 3-ethenyl perylene; (2) 3-ethynyl perylene; and (3) ethylene-m-phenyl BODIPY. | ||
Before investigation of the reaction between SiNc and the dyes, thermal degradation of the dyes in their solutions in toluene was investigated. Fig. S2 in the ESI† displays absorption spectra of dyes (1), (2) and (3) obtained before and after the 240 min thermal treatment at 230 °C. The small difference in the absorption spectra indicates degradation of approximately 6% of the dye (1), whereas the dye (2) persists undegraded – the amplitude of the absorption spectrum of the dye (2) remains almost unchanged. The dye (3) demonstrates the highest degradation rate of 36%. Thus, dyes (1) and (2) are stable enough under the chosen reaction conditions. On the whole, all investigated dyes can be used for the surface modification of SiNc, however stronger degradation rate should be taken into account if the dye (3) is used in the hydrosilylation process.
The thermal hydrosilylation reaction between dyes and H-SiNc described previously in the literature utilized a large excess of a reactive dye (>100 mg of a dye per 5 ml of the reaction mixture).30 This large amount of the dye guarantees the high surface density of attached chromophore, but also leads to the inefficient dye consumption as the reaction yield is relatively low. In order to minimize the amount of reactive dyes (1), (2) and (3) in the reaction with H-SiNc three different methods were investigated:
(i) 2.0 mg of the dye (all dyes from Fig. 1 were tested hereinafter at the same reaction conditions) was dissolved in 5 ml dispersion of H-SiNc in toluene and reacted in the MW reactor for 120 min at 230 °C. However, the resulting product displayed only weak NIR emission originated from the SiNc. We assumed that dye molecules afford only a partial surface coverage of SiNc and weakly-protected SiNc are easily oxidized by oxygen that leads to reduced PL intensity;
(ii) 2.0 mg of the dye was dissolved in 5 ml dispersion of H-SiNc in (C6) and reacted in the MW reactor for 120 min at 230 °C. The product of this reaction displayed only a very small amount of the attached dye. Our results indicate that the dye cannot compete with the excess of (C6) in the reaction mixture and, thus, only limited amount of the dye reacts with the surface of H-SiNc;
(iii) In order to overcome the limitations of the methods (i) and (ii), 2.0 mg of the dye was initially reacted with H-SiNc in toluene for 120 min 230 °C. Then, 5.0 ml of (C6) was injected into solution and the hydrosilylation reaction was performed for another 120 min at 230 °C. The method (iii) allowed us to synthesize SiNc with dye molecules covalently attached to the surface and with strong NIR PL. The obtained nanocrystals exhibit a relatively broad size distribution (Fig. S3, ESI†) with average hydrodynamic diameter measured by dynamic light scattering (DLS) of 4.2 nm (for (C6)-SiNc), 5.2 nm (for (C6)-(1)-SiNc), 4.6 nm (for (C6)-(2)-SiNc) and 6.5 nm (for (C6)-(3)-SiNc).
The UV/VIS absorption and photoluminescence excitation (PLE) spectra of the (C6)-SiNc in hexane exhibit the characteristic behavior of SiNc with the gradual decrease of the absorbance at longer wavelengths (Fig. 2a). As all dyes demonstrate a local absorption minimum at 300 nm (absorption, PL and PLE spectra for dyes (1), (2) and (3) dissolved in hexane are presented in Fig. S4, ESI†), we assumed that the absorbance at 300 nm (A300 nm) characterizes the optical absorption of SiNc without interference with the dye absorption. Therefore, for the quantification of experimental results all absorption spectra were normalized using the A300 nm absorbance with the extinction coefficient of SiNc expressed as
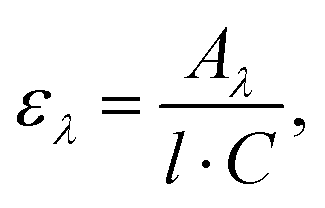 | (1) |
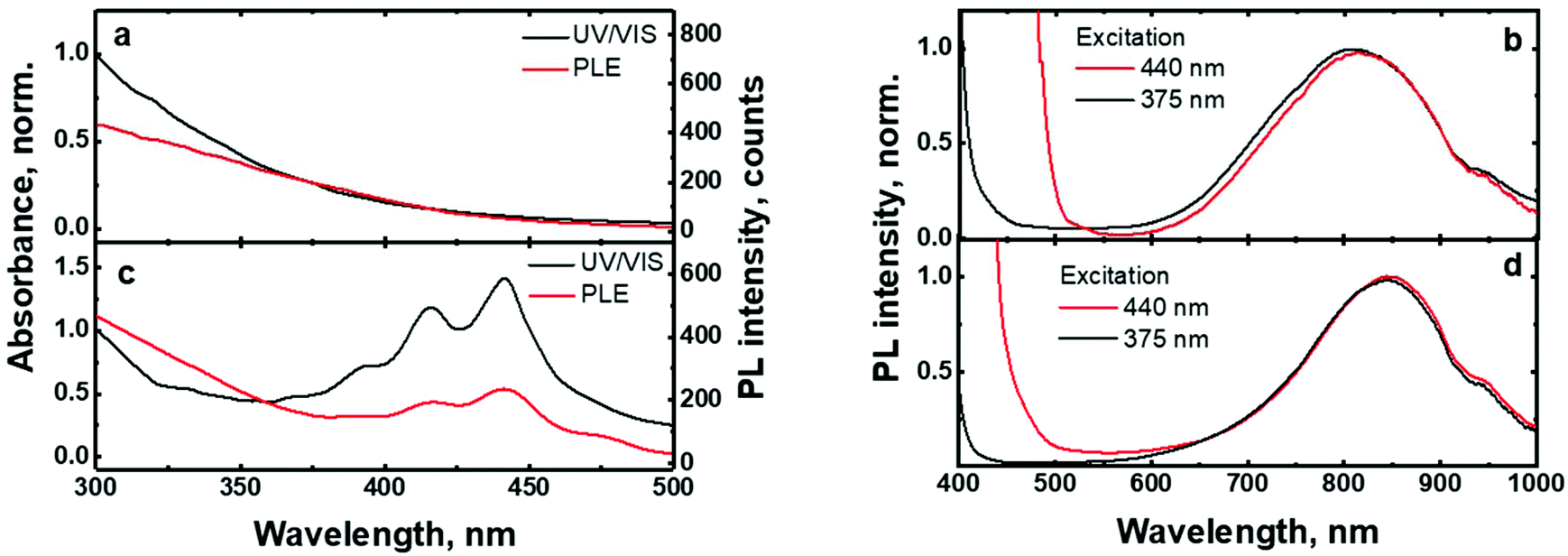 | ||
| Fig. 2 (a): Normalized absorption and PLE spectra of (C6)-SiNc; (b): PL spectra of (C6)-SiNc at different excitation wavelengths. The edge of emission of light emitting diodes (LEDs) used for excitation is observed at λ < 500 nm; (c): normalized absorption and PLE spectra of (C6)-(1)-SiNc; (d): PL spectra of (C6)-(1)-SiNc at different excitation wavelengths. The edge of emission of LEDs used for excitation is observed at λ < 500 nm. PLE spectra were measured at the emission wavelength of 800 nm. | ||
| Excitation (λ, nm) | (C6), ε, cm−1 (×10−4) | (C6)-(1), ε, cm−1 (×10−4) | (C6)-(2), ε, cm−1 (×10−4) | (C6)-(3), ε, cm−1 (×10−4) |
|---|---|---|---|---|
| 300 | 0.72 | 0.73 | 0.75 | 0.71 |
| 375 | 0.17 | 0.26 | 0.36 | 0.17 |
| 405 | 0.09 | 0.34 | 0.47 | 0.09 |
| 440 | 0.05 | 0.49 | 0.65 | 0.04 |
| 515 | 0.01 | 0.03 | 0.07 | 0.03 |
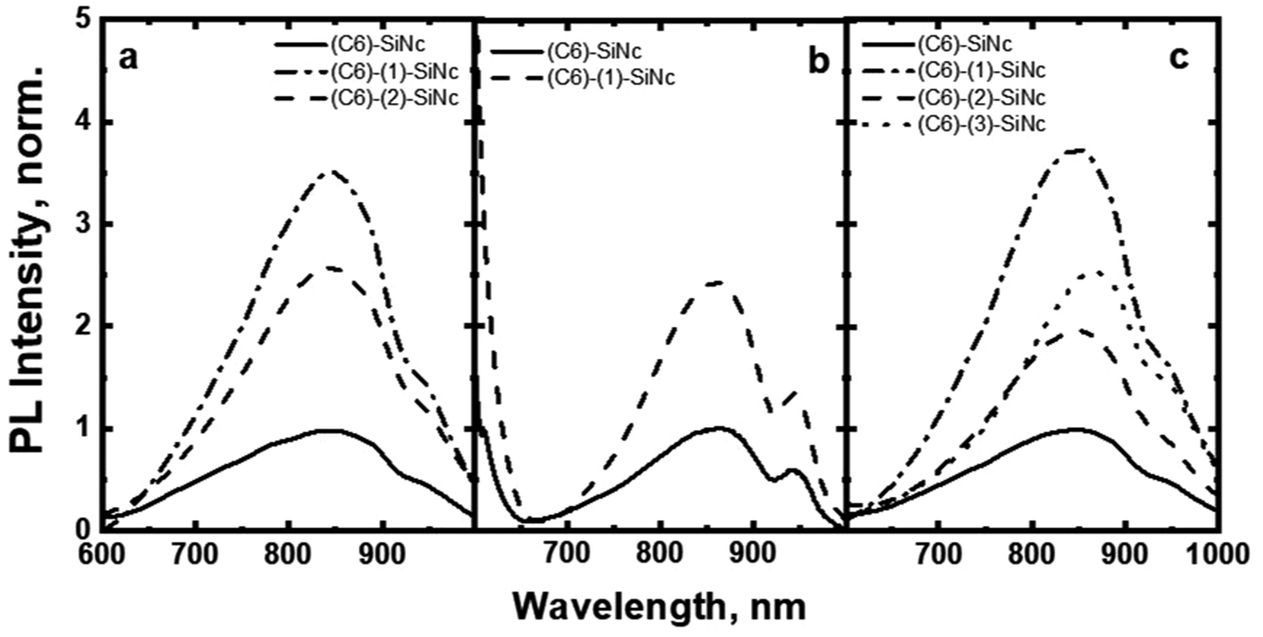
The excitation of (C6)-SiNc using different wavelengths leads to PL spectra with similar shape: full width half maximum (FWHM) of 170 nm and the position of the PL maximum of 810 nm (Fig. 2b). The PLQY values obtained at different excitation wavelengths are slightly different (as shown in the Fig. 3a). The highest PLQY values of 33 ± 6% were measured for the excitation wavelengths of 300 and 375 nm, whereas lower values of 30 ± 5% and 28 ± 5% were found for excitation with wavelengths 405 and 440 nm, respectively (the error bars indicate maximum and minimum values measured for five independently synthesized samples).
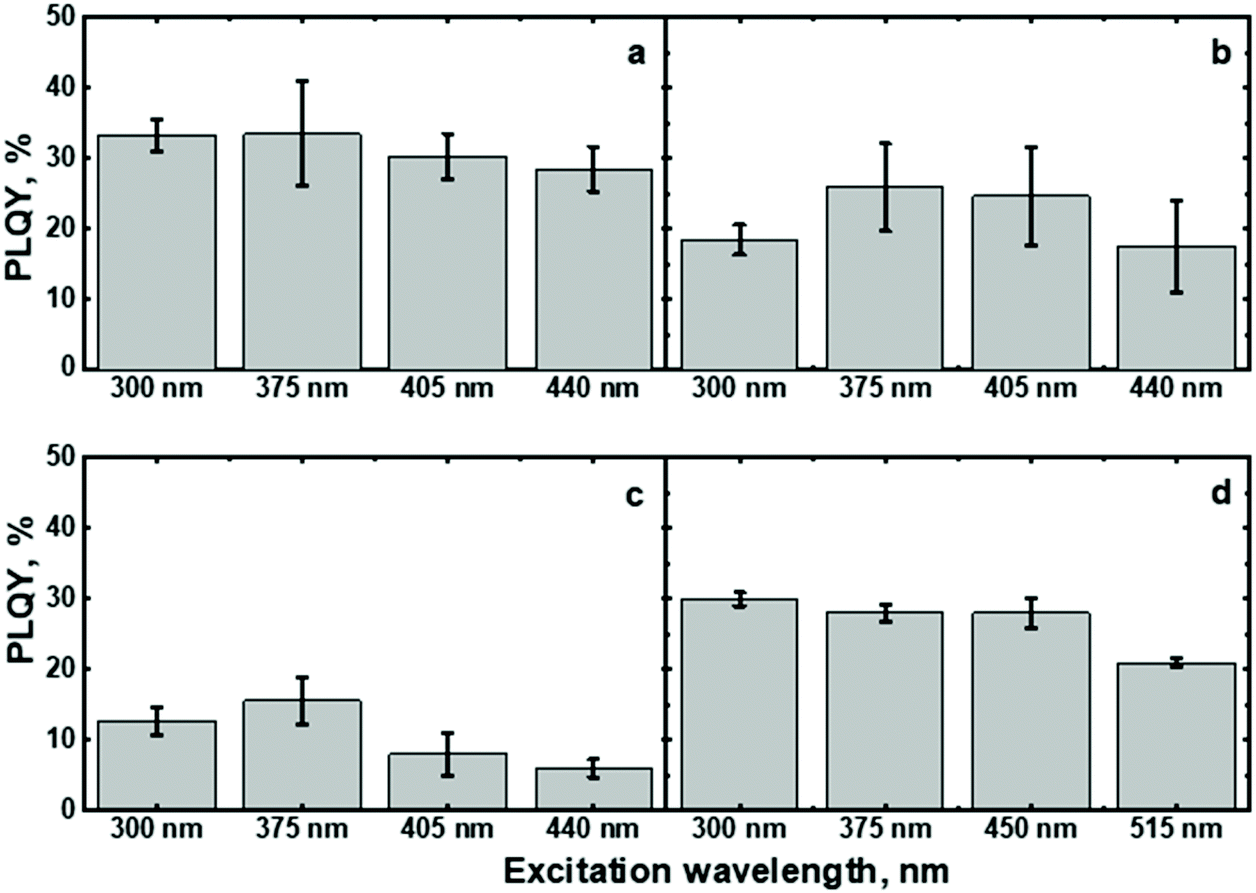 | ||
| Fig. 3 PLQY values of (a): (C6)-SiNc, (b): (C6)-(1)-SiNc, (c): (C6)-(2)-SiNc and (d): (C6)-(3)-SiNc measured at different excitation wavelengths. The PLQY values are the result of averaging for five independent synthetic batches. The error bars indicate maximum and minimum values measured for five independent synthetic batches. | ||
3-Ethenyl perylene
In contrast to the unmodified (C6)-SiNc, absorption and PLE spectra of (C6)-(1)-SiNc demonstrate the shape that is the combination of both SiNc and dye (1) spectra with the maximum of 442 nm (Fig. 2c). The absorption maximum of (C6)-(1)-SiNc at 442 nm exhibits the hypsochromic shift compared to the unreacted dye (1) with the absorption maximum at 453 nm. This effect can be explained by the reduction of the double bond after the hydrosilylation reaction with H-SiNc and, thus, breaking up the conjugation between the perylene core and the double bond. (C6)-(1)-SiNc demonstrate similar with (C6)-SiNc extinction coefficient ε300 nm and enhanced absorption at longer wavelengths (Table 1). For instance, we observed ∼10-fold enhancement of the extinction coefficient at 440 nm (ε440 nm). The PLE spectrum (Fig. 2c) evidently highlights the sensitization of SiNc with the blue-absorbing dye attached to the surface of the nanocrystals. The shape of the PLE spectrum corresponds to the absorption spectrum of the (C6)-(1)-SiNc with significant contribution of the dye absorption.A physical mixture of (C6)-SiNc and the dye (1) displays a similar absorption spectrum, however, the PLE spectrum doesn't indicate any contribution of the dye absorption to the excitation of NIR PL of SiNc (Fig. S6, ESI†). This fact confirms negligible sensitization of SiNc with the unbounded dye (1). The PL spectra of (C6)-(1)-SiNc (Fig. 2d) excited with different excitation wavelengths demonstrate weak PL (between 450 and 500 nm) corresponding to the dye (1) and strong NIR emission of SiNc with the FWHM of 175 nm and the PL maximum of 845 nm. This might be due to the efficient energy transfer from the dye to SiNc, however an additional confirmation for the energy transfer (for instance measurements of PL decay times for dye (1)) is required. A nanosecond PL decay for the attached dye (1) will be discussed in the end of the current section. Earlier we envisioned that light with wavelength of 300 nm allows selective excitation of the SiNc core without contribution of the dye absorption. Unexpectedly, the PLQY value of (C6)-(1)-SiNc (14 ± 5%) measured at 300 nm excitation is smaller than PLQY of the (C6)-SiNc (33 ± 6%) (Fig. 3a and b). Firstly, an enhanced surface oxidation of SiNc after reaction with the dye (1) was considered as a possible reason for the decreased PLQY. The photoluminescence of SiNc is extremely sensitive to the presence of surface chemical defects such as bridged oxygen Si–O–Si and hydroxyl groups Si–OH. To examine this hypothesis, infrared (IR) spectra of (C6)-SiNc and SiNc modified with the dye (1) were measured. There are two characteristic peaks that allowed the comparison of the surface chemical defects. The peak at 800 cm−1 can be attributed to Si–C (stretching) vibration, whereas the broad peak at 960–1140 cm−1 to Si–O (stretching) vibration (Fig. S7, ESI†).25,37,38 One would expect stronger surface oxidation and large number of surface chemical defects for a large R-value (eqn (2))
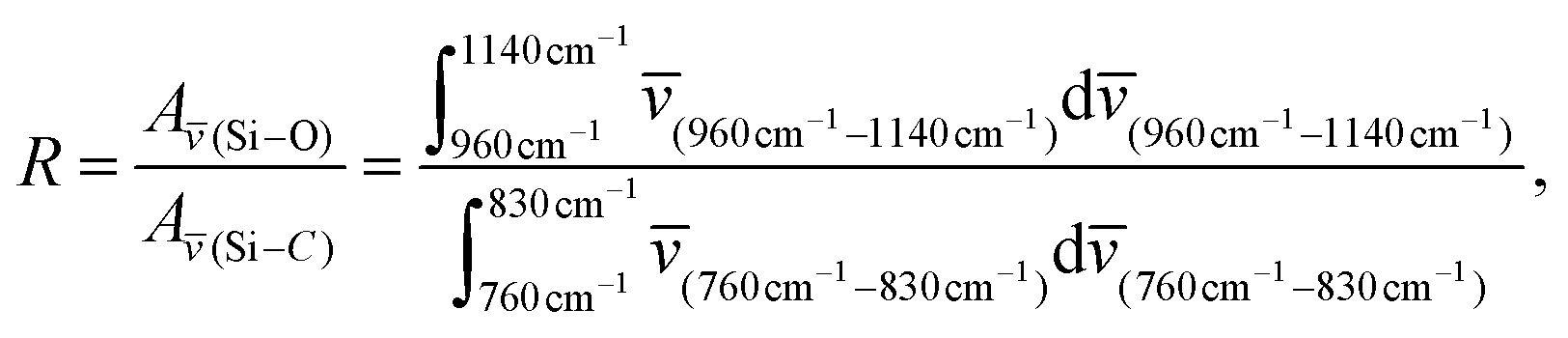 | (2) |
![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) (Si–O) – integrated absorbance area of the Si–O peak, A(Si–C) – integrated absorbance area of the Si–C peak, and – vibration energy.
(Si–O) – integrated absorbance area of the Si–O peak, A(Si–C) – integrated absorbance area of the Si–C peak, and – vibration energy.
Indeed, the (C6)-SiNc and (C6)-(1)-SiNc samples exhibit similar R-values of 2.6 (for (C6)-SiNc) and 2.5 (for (C6)-(1)-SiNc) (Fig. S7†). Thus, R-value indicates that SiNc have similar number of surface chemical defects. Moreover, in the case of the stronger oxidation a strong hypsochromic shift in the PL spectrum of SiNc is usually observed due to emission from trap states.22,39 The (C6)-SiNc and (C6)-(1)-SiNc display, however, similar PL spectra. Therefore, it is unlikely that the reaction with the dye enhances the surface oxidation of SiNc.
In order to explain the decrease of PLQY measured at 300 nm excitation, we assumed that the dye might quench the PL of SiNc. More specifically, the back-energy transfer (BET) from SiNc (donor) to the triplet state of the dye (acceptor) was anticipated. The position of perylene triplet state (∼800–850 nm) coincides well with the broad SiNc NIR emission (with PL maximum at 845 nm).40 Thus, the perylene triplet state might enable such an energy transfer that ultimately reduces the PLQY. The efficient energy transfer between quantum dots and triplet state of dyes attached to its surface was described in literature.41–43 For instance, Mongin et al. reported Dexter-like triplet–triplet energy transfer between CdSe nanocrystals and a surface-anchored polyaromatic dye.43 Recently, Xia et al. described triplet–triplet annihilation upconversion utilizing energy transfer from SiNc to the triplet sate of diphenylanthracene anchored with the SiNc surface.44
To validate this idea, the PL decays of the SiNc emission (Fig. 4a) were measured. All investigated SiNc exhibit PL decays with a multi-exponential behavior due to size polydispersity. Indeed, Mastronardi et al. investigated narrow fractions of SiNc and demonstrated that PL lifetime rises with the increase of the nanocrystal size.16 For simplicity, the PL decays were fitted with double-exponential function and the amplitude-averaged PL lifetime was assessed using eqn (3):
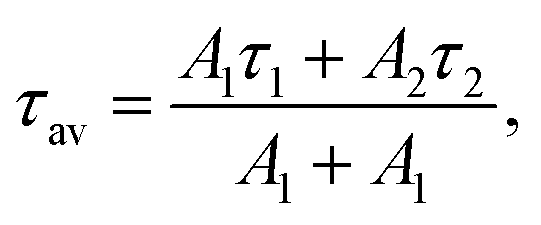 | (3) |
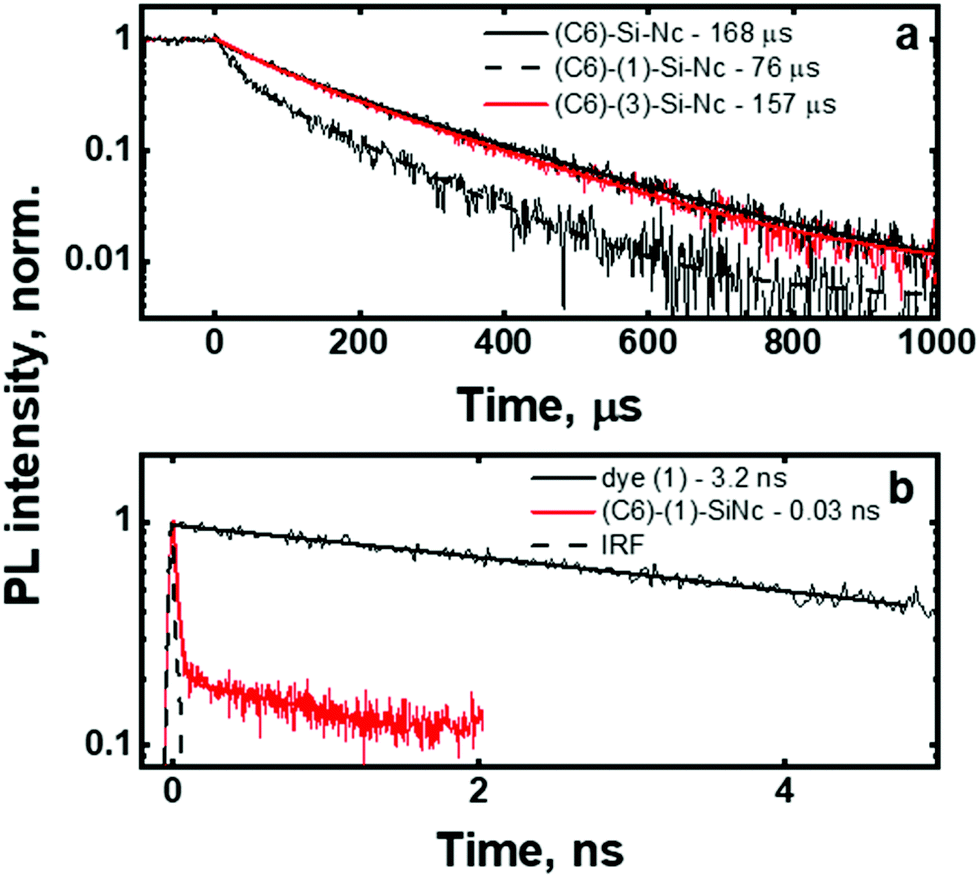 | ||
| Fig. 4 (a): Long-time (μs) PL decays for ligand functionalized SiNc excited at 405 nm and measured at 810 nm. (b): Short-time (ns) PL decays of dye (1) and (C6)-(1)-SiNc; excited at 440 nm and measured at 450–550 nm. IRF is the instrument response function. | ||
The (C6)-SiNc demonstrate relatively long PL lifetime of 168 μs. This value is in good agreement with the previously reported PL lifetime values.25 In contrast, (C6)-(1)-SiNc display significantly shorter lifetime of 76 μs. Thus, quenching of the SiNc NIR PL for (C6)-(1)-SiNc is confirmed. The efficiency of BET can be calculated with eqn (4) or eqn (5):
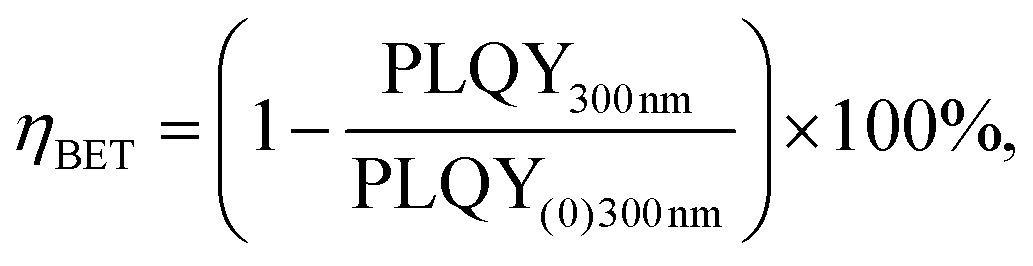 | (4) |
or
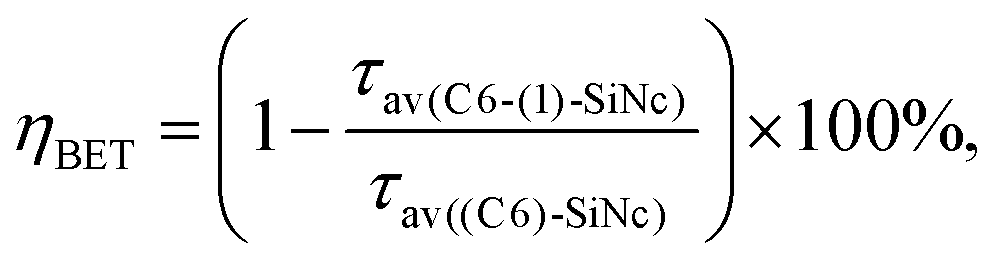 | (5) |
Under the dye excitation (at 440 nm), an additional loss mechanism reducing the PLQY of SiNc needs to be considered. If the efficiency of the direct energy transfer (DET) between the dye (donor) and SiNc (acceptor) is significantly less than 100% the relationship PLQY440 nm < PLQY300 nm is observed. However, Fig. 3b demonstrates that PLQY440 nm ≈ PLQY300 nm. To explain this result and to assess the value of the DET, we focused on the measurements of the ns-decay of the dye (1) before and after reaction with H-SiNc (Fig. 4b). The ns-decay after reaction exhibits a double-exponential behavior with two distinguished lifetimes. The short component with the lifetime of 0.03 ns (and amplitude of 81%) and the long component with the lifetime of 3 ns (and amplitude of 19%) can correspond to quenched and unquenched states of the dye (1), respectively. The observation of the unquenched dye might be due to partial incorporation of the dye molecules into a thin hydrocarbon corona surrounding SiNc.27 Considering the much longer distance between the dye and SiNc surface in this case, we assumed lack of the DET for a small fraction of the dye attached to the surface of SiNc. Excluding this small fraction, the DET efficiency of 99% was calculated using eqn (6):
 | (6) |
Taken together, these results predict an enhancement of ∼350% for NIR PL of SiNc when excited with a blue light source (440 nm). The 350% value is comprised of the 10-fold enhancement due to absorption and the 55% loses due to BET from SiNc to the dye triplet. To demonstrate such an enhancement, the solutions with bare (C6)-SiNc and dye-functionalized (C6)-(1)-SiNc with similar optical density measured at 300 nm were prepared. As the dye demonstrates an absorption minimum at 300 nm, we assumed that solutions with similar optical density at 300 nm have similar concentration of SiNc. The comparison of PL of solutions with similar concentration of SiNc is presented in Fig. 5a, b and c for different excitation sources. A simple normalization using PL of bare SiNc (all solutions were measured at identical conditions for one light source) is an approach to highlight the enhancement achieved via functionalization with the organic dyes. The enhancement of 270% of the NIR PL in case of (C6)-(1)-SiNc (Fig. 5a) was observed, which is in good agreement with the predicted value.
 | ||
| Fig. 5 (a): PL under excitation with LED440 nm of (C6)-SiNc, (C6)-(1)-SiNc and (C6)-(2)-SiNc; (b): PL under excitation with LED 515 nm of (C6)-SiNc and (C6)-(3)-SiNc; (c): PL under broadband excitation (435–550 nm) of (C6)-SiNc, (C6)-(1)-SiNc, (C6)-(2)-SiNc and (C6)-(3)-SiNc. All dispersions have similar absorbance of 0.3 at 300 nm (A300). The PL spectra were normalized to emission of (C6)-SiNc. | ||
3-Ethynyl perylene
We next examined an effect of the triple bond on the hydrosilylation reaction with the dye (2). The absorption and PLE spectra of (C6)-(2)-SiNc display the combination of absorption spectra of SiNc and dye (2) (Fig. 6a) with the maximum at 441 nm. Similar with the dye (1), the physical mixture of (C6)-SiNc and dye (2) doesn't show contribution of thedye (2) absorption to the PLE spectrum (Fig. S6, ESI†). The extinction coefficient ε440 nm for (C6)-(2)-SiNc is slightly higher than the coefficient ε440 nm for (C6)-(1)-SiNc (Table 1). This difference might indicate that 3-ethynylperylene (perylene with triple bond) has higher activities in the hydrosilylation reaction then ethenyl perylene (perylene with double bond). The NIR PL of SiNc exhibits similar spectra under excitation with different wavelengths, for example the PL maximum of 832 nm and FWHM of 170 nm (Fig. 6b). The PLQY value of 12 ± 4% was obtained for (C6)-(2)-SiNc at 300 nm excitation (Fig. 3c). The value of PLQY is lower than PLQY for SiNc modified with dye (1). This observation correlates with the shorter PL lifetime of 72 μs measured for NIR emission of (C6)-(2)-SiNc (Fig. S9a†) as compared with PL lifetime for (C6)-(1)-SiNc. These results confirm more efficient BET from SiNc to the triplet state of the dye (2) caused by the conjugation of the perylene core with the double bond. The value ηBET = 63% was calculated using eqn (4), whereas the value of ηBET calculated with eqn (5) was estimated as 57%.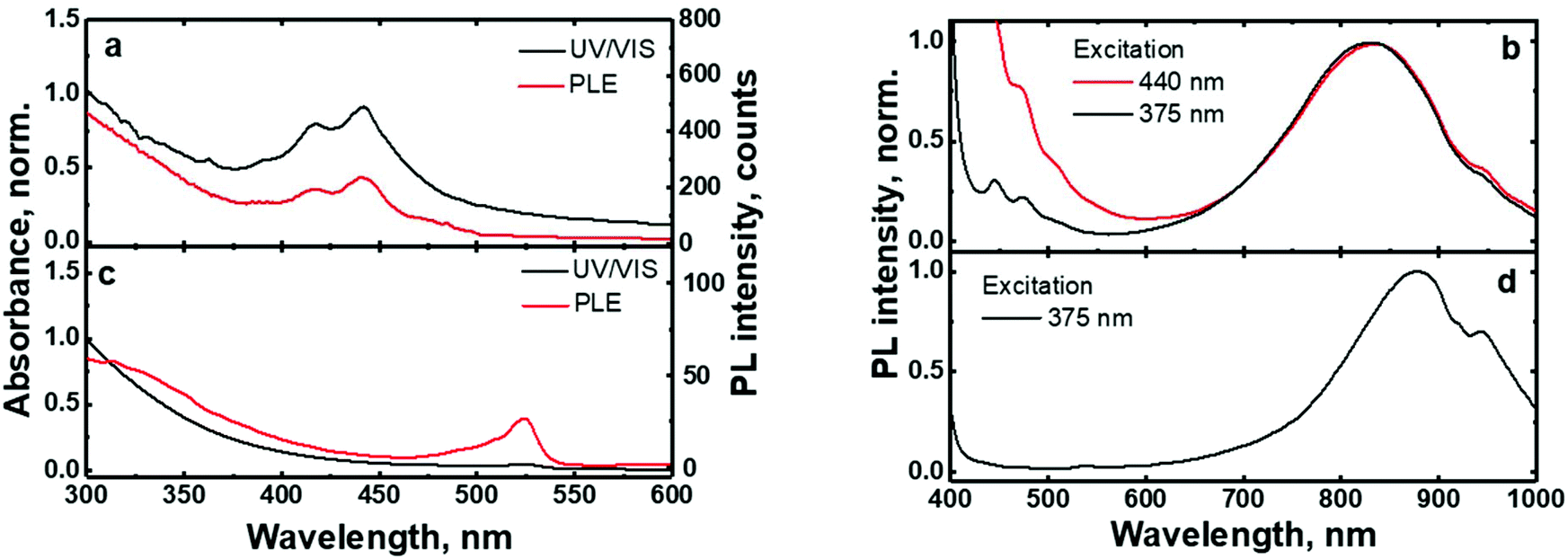 | ||
| Fig. 6 (a): Normalized absorption and PLE spectra of (C6)-(2)-SiNc; (b): PL spectra of (C6)-(2)-SiNc at different excitation wavelengths. The edge of emission of LEDs used for excitation is observed at λ < 500 nm; (c): normalized absorption and PLE spectra of (C6)-(3)-SiNc; (d): PL spectra of (C6)-(3)-SiNc excited at 375 nm. The edge of emission of LED used for excitation is observed at λ < 400 nm. PLE spectra were measured at the emission wavelength of 800 nm. | ||
In contrast to (C6)-(1)-SiNc, the excitation of (C6)-(2)-SiNc leads to the dye emission observed at 450–500 nm (Fig. 6b). The measurement of ns-lifetime (for the dye emission in the range 440–560 nm) displays decay with lifetime of 3.8 ns (Fig. S9b†). This long lifetime indicates that DET from the dye to SiNc is considerably less efficient than DET in the system (C6)-(1)-SiNc. The enhancement factor for the (C6)-(2)-SiNc system should be logically lower than the enhancement in case of (C6)-(1)-SiNc due to less efficient DET and the pronounced BET observed for the (C6)-(2)-SiNc conjugate. Indeed, Fig. 5a demonstrates only 160% enhancement of NIR PL achieved with excitation at 440 nm.
Ethylene-m-phenyl BODIPY
In order to examine the possibility of the DET between a dye absorbing in the green range of the visible spectrum and SiNc, we performed hydrosilylation reaction with the dye (3). The dye (3) demonstrates the absorption maximum at 514 nm which can be clearly seen in the absorption and PLE spectra of (C6)-(3)-SiNc (Fig. 6c). The NIR PL of SiNc exhibits the spectrum with the PL maximum of 878 nm and FWHM of 172 nm (Fig. 6d). It should be noted that we were not able to provide efficient surface anchoring of the dye (3) using the synthetic conditions that were similar with ones for dyes (1) and (2). We obtained only the 3-folds increase of the extinction coefficient at 515 nm. Thus, the synthetic procedure in case of dye (3) requires a further optimization. However, even with this amount of the attached dye we observed NIR PL enhancement of ∼140% for excitation at 515 nm (Fig. 5b). Such a significant enhancement can be explained by two effects. Firstly, the PLQY of SiNc is high (28 ± 5% under 300 nm excitation (Fig. 4d)). This result correlates with the long lifetime of NIR PL (157 μs, as presented in Fig. 5a). Thus, the less pronounced BET with ηBET = 7% (calculated with eqn (5)) is observed in the (C6)-(3)-SiNc system due to the lower dye surface concentration and the higher energy of the BODIPY triplet (∼750 nm).47,48 Secondly, the ns-decay measured for the range 550–600 nm indicates strong quenching of BODIPY luminescence with ηDET = 94%. In contrast to the unreacted dye (3) with lifetime of 7.2 ns (Fig. S8†), the (C6)-(3)-SiNc demonstrates the average PL lifetime of BODIPY emission of 0.4 ns (Fig. S10†). As the reduction of the double bond after the hydrosilylation reaction might lead to only minor change of the lifetime (due to the weak conjugation between the BODIPY core and the double bond), we assumed that the lifetime of 7.2 ns can be used in the calculation of the DET efficiency. Therefore, via having only small losses due to energy transfer processes, the absorption enhancement can be efficiently utilized in case of (C6)-(3)-SiNc.Last-but-not-least, we investigated the NIR PL of sensitized samples under the broad spectral excitation that can mimic, for instance, the excitation with the solar radiation. In order to compare the PL, the concentration of all solutions were tuned to have the similar absorbance at 300 nm. Fig. 5c displays the PL spectra of four solutions under the broadband excitation (we used a white LED together with a 550 nm short-pass filter). The enhancement of 280%, 100%, and 160% for the NIR PL of (C6)-(1)-SiNc, (C6)-(2)-SiNc, and (C6)-(3)-SiNc samples was achieved, respectively.
Conclusions
The two-step hydrosililation reaction performed in a MW reactor allowed us to attach functional dyes (perylene and BODIPY) to the surface of SiNc. A low amount of the functional dyes (2 mg) was used in the reaction in order to achieve 10-fold absorption enhancement at 440 nm (for the product of reaction between 3-ethenyl perylene and H-SiNc) and 3-fold absorption enhancement at 515 nm (for the product of reaction between ethylene-m-phenyl BODIPY and H-SiNc). The efficient direct energy transfer from perylene (ηDET = 99%) and BODIPY (ηDET = 99%) chromophores to SiNc was measured. In general, the dye attachment leads to the decrease of PLQY for NIR PL of SiNc due to the back energy transfer from the SiNc core to the dye. The back energy transfer was stronger in case of perylene, whereas less pronounced in case of the BODIPY chromophore. Overall, the phenomenon of the back energy transfer requires additional investigation and will be described in our future publication. The PL enhancement of 270% and 140% was demonstrated for blue- and green-light excitation of the modified SiNc, respectively. The synthesis of stable SiNc with enhanced NIR PL excited with visible light paves the way for applications of this abundant and non-toxic material in field of solar energy harvesting, optical sensing and bioimaging.Experimental section
Materials
Silicon monoxide (99.9%, 325 mesh) was purchased from Sigma-Aldrich, hydrofluoric acid (48%) was purchased from Fisher Scientific, ethanol 98%, methanol (HPLC grade), toluene (99%+) were purchased from Merck, 1-hexene (99%) was purchased from Acros, n-hexane spectroscopic grade was purchased from Alfa Aesar, 3-ethenylperylene was purchased from Fluorochem. Ltd, UK, and 3-ethynylperylene was purchased from Lumiprobe GmbH, Germany. All chemicals were used without further purification. Ethylene-m-phenyl BODIPY (MS (APCI): m/z (%) 406.3(99.2) M−) was synthesized in agreement with Turshatov et al.40,49 using 3-vinylbenzaldehyde (Sigma-Aldrich) as a precursor.Synthetic method
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used to precipitate SiNc with attached dyes. The obtained cloudy solution was centrifuged at 10000 rpm for 20 minutes; the precipitate was collected, washed three times with methanol and re-dispersed in n-hexane. The dye attachment was controlled using thin-layer chromatography (TLC). The luminescent spot (with Rf = 0.6–0.9) was clearly observed in the control experiment with the unreacted dyes. Under UV excitation of TLC plate with (C6)-(1)-SiNc, (C6)-(2)-SiNc, (C6)-(3)-SiNc conjugates we did not observe any luminescent spots that might correspond to the unreacted dyes.
:1) was used to precipitate SiNc with attached dyes. The obtained cloudy solution was centrifuged at 10000 rpm for 20 minutes; the precipitate was collected, washed three times with methanol and re-dispersed in n-hexane. The dye attachment was controlled using thin-layer chromatography (TLC). The luminescent spot (with Rf = 0.6–0.9) was clearly observed in the control experiment with the unreacted dyes. Under UV excitation of TLC plate with (C6)-(1)-SiNc, (C6)-(2)-SiNc, (C6)-(3)-SiNc conjugates we did not observe any luminescent spots that might correspond to the unreacted dyes.
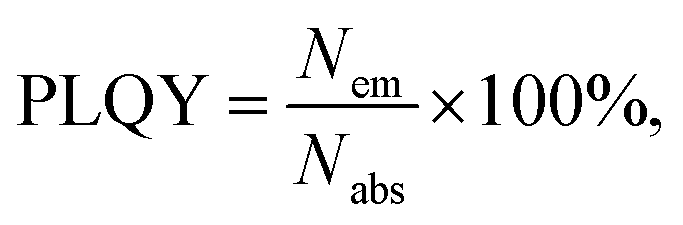 | (7) |
In detail, the LED beam was focused by a lens and directed into an integrating sphere (Labsphere) with a diameter of 15 cm. An optical fiber with a diameter of 1 mm (FP1000URT, Thorlabs) was used to collect the emission from integrating sphere and transfer it to the spectrometer (AvaSpec-ULS2048x64TEC, Avantes). Absorption measurements were performed for both direct and indirect excitation, as well as for the empty sphere. The integration time was adjusted in accordance with the excitation power; longer integration time was used for low power LED300 nm (414.1 μW) and shorter integration time was used for LED375 nm, LED440 nm and LED515 nm with power 1.5, 2.2 and 2.0 mW, respectively. All raw measured spectra were recalculated to give power spectrums using the integration time. The spectral response of the whole system was calibrated using calibration lamp (HL-3plus-INT-Cal, Ocean Optics).
In the experiment with broad excitation via a white-light LED, a short-pass filter with a cut-off edge of 550 nm was used to filter the excitation light. A long-pass filter with a cut-off edge of 600 nm was used in the front of the detector to block the LED emission. All measurements were performed for dispersions with the same absorbance A300 nm = 0.3. The brightness enhancement (Δbrightness) is determined with the following equation (eqn (8)):
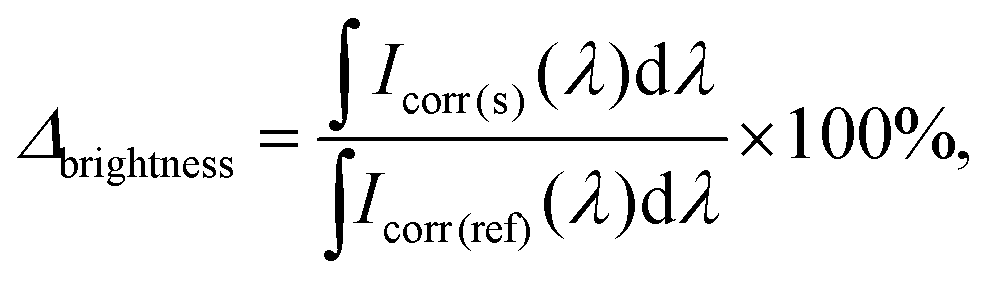 | (8) |
For the NIR PL lifetime measurements, the multichannel scaling (TimeHarp 260, PicoQuant) was used. A modulated 405 nm, 80 mW DL-7146-1012S laser diode from Roithner Laser Technique was used to excite sample in four edge polished cuvette, The TTL signal from laser diode was controlled with a delay generator (DG645, Stanford Research Systems). A double monochromator (DTMS300, Bentham) was used for the collection of photoluminescence at the specific 810 nm wavelength. The emission was detected by a PMT (R928P, Hamamatsu), mounted in cooled housing (CoolOne, Horiba).
Nanosecond PL lifetimes were measured using a streak camera (Hamamatsu C10910) coupled to a spectrometer (Acton SpectraPro SP2300) with an instrument response function of 320 ps at a time base of 2 ns. For the excitation, a mode-locked Ti:sapphire laser (Coherent, Chameleon Ultra) with a pulse width of 140 fs and repetition rate of 80 MHz was used.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support provided by the Helmholtz Association: (i) a Recruitment Initiative Fellowship for BSR; (ii) the funding of chemical synthesis equipment from the Helmholtz Materials Energy Foundry (HEMF); and (iii) the Science and Technology of Nanosystems research programme. DB acknowledges the Directorate General of Higher Education (DGHE) of the Republic of Indonesia for the PhD fellowship 101.2/E4.4/K/2015.Notes and references
- L. T. Canham, Appl. Phys. Lett., 1990, 57, 1046–1048 CrossRef CAS.
- L. Pavesi, L. Dal Negro, C. Mazzoleni, G. Franzo and F. Priolo, Nature, 2000, 408, 440–444 CrossRef CAS PubMed.
- F. Maier-Flaig, J. Rinck, M. Stephan, T. Bocksrocker, M. Bruns, C. Kubel, A. K. Powell, G. A. Ozin and U. Lemmer, Nano Lett., 2013, 13, 475–480 CrossRef CAS.
- F. Priolo, T. Gregorkiewicz, M. Galli and T. F. Krauss, Nat. Nanotechnol., 2014, 9, 19–32 CrossRef CAS PubMed.
- C. Y. Liu, Z. C. Holman and U. R. Kortshagen, Nano Lett., 2009, 9, 449–452 CrossRef CAS PubMed.
- F. Meinardi, S. Ehrenberg, L. Dhamo, F. Carulli, M. Mauri, F. Bruni, R. Simonutti, U. Kortshagen and S. Brovelli, Nat. Photonics, 2017, 11, 177–185 CrossRef CAS.
- G. Konstantatos and E. H. Sargent, Nat. Nanotechnol., 2010, 5, 391–400 CrossRef CAS PubMed.
- S. Chan, Y. Li, L. J. Rothberg, B. L. Miller and P. M. Fauchet, Mater. Sci. Eng., C, 2001, 15, 277–282 CrossRef.
- E. J. Henderson, A. J. Shuhendler, P. Prasad, V. Baumann, F. Maier-Flaig, D. O. Faulkner, U. Lemmer, X. Y. Wu and G. A. Ozin, Small, 2011, 7, 2507–2516 CAS.
- D. Kovalev and M. Fujii, Adv. Mater., 2005, 17, 2531–2544 CrossRef CAS.
- J. Fuzell, A. Thibert, T. M. Atkins, M. Dasog, E. Busby, J. G. C. Veinot, S. M. Kauzlarich and D. S. Larsen, J. Phys. Chem. Lett., 2013, 4, 3806–3812 CrossRef CAS.
- J. H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- F. Erogbogbo, K. T. Yong, I. Roy, G. X. Xu, P. N. Prasad and M. T. Swihart, ACS Nano, 2008, 2, 873–878 CrossRef CAS PubMed.
- M. Dasog, G. B. De los Reyes, L. V. Titova, F. A. Hegmann and J. G. C. Veinot, ACS Nano, 2014, 8, 9636–9648 CrossRef CAS PubMed.
- C. M. Hessel, D. Reid, M. G. Panthani, M. R. Rasch, B. W. Goodfellow, J. W. Wei, H. Fujii, V. Akhavan and B. A. Korgel, Chem. Mater., 2012, 24, 393–401 CrossRef CAS.
- M. L. Mastronardi, F. Maier-Flaig, D. Faulkner, E. J. Henderson, C. Kubel, U. Lemmer and G. A. Ozin, Nano Lett., 2012, 12, 337–342 CrossRef CAS PubMed.
- R. Anthony and U. Kortshagen, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 115407 CrossRef.
- M. Dasog, J. Kehrle, B. Rieger and J. G. C. Veinot, Angew. Chem., Int. Ed., 2016, 55, 2322–2339 CrossRef CAS PubMed.
- M. H. Balcı, R. Sæterli, J. Maria, M. Lindgren, R. Holmestad, T. Grande and M.-A. Einarsrud, Dalton Trans., 2013, 42, 2700–2703 RSC.
- Q. Li, T. Y. Luo, M. Zhou, H. Abroshan, J. C. Huang, H. J. Kim, N. L. Rosi, Z. Z. Shao and R. C. Jin, ACS Nano, 2016, 10, 8385–8393 CrossRef CAS PubMed.
- G. Ledoux, O. Guillois, D. Porterat, C. Reynaud, F. Huisken, B. Kohn and V. Paillard, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 15942–15951 CrossRef CAS.
- R. Sinelnikov, M. Dasog, J. Bearnish, A. Meldrum and J. G. C. Veinot, ACS Photonics, 2017, 4, 1920–1929 CrossRef CAS.
- R. Pandya, R. W. MacQueen, A. Rao and N. J. L. K. Davis, J. Phys. Chem. C, 2018, 122, 22330–22338 CrossRef CAS.
- G. Beane, K. Boldt, N. Kirkwood and P. Mulvaney, J. Phys. Chem. C, 2014, 118, 18079–18086 CrossRef CAS.
- F. Erogbogbo, C. W. Chang, J. May, P. N. Prasad and M. T. Swihart, Nanoscale, 2012, 4, 5163–5168 RSC.
- G. H. Pan, A. Barras, L. Boussekey, X. S. Qu, A. Addad and R. Boukherroub, Langmuir, 2013, 29, 12688–12696 CrossRef CAS PubMed.
- X. Y. Cheng, E. Hinde, D. M. Owen, S. B. Lowe, P. J. Reece, K. Gaus and J. J. Gooding, Adv. Mater., 2015, 27, 6144–6150 CrossRef CAS PubMed.
- M. Rosso-Vasic, L. De Cola and H. Zuilhof, J. Phys. Chem. C, 2009, 113, 2235–2240 CrossRef CAS.
- M. Locritani, Y. X. Yu, G. Bergamini, M. Baroncini, J. K. Molloy, B. A. Korgel and P. Ceroni, J. Phys. Chem. Lett., 2014, 5, 3325–3329 CrossRef CAS PubMed.
- R. Mazzaro, M. Locritani, J. K. Molloy, M. Montalti, Y. X. Yu, B. A. Korgel, G. Bergamini, V. Morandi and P. Ceroni, Chem. Mater., 2015, 27, 4390–4397 CrossRef CAS.
- A. Arrigo, R. Mazzaro, F. Romano, G. Bergamini and P. Ceroni, Chem. Mater., 2016, 28, 6664–6671 CrossRef CAS.
- A. Fermi, M. Locritani, G. Di Carlo, M. Pizzotti, S. Caramori, Y. X. Yu, B. A. Korgel, G. Bergamini and P. Ceroni, Faraday Discuss., 2015, 185, 481–495 RSC.
- L. Ravotto, Q. Chen, Y. G. Ma, S. A. Vinogradov, M. Locritani, G. Bergamini, F. Negri, Y. X. Yu, B. A. Korgel and P. Ceroni, Chem, 2017, 2, 550–560 CAS.
- R. Mazzaro, A. Gradone, S. Angelon, G. Morselli, P. G. Cozzi, F. Romano, A. Vomiero and P. Ceroni, ACS Photonics, 2019, 6, 2303–2311 CrossRef CAS.
- D. Beri, D. Busko, A. Mazilkin, I. A. Howard, B. S. Richards and A. Turshatov, RSC Adv., 2018, 8, 9979–9984 RSC.
- W. Sun, C. X. Qian, X. S. Cui, L. W. Wang, M. A. Wei, G. Casillas, A. S. Helmy and G. A. Ozin, Nanoscale, 2016, 8, 3678–3684 RSC.
- K. Hara, T. Sato, R. Katoh, A. Furube, T. Yoshihara, M. Murai, M. Kurashige, S. Ito, A. Shinpo, S. Suga and H. Arakawa, Adv. Funct. Mater., 2005, 15, 246–252 CrossRef CAS.
- M. Tahir, Muddusir, D. N. Khan, S. Gul, F. Wahab and S. M. Said, J. Mater. Sci.: Mater. Electron., 2019, 30, 19463–19470 CrossRef CAS.
- Z. Yang, G. B. De los Reyes, L. V. Titova, I. Sychugov, M. Dasog, J. Linnros, F. A. Hegmann and J. G. C. Veinot, ACS Photonics, 2015, 2, 595–605 CrossRef CAS.
- A. Turshatov, D. Busko, Y. Avlasevich, T. Miteva, K. Landfester and S. Baluschev, ChemPhysChem, 2012, 13, 3112–3115 CrossRef CAS PubMed.
- K. Okumura, K. Mase, N. Yanai and N. Kimizuka, Chem. – Eur. J., 2016, 22, 7721–7726 CrossRef CAS PubMed.
- M. Mahboub, Z. Y. Huang and M. L. Tang, Nano Lett., 2016, 16, 8037–8037 CrossRef CAS PubMed.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed.
- P. Xia, E. K. Raulerson, D. Coleman, C. S. Gerke, L. Mangolini, M. L. Tang and S. T. Roberts, Nat. Chem., 2020, 12, 137–144 CrossRef PubMed.
- J. C. D. Verhagen, M. A. M. J. van Zandvoort, J. M. Vroom, L. B. A. Johansson and G. van Ginkel, J. Phys. Chem. B, 1997, 101, 10568–10575 CrossRef CAS.
- A. Turshatov, D. Busko, S. Baluschev, T. Miteva and K. Landfester, New J. Phys., 2011, 13, 083035 CrossRef.
- C. S. Zhang, J. Z. Zhao, S. Wu, Z. L. Wang, W. H. Wu, J. Ma, S. Guo and L. Huang, J. Am. Chem. Soc., 2013, 135, 10566–10578 CrossRef CAS PubMed.
- L. J. Patalag, L. P. Ho, P. G. Jones and D. B. Werz, J. Am. Chem. Soc., 2017, 139, 15104–15113 CrossRef CAS PubMed.
- F. Marsico, A. Turshatov, K. Weber and F. R. Wurm, Org. Lett., 2013, 15, 3844–3847 CrossRef CAS PubMed.
- J. C. deMello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- D. O. Faulkner, J. J. McDowell, A. J. Price, D. D. Perovic, N. P. Kherani and G. A. Ozin, Laser Photonics Rev., 2012, 6, 802–806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt04497c |
| This journal is © The Royal Society of Chemistry 2020 |