
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the change in UO2 redox reactivity as a function of H2O2 exposure†
Annika Carolin
Maier
 *a,
Philip
Kegler
b,
Martina
Klinkenberg
b,
Angela
Baena
b,
Sarah
Finkeldei
b,
Felix
Brandt
b and
Mats
Jonsson
a
*a,
Philip
Kegler
b,
Martina
Klinkenberg
b,
Angela
Baena
b,
Sarah
Finkeldei
b,
Felix
Brandt
b and
Mats
Jonsson
a
aDepartment of Chemistry, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, 10044 Stockholm, Sweden. E-mail: acmaier@kth.se
bForschungszentrum Jülich GmbH, Institute of Energy and Climate Research – Nuclear Waste Management and Reactor Safety (IEK-6), 52425 Jülich, Germany
First published on 18th December 2019
Abstract
To assess the long-term leaching behaviour of UO2, the main constituent of spent nuclear fuel, the oxidative dissolution of UO2 pellets was studied at high H2O2 exposures ranging from 0.33 mol m−2 to 1.36 mol m−2. The experiments were performed in aqueous media containing 10 mM HCO3− where the pellets were exposed to H2O2 three consecutive times. The results indicate that the dissolution yield (amount of dissolved uranium per consumed H2O2) at high H2O2 exposures is significantly lower compared to previous studies of both pellets and powders and decreases for each H2O2 addition for a given pellet. This implies a change in redox reactivity, which is attributed to irreversible alteration of the pellet surface. Surface characterization after the exposure to H2O2, by SEM, XRD and Raman spectroscopy shows, that the surface of all pellets is significantly oxidized.
Introduction
After its use in a reactor, nuclear fuel is planned to be stored in deep geological repositories in many countries.1 A deep repository must be designed to prevent groundwater intrusion in order to remain intact for long time spans.2 Predictive modelling as well as studies of natural analogues are crucial for its safety assessment.3–5 Under the reducing groundwater conditions prevailing at deep repository sites, the UO2 matrix, which makes up most of the spent nuclear fuel, has very low solubility.6 At the time when water intrusion can be expected to occur, the predominant radiation from the spent nuclear fuel will be alpha-radiation.7,8 The radiation will induce radiolysis of water in close proximity to the fuel. This can generate oxidizing conditions at the fuel surface under which UO2 is considerably more soluble6 thereby facilitating the migration of U and other radionuclides in the environment.9 Among the radiolysis products, H2O2 has been demonstrated to be the main oxidant responsible for uranium dissolution in systems exposed to alpha-radiation.10H2O2 can react in two different ways on the surface of UO2, via catalytic decomposition producing water and molecular oxygen ((1)–(3))11 and by oxidizing U(IV) ((4) and (5))12,13 respectively.
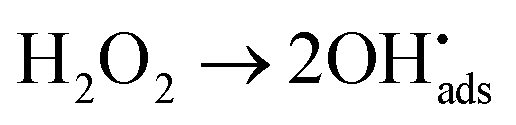 | (1) |
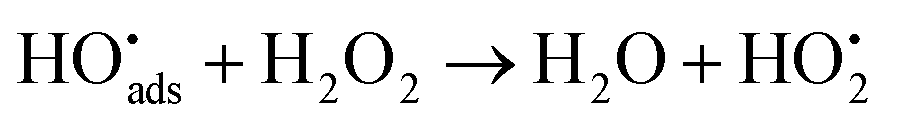 | (2) |
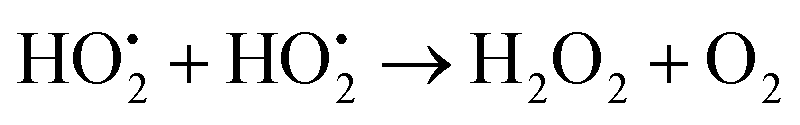 | (3) |
| H2O2 + UO2(S) → UO2 ads2+ + 2OH− | (4) |
| UO2 ads2+ → UO2 dissolved2+ | (5) |
It is important to note that the hydroxyl radicals formed in reaction (1) are stabilized by adsorption to the UO2 surface, adsorption being a prerequisite for this reaction to occur spontaneously. Under certain conditions, reaction (5) can be rate limiting but in the presence of the common groundwater constituent HCO3−, dissolution of U(VI) is facilitated by the formation of soluble complexes.14–16
To quantify the competition between oxidation of UO2 by H2O2 and catalytic decomposition of H2O2 on the UO2 surface, the term dissolution yield has been introduced.17 It is defined as the ratio between the amount of U(VI) dissolved from a pellet or a given amount of powder and the amount of H2O2 consumed on the same solid specimen 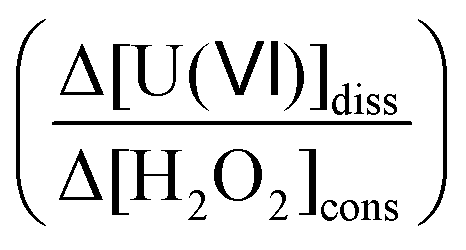 . As the surface area from which the uranium is dissolved is identical to the surface area on which H2O2 is consumed, the dissolution yield can be regarded as independent of the UO2 surface area that was exposed to the solution. This entity offers a straightforward way to compare experiments on specimens with different surface areas or where surface areas are difficult to determine experimentally.
. As the surface area from which the uranium is dissolved is identical to the surface area on which H2O2 is consumed, the dissolution yield can be regarded as independent of the UO2 surface area that was exposed to the solution. This entity offers a straightforward way to compare experiments on specimens with different surface areas or where surface areas are difficult to determine experimentally.
It was recently shown that the dissolution yield for UO2 powder ranges from 44% to 100% depending on the initial H2O2 concentration.18 For UO2 pellets the dissolution yield was reported to be around 15%.19–21 The range of dissolution yields observed for UO2 powder was rationalized from the mechanism for catalytic decomposition of H2O2. Hydrogen peroxide acts as radical scavenger for surface bound hydroxyl radicals (reaction (2)) and the higher the concentration of H2O2, the larger is the fraction of H2O2 consumed through catalytic decomposition. In fact, the key-competing reactions involving the surface-bound hydroxyl radical 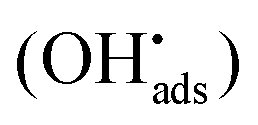 can be described as follows ((6) and (7)):18
can be described as follows ((6) and (7)):18
| UO2 − OH˙ → UO2+ + OH− | (6) |
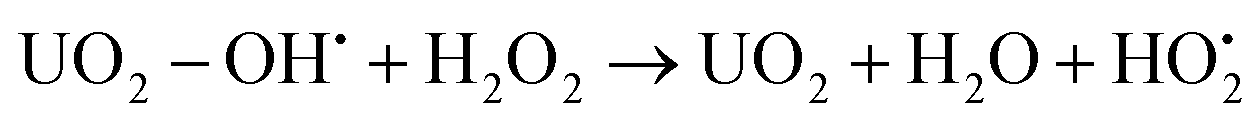 | (7) |
From this mechanism a kinetic definition of the dissolution yield accounting for the H2O2-concentration dependence can be derived (8):
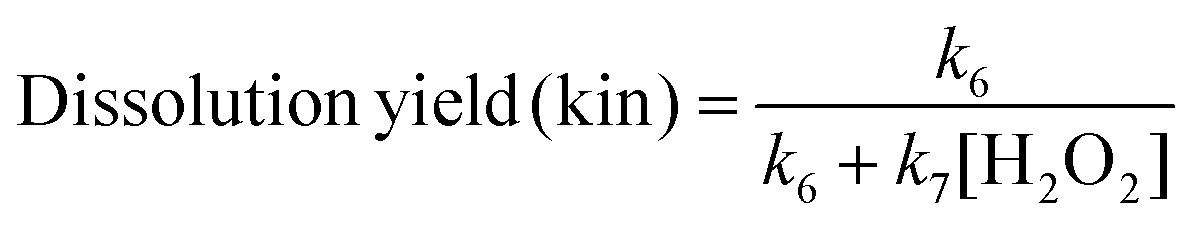 | (8) |
It should be noted that the U(V) species formed in reaction (6) must be oxidized one step further before dissolution takes place.
It was recently observed, that the reaction between H2O2 and UO2 powder does not follow strict first order kinetics.18,22 Instead, it turned out that the rate for H2O2 consumption at a given H2O2 concentration depends on the initial H2O2 concentration. In other words, the overall kinetics changes with turnover of H2O2. This change in surface reactivity can only be attributed to a surface alteration process, possibly due to passivation of reactive sites by oxidized uranium or surface bound hydroxyl radicals. In the experiments mentioned above, the surface area to solution volume ratios were between 2700 m−1 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 m−1 for powder experiments18 while they were around 22 m−1 for pellets.19,20 This means that the UO2 surfaces are exposed to very different amounts of H2O2 in powder experiments as compared to pellet experiments. In the powder experiments the H2O2 exposure ranges from 1.8 × 10−5 mol m−2 to 7.4 × 10−4 mol m−2 while in the pellet experiments the H2O2 exposure is around 0.08 mol m−2. In view of the time spans relevant for a deep repository, the very low exposures of the powder experiments correspond to an extremely short leaching time and the results may therefore not be relevant for a long-term safety assessment. To explore the possible change in surface reactivity of spent nuclear fuel, considerably higher exposures are needed.
800 m−1 for powder experiments18 while they were around 22 m−1 for pellets.19,20 This means that the UO2 surfaces are exposed to very different amounts of H2O2 in powder experiments as compared to pellet experiments. In the powder experiments the H2O2 exposure ranges from 1.8 × 10−5 mol m−2 to 7.4 × 10−4 mol m−2 while in the pellet experiments the H2O2 exposure is around 0.08 mol m−2. In view of the time spans relevant for a deep repository, the very low exposures of the powder experiments correspond to an extremely short leaching time and the results may therefore not be relevant for a long-term safety assessment. To explore the possible change in surface reactivity of spent nuclear fuel, considerably higher exposures are needed.
In this work, we have studied the oxidative dissolution of UO2 pellets at H2O2 exposures ranging from 0.33 mol m−2 to 1.36 mol m−2. Based on the experimental results of this and previous studies,18,20,22 the effect of H2O2 exposure on the UO2 surface reactivity is analysed and discussed.
Material and methods
Pellet fabrication and characterisation
Caution! Although natural uranium was used in this study and the radioactivity of the material is low due to its long half-life, safety precautions regarding the work with radioactive materials should be followed. The work with radioactive materials should only be conducted by trained staff and take place in appropriate facilities. Precursor powders for the pellets were produced by co-precipitation, where ammonium di-uranate (ADU) is precipitated from an aqueous solution containing 16.5 M ammonia and 2 M uranyl-nitrate. The [UO2(NO3)2·6H2O] ≥ 99% was supplied by Merck. After precipitation the powder was washed four times with high purity water to remove NH4OH residuals. During the last washing step water was replaced with ethanol and the powder was left to dry. It was then calcined in air at 600 °C for 5 h for de-nitration, de-hydration, and transformation to U3O8. In a second step, the U3O8 powder was reduced to stoichiometric UO2 during thermal treatment for 5 h at 600 °C in a tube furnace flushed with a 4% H2–96% Ar mixture (HYTEC). Afterwards, the calcined powder was compacted (for pressures see ESI S1†) to disk shaped green bodies (10 mm diameter) and pellets were sintered at 1700 °C for 10 h in a 4% H2 in Ar atmosphere to stoichiometric UO2 according to ref. 23. After sintering, the pellets were polished in several steps, the last step with a 0.04 μm colloidal SiO2 paste, and then thermally treated at 1350 °C for 180 min in reducing atmosphere (HYTEC). Total densities (Table 1) of the pellets were determined by a modified Archimedes method as described in ref. 24, where paraffin is used to cover all surfaces including open porosity and to avoid an uptake of water into the ceramic during the measurement. Additionally, the porosity of the pellets was determined from SEM observations via image analyses (ImageJ). A comparison of the two measurement methods shows coherent results, i.e. the porosity observed at the surface is representative for the volume of the pellets (SEM images see ESI S2†). Grain sizes were determined by measuring and averaging the length of the long and short axes of at least 100 grains per pellet. The average grain size of all samples was 12 ± 1 μm and the average weight of the pellets is 0.97 ± 0.03 g. The surface areas of the pellets were determined geometrically and amounts (1.53 ± 0.05) × 10−4 m2.| Pellet ID | Density/theoretical density × 100% | |
|---|---|---|
| Archimedes | SEM | |
| P1 | 92.9 | 92.2 |
| P2 | 93.5 | n.d. |
| P3 | 94.0 | n.d. |
| P4 | 94.6 | n.d. |
| P5 | 95.0 | 95.7 |
Changes to the microstructure during the exposure to H2O2 where analysed using a Quanta 200 FEG SEM (Thermo Fisher/FEI). Images were recorded before and after dissolution using both secondary electron (SE) and backscatter electron (BSE) detectors at an acceleration voltage of 20 kV in low vacuum mode.
XRD patterns of the pellets were recorded on a Bruker D4 Endeavor diffractometer, 40 kV and 40 mA, in Bragg–Brentano geometry. The diffractometer is equipped with a copper X-ray tube and a primary nickel filter producing graphite monochromized CuKα1 radiation (λ = 1.54187 Å). A linear silicon strip LynxEye detector (Bruker – AXS) was used. The patterns were recorded in the range of 2θ = 5–130° with a 0.02° increment and a scan speed of 2 s per step. The aperture of the fixed divergence slit was set to 0.2 mm and the receiving slit to 8.0 mm respectively. To determine the reaction products that form on the surface of the pellets during the exposure to H2O2, non-polarized Raman spectra were recorded after three consecutive exposures to H2O2 on a Horiba LabRAM HR spectrometer using a Peltier cooled multichannel CCD detector. An objective lens with a 50× magnification was linked to the spectrometer, allowing the analysis of samples as small as 2 μm in diameter. The incident radiation was produced by a He–Ne laser at a power of 17 mW (λ = 632.8 nm). The focal length of the spectrometer was 800 mm and a 1800 gr mm−1 grating was used. The spectral resolution was approximately 1 cm−1 with a slit of 100 μm. All spectra were recorded in the range between 200–2000 cm−1. For each pellet three spectra were recorded at three different locations.
Dissolution experiments
Prior to the dissolution experiments each pellet was washed in de-aerated 10 mM bicarbonate solution to remove pre-oxidized uranium from the surface, as pre-oxidized uranium might appear during production and handling of the pellet. The washing was carried out with the following procedure: first, each pellet was immersed in de-aerated bicarbonate solution for 10 min. In a second step, the bicarbonate solution was replaced several times after 10 min, 12 h and again after 10 min. After washing, the bicarbonate solution was replaced once more and H2O2 was added immediately to a concentration of 2.25 mM and a total sample volume of 40 ml. Blank experiments without UO2 pellets were carried out in parallel to the pellet experiments to correct for H2O2 consumption on the reaction vessels. The dissolution experiments were carried out at room temperature and the pH of the leaching solution was approximately 8.2.H2O2 and U(VI) concentrations were measured using a Lambda 19 PerkinElmer UV/VIS/NIR spectrophotometer. H2O2 was measured at 360 nm using the Ghormley triiodide method25 whereas U(VI) was measured at 653 nm using the Arsenazo III method.26 The samples were purged with Ar (≥99.999%, Air Liquide) throughout the experiments.
Chemicals used in all experiments were of reagent grade or higher unless otherwise stated. Purified water (18.2 MΩ cm, Merck MilliQ) was used throughout. Between individual H2O2 exposures each pellet was rinsed with water and sonicated to remove reaction products (i.e. U(VI)) from the previous experiment. It was then repeatedly washed with 10 mM de-aerated bicarbonate solution using the same procedure as stated above.
After the last experiment the pellet was washed with de-aerated MilliQ water and then left to dry in an Ar atmosphere.
Results and discussion
Dissolution experiments
Fig. 1 shows the results of dissolution experiments for pellet P5 during three consecutive H2O2 exposures. For pellets P1 to P4 the trends are similar (see ESI S3†). From the figure it becomes clear that the amount of uranium released decreases in every consecutive exposure to H2O2. In contrast, the consumption of H2O2 is almost identical for each exposure. It should be noted that the data in Fig. 1 is not corrected for the background consumption of H2O2 on the glass surfaces of the reaction vessels. Data corrected for the background consumption can be found in Table 2 along with the corresponding dissolution yields. It is clear that the dissolution yields are quite low compared to previous studies and that for a given pellet, it decreases for every new exposure to H2O2. As mentioned above, previously reported dissolution yields for UO2 pellets are around 15% (ref. 19–21) while for powders the dissolution yield is even higher.18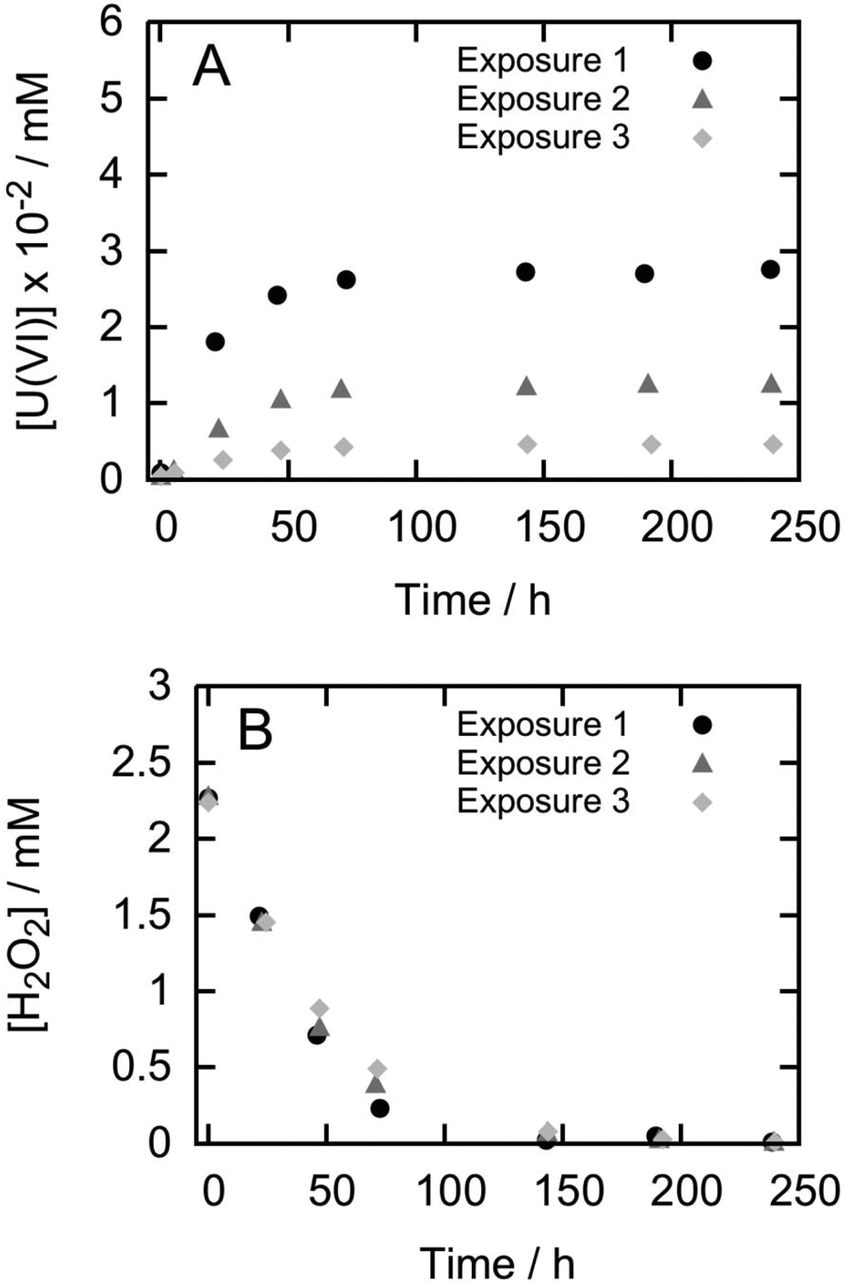 | ||
| Fig. 1 Uranium release (A) and H2O2 consumption (B) from pellet P5 during three consecutive exposures to H2O2. | ||
| Sample ID | H2O2 exposure no. | H2O2 consumed on UO2 (mM) | Dissolution yield (%) |
|---|---|---|---|
| P1 | # 1 | 1.20 | 1.51 |
| # 2 | 1.74 | 1.08 | |
| # 3 | 1.63 | 0.53 | |
| P2 | # 1 | 1.72 | 1.98 |
| # 2 | 1.80 | 0.77 | |
| # 3 | 1.71 | 0.31 | |
| P3 | # 1 | 1.54 | 2.33 |
| # 2 | 1.28 | 0.79 | |
| # 3 | 1.77 | 0.25 | |
| P4 | # 1 | 1.72 | 1.97 |
| # 2 | 1.76 | 0.91 | |
| # 3 | 1.56 | 0.26 | |
| P5 | # 1 | 1.74 | 1.62 |
| # 2 | 1.60 | 0.76 | |
| # 3 | 1.47 | 0.28 |
The results of the consecutive H2O2 exposures presented here provide some new insights since the systematic decrease in dissolution yield strongly indicates that the pellet surface is being altered, even in the presence of 10 mM bicarbonate and thorough washing with HCO3− solution between the exposures. This behaviour has, to the best of our knowledge, not been reported previously.
It was previously suggested that the dissolution yield depends on the initial H2O2 concentration and that this could be explained by the mechanism for catalytic H2O2 decomposition.18 The results presented here do not support the previous conclusion. Instead, the dissolution yield appears to depend on the total exposure to H2O2 expressed in mol m−2 of UO2 surface.
As already pointed out, there is a significant difference in the solid surface area to solution volume ratio between pellet experiments and powder experiments. In the current experiments, the surface area to volume ratio is around 3.5–4.0 m−1 while in powder experiments the corresponding ratio is often around 5000 m−1 or higher. This means that a pellet is exposed to about three orders of magnitude more H2O2 per surface area of material compared to the powder at the same initial H2O2 concentration. To analyse the possible correlation between the dissolution yield and H2O2-exposure per surface area, we have plotted the dissolution yields reported above as function of total H2O2-exposure per surface area (Fig. 2). For the second and third exposures, the accumulated H2O2 exposure is used.
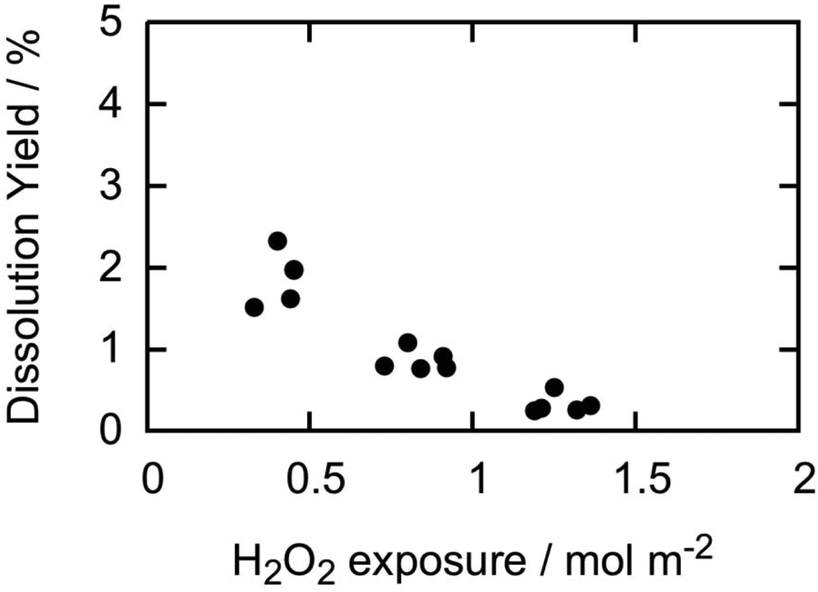 | ||
| Fig. 2 Dissolution yield vs. accumulated H2O2 consumption per m2 of exposed UO2 for pellets in this study. | ||
The results indicate a clear trend where the dissolution yield decreases with increasing H2O2-exposure per surface area. Again, this implies that the surface is continuously being altered in what seems to be an irreversible way. Although the overall reactivity of H2O2 towards the pellets is the same in all three exposures, the change in dissolution yield is a direct consequence of a change in the redox reactivity by as much as a factor of 3 to 4. This is in line with the change in kinetics observed in the powder experiments discussed above.18,22 Since the dissolution yields are considerably higher for powders, a change in the redox reactivity also has a significant impact on the overall reactivity.
Compared to the results from pellet experiments (Fig. 2), powder experiments represent the other extreme in terms of solid surface area to solution volume ratio. In recently published powder experiments18 the same general trend was observed, despite the quite obvious H2O2 concentration dependence for high surface area experiments. In order to connect the low and high solid surface area to solution volume experiments, we plot the results from powder experiments18 and results from the pellet experiments in this study in the same graph (Fig. 3a). In addition, the result of a previously published pellet experiment is included.20
 | ||
| Fig. 3 Dissolution yield vs. accumulated H2O2 consumption per m2 of exposed UO2. UO2 pellets from this study (blue triangles), UO2 pellet20 (red dot), as well as powders18 (green diamonds). (A) Linear axes and (B) logarithmic axes. | ||
As can be seen, the data from the previously published pellet experiment performed at a slightly higher surface to volume ratio than in the present study connects the data from this work with the data for powder experiments. Given the wide range in H2O2-exposure per surface area, we have also made the same plot with logarithmic axes (Fig. 3b).
Again, we observe a more or less continuous trend that clearly shows how the pellet becomes increasingly resistant to oxidative dissolution induced by H2O2. It is interesting to compare the present results to the expected exposure conditions in a deep repository. The rate of H2O2 consumption on a 1000 years old fuel with a burn-up of 55 MWd/kgU has been calculated to be 1.21 × 10−10 mol m−2 s−1 according to ref. 27. Consequently, a total H2O2 exposure of 1.36 mol m−2, as in the present work, corresponds to 356 years of exposure for the 1000 year old fuel. This is a fairly short time span compared to the time span that has to be considered for a deep geological repository. For the same fuel at 100000 years age the time span to reach the same H2O2 exposure is slightly above 14000 years.
For lab experiments aiming at elucidating the kinetics of spent nuclear fuel dissolution, the results presented above demonstrate the importance of the history of the specimens used in leaching experiments.
It should be noted that under the conditions used in the experiments presented here (10 mM HCO3−), studtite formation is thermodynamically favourable.28 However, when considering the equilibrium constants for uranyl carbonate- and uranyl peroxo carbonate complexes,29,30 the solubility under the present conditions is still significant and therefore Studtite precipitation is not to be expected. Studtite formation is usually confirmed in systems where UO2 has been exposed to H2O2 in the absence of HCO3−.31,32 In a previous study,33 UO2 powder, a UO2 pellet and a SIMFUEL pellet (UO2 doped with 11 nonradioactive isotopes of fission products to mimic real spent nuclear fuel) were exposed to high concentrations of H2O2 in aqueous solutions not containing HCO3−. High H2O2 concentrations were used to favour studtite formation. Post exposure investigation of the pellet surfaces with Raman spectroscopy confirmed the formation of studtite/metastudtite on UO2 powder and on the UO2 pellet. Interestingly, studtite/metastudtite formation was not observed on SIMFUEL. For SIMFUEL, the dissolution yield is very low and the reactivity of H2O2 is dominated by catalytic decomposition.
It has been reported that the presence of HCO3− efficiently removes oxidized uranium from the surface in oxidative dissolution experiments.34,35 However, post exposure surface characterization showing an unaltered surface, have only been reported for HCO3− containing systems where the oxidant is molecular oxygen. H2O2 containing systems still remain to be explored.
To shed some light on the nature of the solid phase alteration caused by the exposure of UO2 to H2O2 in this study, surface characterization of the pellets was performed using SEM, XRD, and Raman spectroscopy.
Scanning electron microscopy
BSE-SEM images of pellet P3 before (A) and after (B) three consecutive exposures to H2O2 are shown in Fig. 4. A significant change in the surface morphology is visible after the exposure to H2O2, mainly at the grain boundaries, but also at the grain surfaces. Already before the experiments, the grain boundaries are slightly etched due to the thermal treatment. When looking at the grains themselves, small square shaped structures appear after thermal etching. We ascribe them to a surface relaxation feature that appears at elevated temperatures. After the exposure to H2O2 these features become more pronounced, indicating dissolution, even from the grains themselves. After the dissolution experiment, some grains are liberated due to etching of the complete grain boundary. | ||
| Fig. 4 BSE-SEM images of P3, (A) before the exposure to H2O2 and (B) after the third exposure to H2O2. | ||
A closer view of P3 after dissolution is provided in Fig. 5 to reveal the details of individual grains. A significant surface roughness of the grain surfaces appears after the dissolution experiments (SE-image, Fig. 5A), which coincides with a new contrast appearing in the BSE image (Fig. 5B). The increased contrast and surface roughness may be due to differences in reactivity of the surface, leading to the preferential dissolution of parts of the surface. These irregularities may be a result of orientational or stoichiometrical effects. The latter will be discussed in the Raman spectroscopy section.
 | ||
| Fig. 5 (A) SE-SEM of P3 after the third exposure to H2O2 and (B) BSE-SEM of the same area. | ||
It becomes obvious that dissolution mainly occurs on certain grain boundaries while other grain boundaries remain intact. Also grain boundaries from just under the pellet surface dissolve, opening up small crack-like structures throughout different grains. Additionally, individual grains are loosened up from the matrix during the exposure to H2O2 leaving holes behind.
X-ray diffractometry
XRD patterns recorded prior to the dissolution experiments (Fig. 6a) confirm the stoichiometry of UO2 in a single cubic phase.36 The lattice parameters for samples P1 and P5 were calculated to be 5.477 ± 0.002 Å and 5.479 ± 0.003 Å based on the Debye–Scherrer method, which is in good agreement with literature.23 XRD results from after the exposure to H2O2 (Fig. 6b) reveal, that hyper-stoichiometric UO2 was formed which remains on the surface during the experiments; i.e. a smaller secondary peak appears at higher 2Θ angles next to the primary UO2 peak. Broadening of this secondary peak can be due to both, a decrease in crystallinity based on the formation of interstitial oxygen defects as well as a range of different oxidation states. Assuming that different states of oxidized uranium form during the reaction with H2O2, the range of hyper-stoichiometry can be narrowed down to x ≤ 0.25 in UO2+x. The highest oxidation state therefore matches the U4O9 phase as indexed in purple.37 It should be noted that no signs of studtite formation can be observed.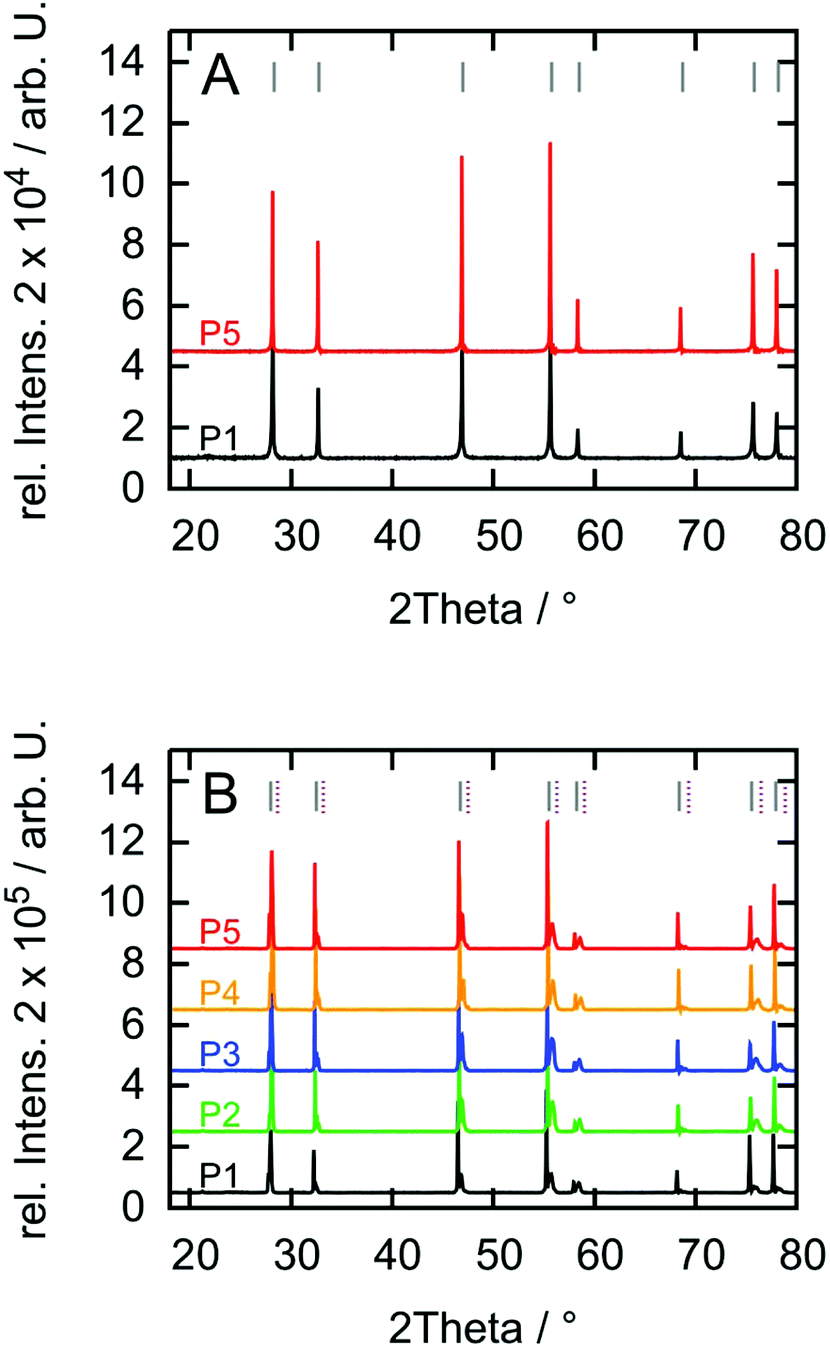 | ||
| Fig. 6 (A) Diffractograms before the first H2O2 exposure and (B) after the third H2O2 exposure. Y-Axis offset is added for clarity. Indices for UO2 are shown as gray lines36 and for U4O9 as purple dashed lines.37 | ||
Raman spectroscopy
The Raman spectrum of a sintered pellet which is a twin of the pellets which were used in the H2O2 experiments, revealed the stoichiometric UO2 bands at 445 cm−1 and 1150 cm−1.38 Also a small band at 918 cm−1 was observed.39 Spectra of samples P1 and P5 after the third exposure to H2O2 are shown in Fig. 7.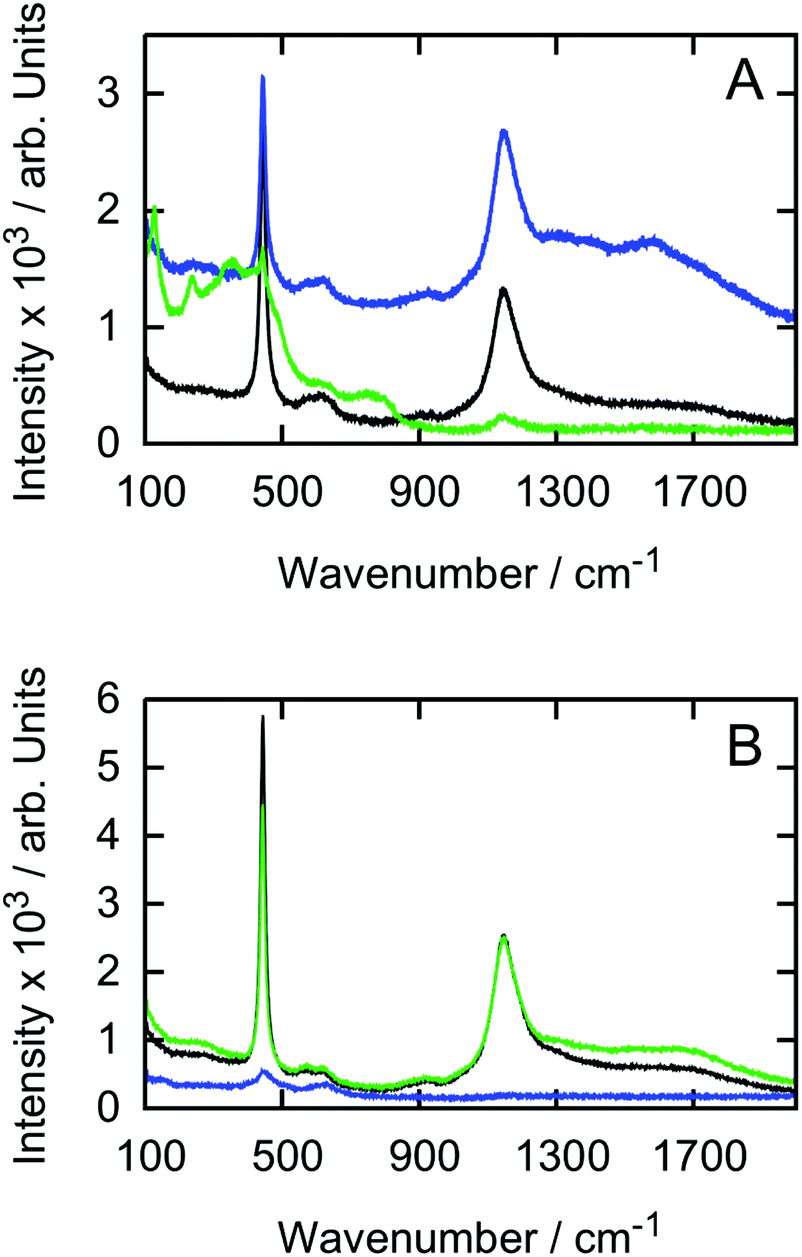 | ||
| Fig. 7 Raman spectra recorded at three different locations after the dissolution experiments for P1 (A) and P5 (B). | ||
New bands appear for all samples after they were exposed to H2O2. These bands can be ascribed to various oxidation states of hyper-stoichiometric UO2 as shown in Table 3. Their intensities vary for each pellet and location where they were recorded. Even oxidation states up to U3O8 were found. As expected, no signs of studtite formation were observed using this technique either. It should be pointed out that studtite formation was previously observed using Raman on UO2 specimens exposed to radiation in solutions without HCO3−41,42
As compared to the hyper-stoichiometry found by XRD, the Raman results reveal higher oxidation states. Since the XRD is insensitive to distortions in the anion sub lattice, transitional phenomena regarding distortions to the oxygen sub lattice remain unnoticed with XRD when cubic UO2 is gradually oxidized to tetragonal U3O7. Based on the attenuation length of photons penetrating UO2, the 8 keV X-rays penetrate the surface of the pellet much deeper as compared to the 1.95 eV Raman laser. Therefore one can assume, that the oxidation gradually decreases from the surface towards the center of the pellet as higher oxidation states than U4O9 were only measured by Raman spectroscopy.
Low frequency vibrations (below 343 cm−1), which only occur very locally, could not be identified in detail. However, we assume that they are an indication of distortion to the cationic sub lattice.
Conclusions
The experimental results presented in this work show that the redox reactivity of UO2 decreases quite dramatically with exposure to H2O2 in HCO3− containing aqueous solutions. Surface alteration to which this change in redox reactivity is attributed appears to be permanent and cannot be reverted merely by exposure to bicarbonate. The nature of this alteration is an increase in oxidation state; however, the degree of oxidation is not straightforward to assess. Surface characterization techniques indicate that an oxidation state gradient evolves upon high exposure to H2O2.These finding demonstrate the importance of keeping track of and reporting the H2O2 exposure history when performing lab experiments on oxidative dissolution of UO2 specimens in general and in particular when extrapolating these results to repository conditions.
In case of groundwater intrusion into a deep repository for spent nuclear fuel, the observed change in redox reactivity would lead to a fairly drastic inhibition of radiation induced fuel matrix dissolution and thereby also limit the radionuclide release. In fact, by numerically integrating the curve presented in Fig. 3 we can estimate the fraction of dissolved fuel matrix before reaching complete inhibition of oxidative matrix dissolution to less than 0.1% of the total inventory. This would have a tremendous impact on the safety assessment of a repository for spent nuclear fuel.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Nuclear Fuel and Waste Management Company (SKB) is gratefully acknowledged for financial support. This project has received funding from the European Union's Horizon 2020 research and training program of the European Atomic Energy Community (EURATOM) (H2020-NFRP2016/2017) under grant agreement no. 755443 (DisCo).References
- International Approaches for Deep Geologic Disposal of Nuclear Waste: Geological Challenges in Radioactive Waste Isolation, Fifth World Wide Review, LBNL Report 1006984, ed. B. Faybishenko, J. Birkholzer, D. Sassani and P. Swift, Lawrence Berkeley National Laboratory, Sandia National Laboratories, University of California, Berkeley, 2016 Search PubMed.
- J.-S. Kim, S.-K. Kwon, M. Sanchez and G.-C. Cho, Geological Storage of High Level Nuclear Waste, KSCE J. Civ. Eng., 2011, 15, 721–737 CrossRef.
- C. Poinssot and S. Gin, Long-term Behavior Science: The cornerstone approach for reliability assessing the long-term performance of nuclear waste, J. Nucl. Mater., 2012, 420, 182–192 CrossRef CAS.
- J. Bruno and K. Spahiu, The long-term effect of hydrogen on the UO2 spent fuel stability under anoxic conditions: Findings from the Cigar Lake Natural Analogue study, Appl. Geochem., 2014, 49, 178–183 CrossRef CAS.
- T. Eriksen, D. W. Shoesmith and M. Jonsson, Radiation induced dissolution of UO2 based nuclear fuel – A critical review of predictive modelling approaches, J. Nucl. Mater., 2012, 420, 409–423 CrossRef CAS.
- A. S. Kertes and R. Guillaumont, Solubility of UO2. A Comparative Review, Nucl. Chem. Waste Manage., 1985, 5, 215–219 CrossRef CAS.
- R. Håkansson, Beräkning av nuklidinnehåll resteffekt, aktivitet samt doshastighet för utbänt kärnsbränsle, SKB Rapport R-99-74, Svensk Kärnsbränslehantering AB, 2000 Search PubMed.
- R. Ewing, Long-term storage of spent nuclear fuel, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
- N. E. Jemison, A. E. Shiel, T. M. Johnson, C. C. Lundstrom, P. E. Long and K. H. Williams, Field Application of 238U/235U Measurements To Detect Reoxidation and Mobilization of U(IV), Environ. Sci. Technol., 2018, 52, 3422–3430 CrossRef CAS PubMed.
- E. Ekeroth, O. Roth and M. Jonsson, The relative impact of radiolysis products in radiation induced oxidative dissolution of UO2, J. Nucl. Mater., 2006, 355, 38–46 CrossRef CAS.
- A. Hiroki and J. A. LaVerne, Decomposition of Hydrogen Peroxide at Water-Ceramic Oxide Interfaces, J. Phys. Chem. B, 2005, 109, 3364–3370 CrossRef CAS PubMed.
- E. Ekeroth and M. Jonsson, Oxidation of UO2 by radiolytic oxidants, J. Nucl. Mater., 2003, 322, 242–248 CrossRef CAS.
- C. M. Lousada, M. Trummer and M. Jonsson, Reactivity of H2O2 towards different UO2-based materials: The relative impact of radiolysis products revisited, J. Nucl. Mater., 2013, 434, 434–439 CrossRef CAS.
- I. Grenthe, D. Ferri, F. Salvatore and G. Riccio, Studies on metal carbonate equilibria. Part 10. A solubility study of the complex formation in the uranium(VI)–water–carbon dioxide (g) system at 25 °C, Dalton Trans., 1984, 2439–2443 RSC.
- J. de Pablo, I. Casas, J. Giménez, M. Molera, M. Rovira, L. Duro and J. Bruno, The oxidative dissolution mechanism of uranium dioxide. I. The effect of temperature in hydrogen carbonate medium, Geochim. Cosmochim. Acta, 1999, 63, 3097–3103 CrossRef CAS.
- M. M. Hossain, E. Ekeroth and M. Jonsson, Effects of HCO3− on the kinetics of UO2 oxidation by H2O2, J. Nucl. Mater., 2006, 358, 202–208 CrossRef CAS.
- M. Jonsson, E. Ekeroth and O. Roth, Dissolution of UO2 by One- and Two-Electron Oxidants, Mater. Res. Soc. Symp. Proc., 2004, 807, 77–82 CrossRef CAS.
- A. Barreiro Fidalgo, Y. Kumagai and M. Jonsson, The role of surface-bound hydroxyl radicals in the reaction between H2O2 and UO2, J. Coord. Chem., 2018, 71, 1799–1807 CrossRef CAS.
- A. Barreiro Fidalgo and M. Jonsson, Radiation induced dissolution of (U, Gd)O2 pellets in aqueous solution - A comparison to standard UO2 pellets, J. Nucl. Mater., 2019, 514, 216–223 CrossRef CAS.
- S. Nilsson and M. Jonsson, H2O2 and radiation induced dissolution of UO2 and SIMFUEL pellets, J. Nucl. Mater., 2011, 410, 89–93 CrossRef CAS.
- K. Nilsson, O. Roth and M. Jonsson, Oxidative dissolution of ADOPT compared to standard UO2 fuel, J. Nucl. Mater., 2017, 488, 123–128 CrossRef CAS.
- Y. Kumagai, A. Barreiro Fidalgo and M. Jonsson, Impact of Stoichiometry on the Mechanism and Kinetics of Oxidative Dissolution of UO2 Induced by H2O2 and γ-Irradiation, J. Phys. Chem. C, 2019, 123, 9919–9925 CrossRef CAS.
- G. Leinders, T. Cardinaels and T. Verwerft, Accurate lattice parameter measurements of stoichiometric uraniumdioxide, J. Nucl. Mater., 2015, 459, 135–142 CrossRef CAS.
- A. E. E. McKenzie, General Physics, Cambridge University Press, Cambridge, 1965 Search PubMed.
- A. O. Allen, T. W. Davis, G. V. Elmore, J. A. Ghormley, B. M. Haines and C. J. Hochanadel, Decomposition of Water and Aqueous Solutions Under Pile Radiation, ORNL Publications 130, Oak Ridge National Laboratory, Tennessee, 1949 Search PubMed.
- S. B. Savvin, Analytical Use of Arsenazo III Determination of Thorium, Zirconium, Uranium and Rare Earth Elements, Talanta, 1961, 8, 673–685 CrossRef CAS.
- M. Jonsson, F. Nielsen, O. Roth, E. Ekeroth, S. Nilsson and M. M. Hossain, Radiation Induced Spent Nuclear Fuel Dissolution under Deep Repository Conditions, Environ. Sci. Technol., 2007, 41, 7087–7093 CrossRef CAS.
- D. Gorman-Lewis, P. C. Burns and J. B. Fein, Review of uranyl mineral solubility measurements, J. Chem. Thermodyn., 2008, 40, 335–352 CrossRef CAS.
- R. Guillaumont, T. Fanghändel, J. Fuger, I. Grenthe, V. Neck, D. A. Palmer and M. H. Rand, Update on the Chemical Thermodynamics of Uranium, Neptunium, Americium and Technetium, tech. rep. 5, OECD Nuclear Energy Agency, Data Bank Issy-les-Moulineaux, 2003 Search PubMed.
- P. L. Zanonato, P. Di Bernardo, Z. Szabó and I. Grenthe, Chemical Equilibria in the Uranyl(VI)-Peroxide-Carbonate System; Identification of Precursors for the Formation of Poly-Peroxometallates, Dalton Trans., 2012, 41, 11635–11641 RSC.
- P. Díaz-Arocas, J. Quinoñes, C. Maffiotte, J. Serrano, J. Garcia, J. R. Almazán and J. Esteban, Effects of Secondary Phases Formation in the Leaching of UO2 Under Simulated Radiolytic Products, Mater. Res. Soc. Symp. Proc., 1994, 353, 641–646 CrossRef.
- B. D. Hanson, B. McNamara, E. C. Buck, J. I. Friese, E. Jenson, K. Krupka and B. W. Arey, Corrosion of commercial spent nuclear fuel. 1. Formation of studtite and metastudtite, Radiochim. Acta, 2005, 93, 159–168 CAS.
- S. Sundin, B. Dahlgren, O. Roth and M. Jonsson, H2O2 and radiation induced dissolution of UO2 and SIMFUEL in HCO3−deficient aqueous solution, J. Nucl. Mater., 2013, 443, 291–297 CrossRef CAS.
- I. Casas, J. de Pablo, F. Clarens, J. Giménez, J. Merino, J. Bruno and A. Martinez-Esparza, Combined effect of H2O2 and HCO3− on UO2 (s) dissolution rates under anoxic conditions, Radiochim. Acta, 2009, 97, 485–490 CAS.
- J. Bruno, I. Casas, E. Cera, J. de Pablo, J. Giménez and M. E. Torrero, Uranium(IV) Dioxide and SIMFUEL as Chemical Analogues of Nuclear Spent Fuel Matrix Dissolution. A Comparison of Dissolution Results in a Standard NaCl/NaHCO3 Solution, Mater. Res. Soc. Symp. Proc., 1995, 353, 601–608 CrossRef CAS.
- L. Desgranges, G. Baldinozzi, G. Rousseau, J.-C. Nièpce and G. Calvarin, Neutron Diffraction Study of the in Situ Oxidation of UO2, Inorg. Chem., 2009, 48, 7585–7592 CrossRef CAS PubMed.
- N. Masaki and K. Doi, Analysis of the Superstructure of U4O9 by Neutron Diffraction, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 785–791 CrossRef CAS.
- J. M. Elorrieta, L. J. Bonales, N. Rodríguez-Villagra, V. G. Baonza and J. Cobos, A detailed Raman and X-ray study of UO2+x oxides and related structure transitions, Phys. Chem. Chem. Phys., 2016, 18, 28209–28216 RSC.
- T. Livneh and E. Sterer, Effect of pressure on the resonant multiphonon Raman scattering in UO2, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 085118 CrossRef.
- M. L. Palacios and S. H. Taylor, Characterization of Uranium Oxides in Situ Micro-Raman Spectroscopy, Appl. Spectrosc., 2000, 54, 1372–1378 CrossRef CAS.
- G. Guimbretière, L. Desgranges, C. Jegou, A. Canizarès, P. Simon, R. Carabello, N. Raimboux, M.-F. Barthe, M.-R. Ammar, O. A. Maslova, F. Duval and R. Omnée, Characterization of Nuclear Materials in Extreme Conditions: Raman Spectroscopy Approach, IEEE Trans. Nucl. Sci., 2014, 61, 2045–2051 Search PubMed.
- A. Traboulsi, J. Vandenborre, G. Blain, B. Humbert, J. Barbet and M. Fattahi, Radiolytic Corrosion of Uranium Dioxide: Role of Molecular Species, J. Phys. Chem. C, 2014, 54, 1071–1080 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt04395k |
| This journal is © The Royal Society of Chemistry 2020 |