
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and properties of anionic ruthenium thionitrosyl and selenonitrosyl complexes that contain tetraanionic 2-hydroxybenzamidobenzene ligands†
Chi-Wai
Fung
a,
Go
Fukada
a,
Yuichiro
Mutoh
 *a,
Noriko
Tsuchida
b and
Shinichi
Saito
*a
*a,
Noriko
Tsuchida
b and
Shinichi
Saito
*a
aDepartment of Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: ymutoh@rs.tus.ac.jp; ssaito@rs.kagu.tus.ac.jp
bDepartment of Liberal Arts, Faculty of Medicine, Saitama Medical University, 38 Morohongo, Moroyama-machi, Iruma-gun, Saitama 350-0495, Japan
First published on 10th December 2019
Abstract
Although transition-metal complexes that contain thiocarbonyl (CS) and selenocarbonyl (CSe) ligands have been well studied, only three neutral or cationic selenonitrosyl (NSe) complexes have been reported, while anionic NSe complexes remain elusive. Herein, we report the first examples of anionic NSe-ligated ruthenium complexes, which were obtained from the reaction of anionic ruthenium nitrido complexes, elemental selenium, and 4-(N,N-dimethylamino)pyridine (DMAP). The structures of one of these ruthenium NSe complexes, as well as of the corresponding thionitrosyl (NS) and nitrosyl (NO) complexes, were systematically examined by X-ray diffraction analyses and theoretical calculations. In contrast to previous reportes, the NSe ligand in these complexes is a better π-acceptor than the NO and NS ligands and exhibits a stronger trans influence.
Introduction
The chemistry of nitric oxide (NO) is well investigated, primarily due to the importance of NO in a biological context, e.g. in cell signalling and other physiological functions in living organisms.1 Accordingly, it is hardly surprising that numerous transition-metal complexes of NO have been studied in depth.2 Yet, the chemistry of the heavier isologues of NO, i.e., nitric sulfide (NS) and nitric selenide (NSe), has attracted less attention, as these species can only be detected in harsh environments such as low-temperature matrixes.3 Stabilizing these fleeting species in transition-metal complexes would afford an opportunity to study the properties of these thionitrosyl (M–NS) and selenonitrosyl complexes (M–NSe) and thus broaden our understanding of the chemistry of NS and NSe.In contrast to the chemistry of thiocarbonyl (CS) and selenocarbonyl (CSe) transition-metal complexes,4 that of NS and NSe complexes has been much less developed, which is probably due to the scarcity of NS and NSe complexes. The first transition-metal complexes of NS was obtained from the reaction of a molybdenum nitrido complex with elemental sulfur,5 and other NS complexes have also been prepared by the treatment of the corresponding transition-metal nitrido complexes6 with elemental sulfur7 or its equivalents.8,9 Moreover, reactions of metal complexes with trithiazyl chloride (N3S3Cl3) as the NS source have been reported.10 Following the successful synthesis of NS complexes from the corresponding nitrido complexes, a few NSe complexes were prepared by the reaction of nitrido complexes with elemental selenium,7a,c,d albeit that this approach is less generic for NSe than for NS. For example, cis-[Ru(N)(Cl)L2] (L = 2-[(2,6-diisopropylphenyl)imino]methyl-4,6-dibromophenolato), did not afford the corresponding NSe complex under reaction conditions similar to those for the reaction with NS.7e
To investigate the properties of the chalcogenonitrosyls (NE; E = O, S, Se) in transition-metal complexes, systematic studies on a series of NE complexes would be highly desirable.11 So far, only three series of NE complexes have been reported, which include neutral and cationic metal complexes of [Os(NE)Cl2Tp] (Tp = hydrotris(1-pyrazolyl)borate),7a [Ru(NE)Cl3(AsPh3)2]7d,12 and [Ir(NE){N(CHCHPtBu2)2}][PF6]7c,13 (Fig. 1). Conversely, a series of anionic transition-metal NE complexes has not yet been reported. Examples of anionic NS complexes remain scarce,14 while anionic NSe complexes remain elusive. The synthesis of such a series of anionic NE (E = O, S, Se) complexes should thus be important to gain deeper insight into the chemistry of NE, particularly with respect to the question how the overall charge influence the properties of such NE complexes. Herein, we report the synthesis of anionic ruthenium NE (E = O, S, Se) complexes that bear tetraanionic 2-hydroxybenzamidobenzene ligands, i.e., [Ph4P][Ru(NE)(hybebR1,R2)(dmap)] (E = O, S, Se; hybeb = N,N′-(1,2-phenylene)bis(2-hydroxybenzamide), R1 = H, Cl; R2 = H, Cl, CF3; dmap = 4-(N,N-dimethylamino)pyridine). The structures of some of these NE complexes were determined in detail by a combination of single-crystal X-ray diffraction analyses and theoretical calculations. All the Ru–NE complexes obtained in this study are classified as {RuNE}6 according to the Enemark-Feltham notation.15
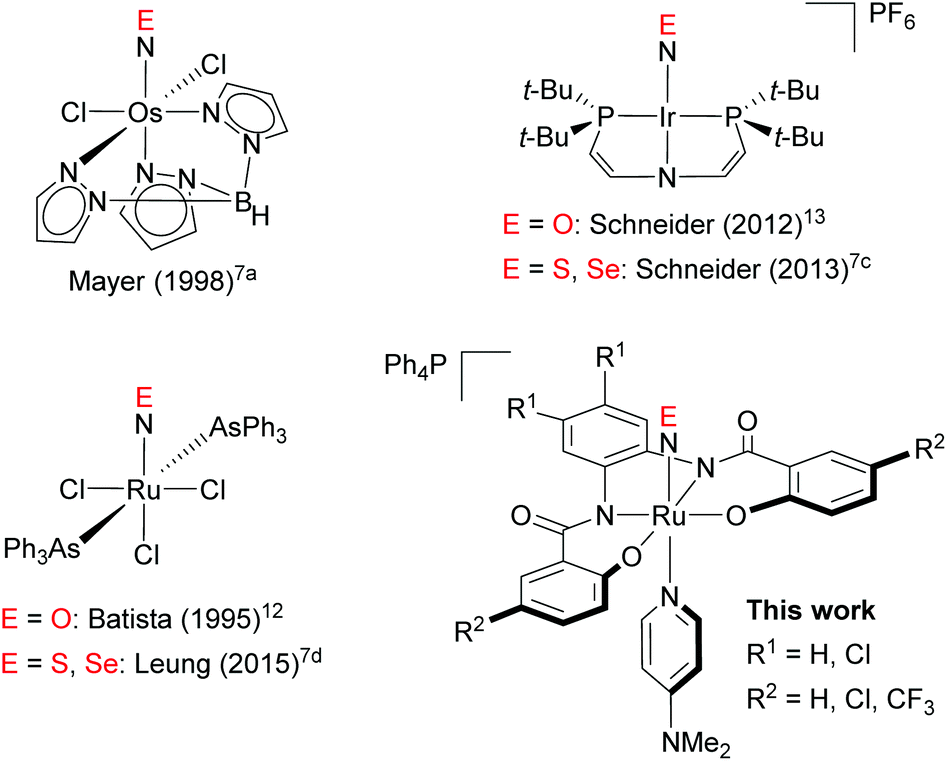 | ||
| Fig. 1 Examples of a series of transition-metal NE (E = O, S, Se) complexes. | ||
Results and discussion
Syntheses of 2-hydroxybenzamidobenzene derivatives (H4hybebR1,R2)
We selected the 2-hydroxybenzamidobenzene derivatives H4hybebR1,R2 (1) as tetradentate ligands for the synthesis of anionic NE complexes of ruthenium, as they can be readily prepared, and offer an opportunity to study the electronic effect on the properties of the complexes. Moreover, when such ligands coordinate to transition metals, a rigid structure is usually formed. Unsubstituted H4hybebH,H (1a) was prepared in 92% yield by the Ph3PCl2-mediated condensation16 of salicylic acid and 1,2-phenylenediamine (Scheme 1), and substituted H4hybebH,Cl (1b) and H4hybebCl,Cl (1c) were prepared in good yield under similar reaction conditions.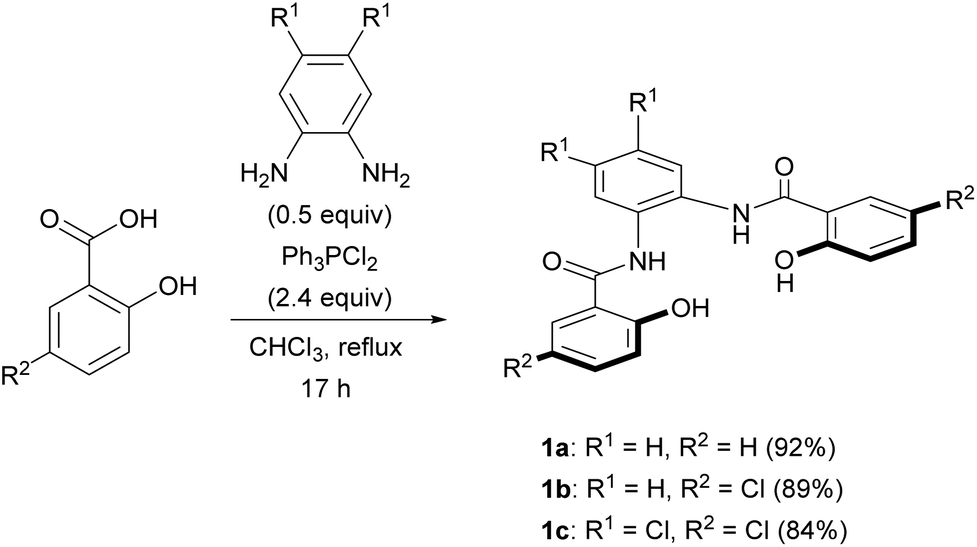 | ||
| Scheme 1 Synthesis of H4hybebR1,R2 ligands 1a–c. | ||
Unexpectedly, the attempted synthesis of H4hybeb derivatives with a CF3 group at R2 [1d: H4hybebH,CF3; 1e: H4hybebCl,CF3] failed under the conditions shown in Scheme 1. To prepare these compounds, we treated THP-protected 4-(trifluoromethyl)phenol with BuLi and ethyl chloroformate to afford ester 2 (Scheme 2).17 Subsequently, 2 was treated with 1,2-phenylenediamine derivatives in the presence of sodium bis(trimethylsilyl)amide (NaHMDS)18 to furnish the corresponding THP-protected H4hybebR1,CF3. Finally, the THP group was removed under acidic conditions to generate 1d and 1e in good yield. The H4hybebR1,R2 ligands 1a–e contained a small amount of solvent molecules (hexane or EtOAc) that could not be removed (Fig. S1–S4 and S7–S10, ESI†).
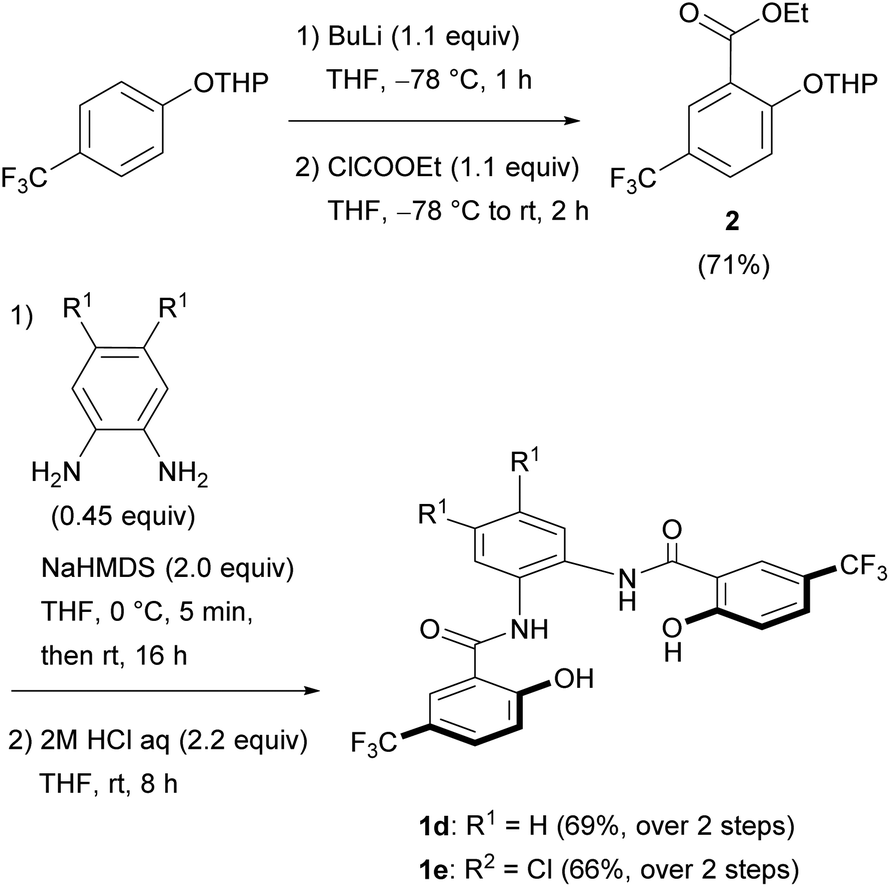 | ||
| Scheme 2 Synthesis of H4hybebR1,CF3 ligands 1d and 1e. | ||
Syntheses of the nitrido complexes [Ph4P][Ru(N)(hybebR1,R2)] (3)
With H4hybebR1,R2 ligands 1a–e in hand, we focused on the preparation of nitrido complexes that bear tetraanionic hybebR1,R2 ligands as precursors for the synthesis of the corresponding NS and NSe complexes. Based on a modified literature procedure (Scheme 3),19a the reaction of 1a and [Bu4N][Ru(N)Cl4]20 in the presence of an excess 2,6-lutidine in a mixture of MeOH/THF (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) afforded the tetrabutylammonium salt [Bu4N][Ru(N)(hybebH,H)],19 which was treated with [Ph4P]Br to afford nitrido complex [Ph4P][Ru(N)(hybebH,H)] (3a) in 72% yield. The cation metathesis was necessary to obtain single crystals suitable for an X-ray diffraction analysis. Nitrido complexes 3b–e were prepared in a similar fashion and obtained as orange/red crystals, which are air- and moisture-stable in both the solid state and in solution.
:1, v/v) afforded the tetrabutylammonium salt [Bu4N][Ru(N)(hybebH,H)],19 which was treated with [Ph4P]Br to afford nitrido complex [Ph4P][Ru(N)(hybebH,H)] (3a) in 72% yield. The cation metathesis was necessary to obtain single crystals suitable for an X-ray diffraction analysis. Nitrido complexes 3b–e were prepared in a similar fashion and obtained as orange/red crystals, which are air- and moisture-stable in both the solid state and in solution.
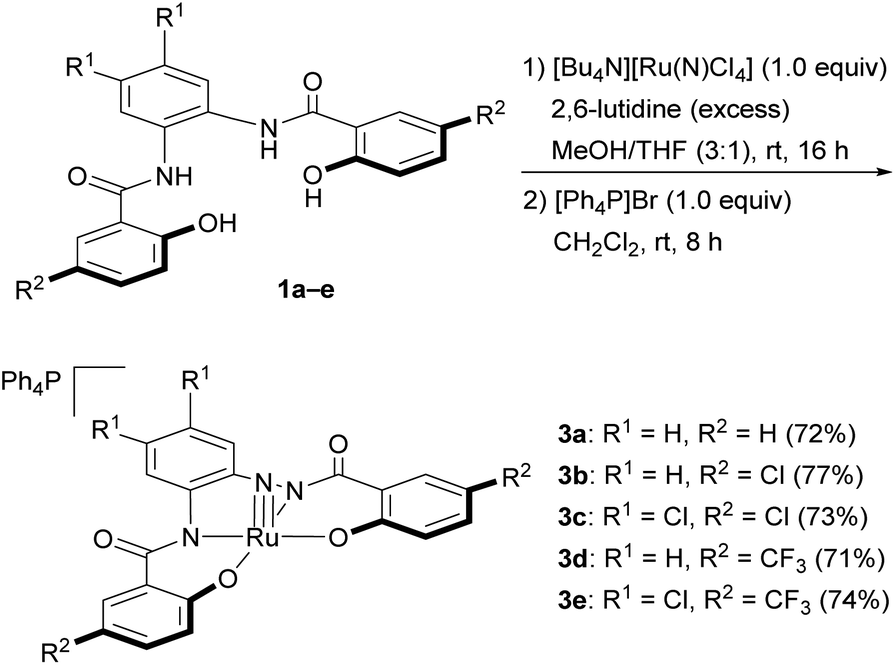 | ||
| Scheme 3 Synthesis of nitrido complexes 3. | ||
The molecular structure of nitrido complex [Ph4P][Ru(N)(hybebH,CF3)] (3d) was unambiguously determined by a single-crystal X-ray diffraction analysis. Complex 3d exhibits a distorted square-pyramidal coordination geometry in which the nitride ligand is located at the apical position (Fig. 2). Based on this analysis, the ruthenium center in 3 exhibits a 16-electron configuration with the formal oxidation state of +6. The length of the Ru1–N1 bond in 3d (1.6040(15) Å) falls in the range of previously reported five-coordinate anionic ruthenium nitrido complexes such as [Na(dme)][Ru(N)(meso-octamethylporphyrinogen)] (1.569(6) Å (ref. 21)), [Bu4N][Ru(N)(hybebH,H)] (1.594(4) Å (ref. 19a)), [Bu4N][Ru(N)(O2C6H4)2] (1.603(4) Å (ref. 22)), and [Bu4N][Ru(N)(S2C6H4)2] (1.613(5) Å (ref. 23)).
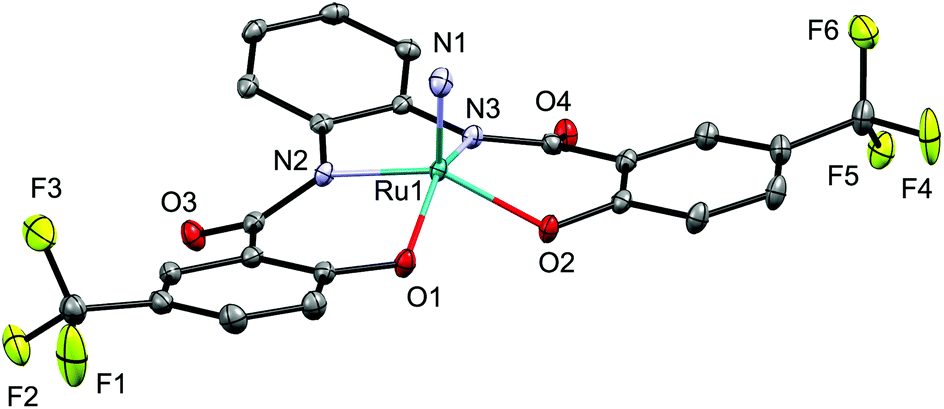 | ||
| Fig. 2 Molecular structure of the anionic part of nitrido complex 3d. Hydrogen atoms are omitted for clarity and only selected atoms are labelled. Selected bond length (Å): Ru1–N1 1.6040(15). | ||
Syntheses of the NS complexes [Ph4P][Ru(NS)(hybebR1,R2)(dmap)] (4)
Subsequently, we synthesized NS complexes via the reaction of 3 with elemental sulfur (Table 1). When 3a was treated at room temperature with 1/8 S8 (10 equiv.) in the presence of DMAP (10 equiv.), the colour of the mixture gradually turned from orange to black. The reaction reached completion after 40 h, and the NS complex [Ph4P][Ru(NS)(hybebH,H)(dmap)] (4a) was isolated in 67% yield in the form of black plates (entry 1). An acceleration of the reaction progress was observed when nitrido complexes with electron-withdrawing groups on the hybeb ligands were used (entries 2 and 3). After 0.5–1.5 h, the reactions of 3b and 3c with sulfur and DMAP smoothly furnished good yields of 4b and 4c, respectively. The reactivity of CF3-substituted complexes 3d and 3e was very high, and the reactions were complete after 15 min (entries 4 and 5). The formation of NS complexes was not observed when the reaction was carried out in the absence of DMAP.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 3 | R1 | R2 | Time (h) | Product | Yield (%) |
| 1 | 3a | H | H | 40 | 4a | 67 |
| 2 | 3b | H | Cl | 1.5 | 4b | 72 |
| 3 | 3c | Cl | Cl | 0.5 | 4c | 73 |
| 4 | 3d | H | CF3 | 0.25 | 4d | 74 |
| 5 | 3e | Cl | CF3 | 0.25 | 4e | 69 |
Two possible pathways can be considered for the formation of these NS complexes. A sulfurization of the nitrido complexes would most likely result in the formation of unisolable intermediates of the type [Ph4P][Ru(NS)(hybebR1,R2)],14e followed by coordination of DMAP to stabilize these unsaturated complexes, which would lead to the coordinatively saturated product 4. Alternatively, the reactivity of the nitrido ligand in 3 would be increased upon coordination of DMAP trans to the nitrido ligand prior to the formation of the nitrogen–sulfur bond.6d In order to examine the pathway and the role of DMAP toward the formation of 4, we monitored the reactions of 3 with (a) sulfur in the absence of DMAP, and with (b) DMAP in the absence of sulfur by 1H NMR spectroscopy. However, changes were not observed in these 1H NMR spectra: the mechanism of this reaction and the role of DMAP remains unclear at this stage.
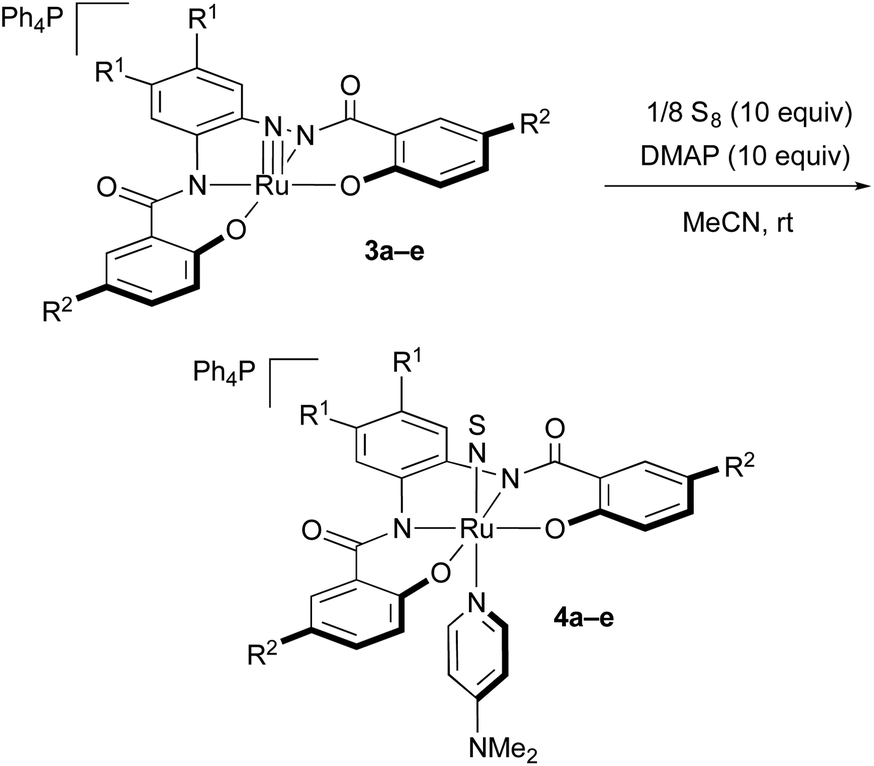
The molecular structure of 4d was unequivocally determined by a single-crystal X-ray diffraction analysis (Fig. 3). The six-coordinate octahedral geometry of the ruthenium center was confirmed, whereby the DMAP ligand is located trans to the NS ligand. The Ru–N–S angle is 171.0(3)/172.2(3)°, which confirms a terminal coordination and linear alignment of the NS ligand. The N–S bond length in 4d [1.511(4)/1.516(6)] is similar to those in other NS complexes such as mer-[Ru(NS)Cl3(AsPh3)2] (1.502(4) Å (ref. 7d)), [Ph4P][Ru(NS)Cl4(H2O)] (1.504(4) Å (ref. 14a)), [Ph4P][Os(NS)Cl4(H2O)] (1.514(5) Å (ref. 14b)), and [(Ir(NS){N(CHCHPtBu2)2}][PF6] (1.522(2) Å (ref. 7c)).
 | ||
| Fig. 3 Molecular structure of the anionic part of NS complex 4d. Only one of the two independent anions per unit cell is shown. Hydrogen atoms are omitted for clarity and only selected atoms are labelled. Selected bond lengths (Å) and angles (°): S1–N1 1.511(4); N1–Ru1 1.746(4); Ru1–N2 2.155(4); S1–N1–Ru1 171.0(3); N1–Ru1–N2 173.53(19). | ||
Syntheses of the NSe complexes [Ph4P][Ru(NSe)(hybebR1,R2)(dmap)] (5)
Next, we attempted to synthesize NSe complexes via a similar approach, using elemental selenium instead of sulfur (Table 2). As expected, in accordance with the aforementioned low reactivity of 3a and 3b, the corresponding NSe complexes [Ph4P][Ru(NSe)(hybebR1,R2)(dmap)] (5a: R1 = R2 = H; 5b: R1 = H, R2 = Cl) were not formed (entries 1 and 2). The reaction of 3c with selenium and DMAP proceeded sluggishly and led only to the formation of an equilibrium mixture between 3c and the product [Ph4P][Ru(NSe)(hybebCl,Cl)(dmap)] (5c) in a ratio of 60:40 after 36 h. From the mixture, 5c could be isolated in 38% yield in the form of black microcrystals (entry 3). Considering the electrophilic nature of the nitrido ligand in high-valent ruthenium complexes,24 we anticipated that hybebR1,CF3 ligands with a CF3 group should enhance the reactivity of the nitrido complexes. In fact, when 3d and 3e were treated with selenium in the presence of DMAP, full conversion of 3d and 3e was observed after 12 h. The corresponding anionic ruthenium NSe complexes [Ph4P][Ru(NSe)(hybebH,CF3)(dmap)] (5d) and [Ph4P][Ru(NSe)(hybebCl,CF3)(dmap)] (5e) were obtained in 46% and 48% yields, respectively (entries 4 and 5), and these represent the first examples of anionic transition-metal NSe complexes.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 3 | R1 | R2 | Time (h) | Product | Yield (%) |
| 1 | 3a | H | H | 72 | 5a | 0 |
| 2 | 3b | H | Cl | 72 | 5b | 0 |
| 3 | 3c | Cl | Cl | 36 | 5c | 38 |
| 4 | 3d | H | CF3 | 12 | 5d | 46 |
| 5 | 3e | Cl | CF3 | 12 | 5e | 48 |
NSe complexes 5c–e are stable in the solid state under air and at low temperature in solution. However, 5c–e decompose in solution at room temperature, even under an atmosphere of argon. When a CDCl3 solution of 5d was kept standing at room temperature (Scheme 4), the gradual liberation of DMAP under concomitant formation of nitrido complex 3d and a black precipitate (presumably elemental selenium) was observed. The addition of DMAP (10 equiv.) or selenium (10 equiv.) to the CDCl3 solution inhibited the decomposition, and only ca. 40% of 5d was converted into 3d after 24 h. An intermediate was not observed in the 1H NMR spectrum of the reaction mixture. Due to the instability of the NSe complexes, it was necessary to work up the reaction rapidly, and the recrystallization had to be conducted at low temperature.
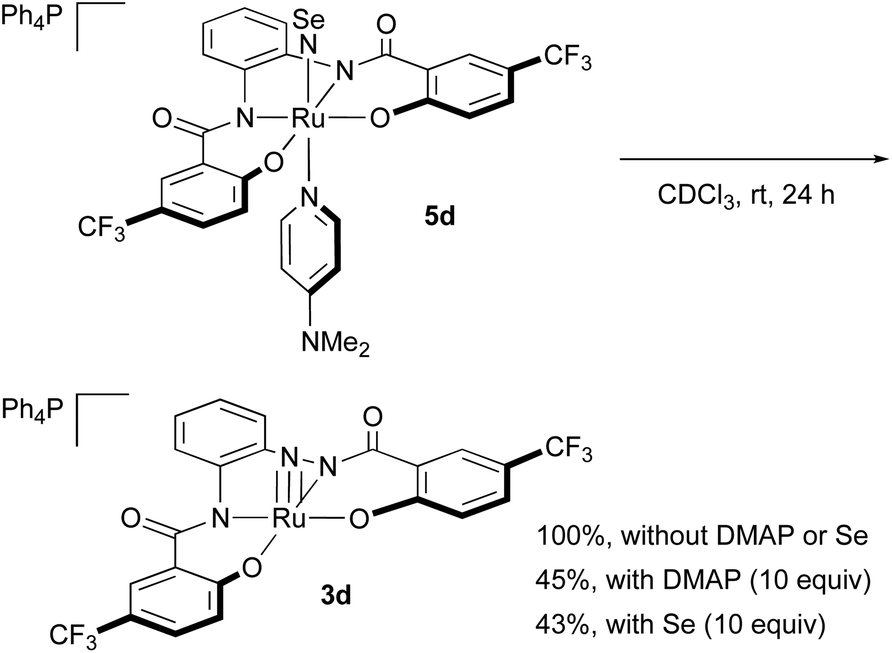 | ||
| Scheme 4 Decomposition of NSe complex 5d in CDCl3 into nitrido complex 3d. | ||
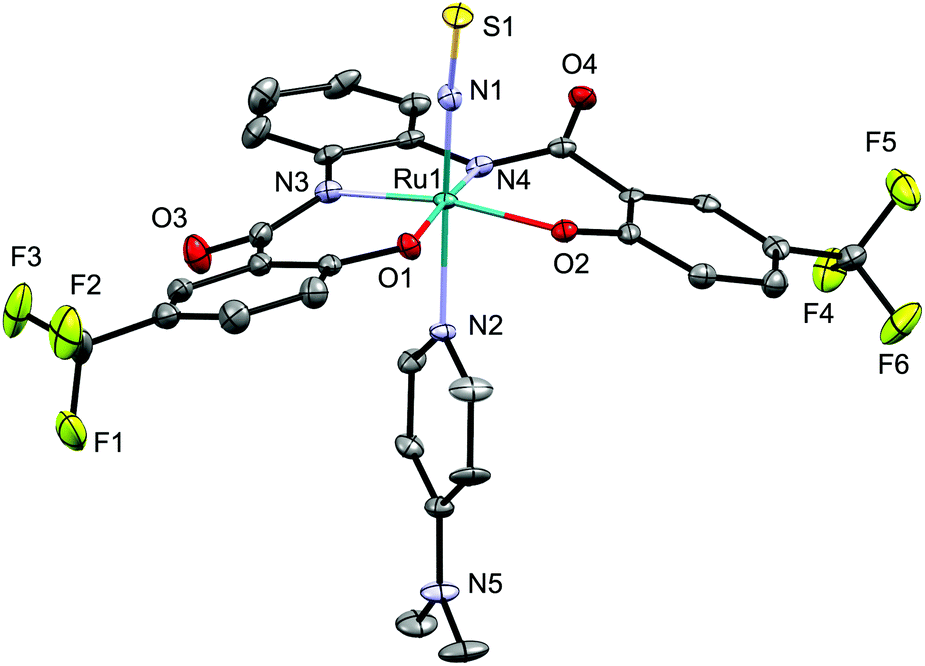
The molecular structure of 5d was determined by crystallographic measurements (Fig. 4). Structural features similar to those of NS complex 4d were observed for NSe complex 5d, which exhibits a six-coordinate octahedral coordination geometry with a linear Ru–N–Se linkage [171.2(3)/169.9(3)°] and short Ru–NSe and N–Se distances. The N–Se bond distance in 5d [1.670(4)/1.671(4) Å] is longer than those in [Os(NSe)Cl2Tp] (1.629(10) Å (ref. 7a)) and mer-[Ru(NSe)Cl3(AsPh3)2] (1.650(3) Å (ref. 7d)), but similar to that in [(Ir(NSe){N(CHCHPtBu2)2}][PF6] (1.678(4) Å (ref. 7c)).
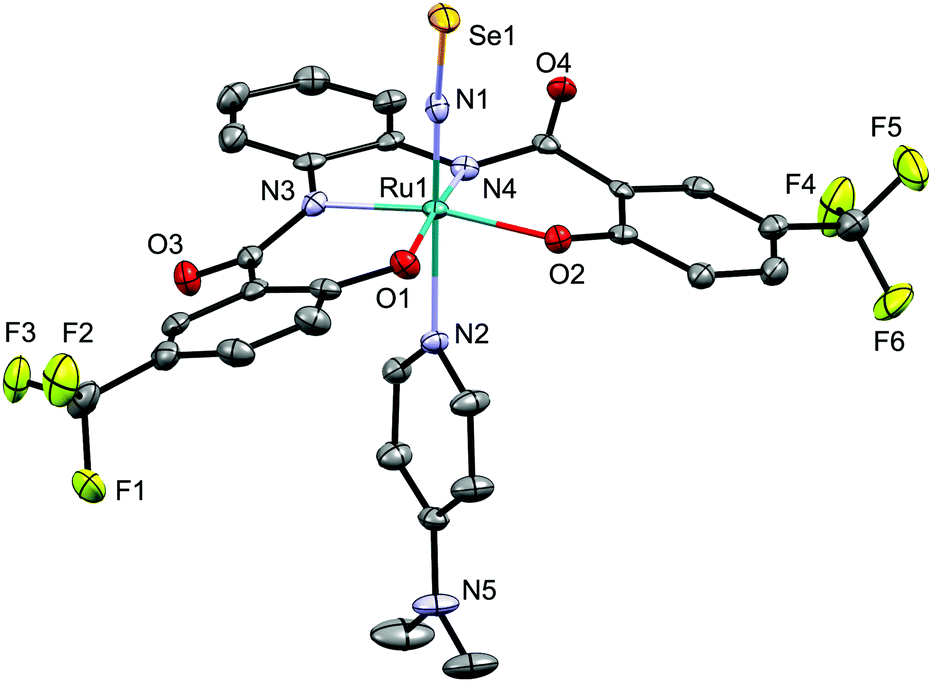 | ||
| Fig. 4 Molecular structure of the anionic part of NSe complex 5d. Only one of the two independent anions per unit cell is shown. Hydrogen atoms are omitted for clarity and only selected atoms are labelled. Selected bond lengths (Å) and angles (°): Se1–N1 1.670(4); N1–Ru1 1.734(4); Ru1–N2 2.173(4); Se1–N1–Ru1 171.2(3); N1–Ru1–N2 173.79(16). | ||
Encouraged by the successful synthesis of these NSe complexes, we further studied the synthesis of hitherto unprecedented tellurium analogues of NO complexes, i.e., telluronitrosyl (NTe) complexes. However, all attempts to synthesize NTe complexes via the reaction of 3 and elemental tellurium in the presence of pyridines failed, which might be attributed to the low solubility of elemental tellurium in common organic solvents. It should be noted here that this result is consistent with previously reported results on the reactions of osmium and neutral ruthenium nitrido complexes.7a,d
Synthesis of the NO complex [Ph4P][Ru(NO)(hybebH,CF3)(dmap)] (6)
To compare the structures of NS complex 4d and NSe complex 5d with the corresponding NO complex [Ph4P][Ru(NO)(hybebH,CF3)(dmap)] (6), we attempted to synthesize 6via the oxidation of 3d using Me3NO,7a,e,13 H2O225 or O2 with Et3B·DMAP,4e but all of these attempts were unsuccessful. Therefore, 6 was synthesized according to a modified literature method (Scheme 5):261d was treated with sodium hydride, followed by [RuNOCl3]26 and DMAP to generate the sodium salt Na[Ru(NO)(hybebH,CF3)(dmap)]. A subsequent cation metathesis with [Ph4P]Br afforded the targeted NO complex 6 in the form of black crystals (34% yield over two steps).
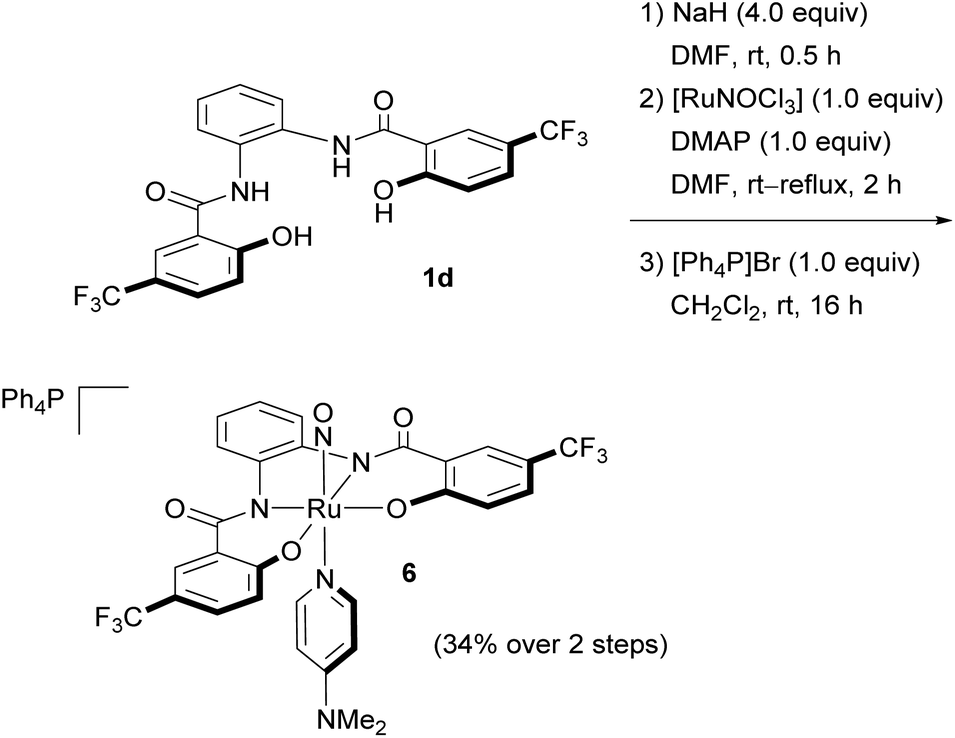 | ||
| Scheme 5 Synthesis of NO complex 6. | ||
In its IR spectrum, NO complex 6 exhibits a ν(NO) absorption at 1810 cm−1, while the ν(NS) and the ν(NSe) bands of 4d and 5d, respectively could not be clearly identified due to the overlap with vibrational stretches arising from the hybebR1,R2 ligand and the [Ph4P]+ ion. The molecular structure of 6 was determined by a single-crystal X-ray diffraction analysis (Fig. 5). The alignment of the Ru–N–O moiety is almost linear [174.9(2)/177.27(19)°] with an N–O bond distance of 1.144(3)/1.149(3) Å, which is similar to that in other NO complexes such as mer-[Ru(NO)Cl3(AsPh3)2] (1.151(9) Å (ref. 12)) and [(Ir(NO){N(CHCHPtBu2)2}][PF6] (1.168(3) Å (ref. 7c)), but shorter than that in [Os(NO)Cl2Tp] (1.19(4) Å (ref. 7a)).
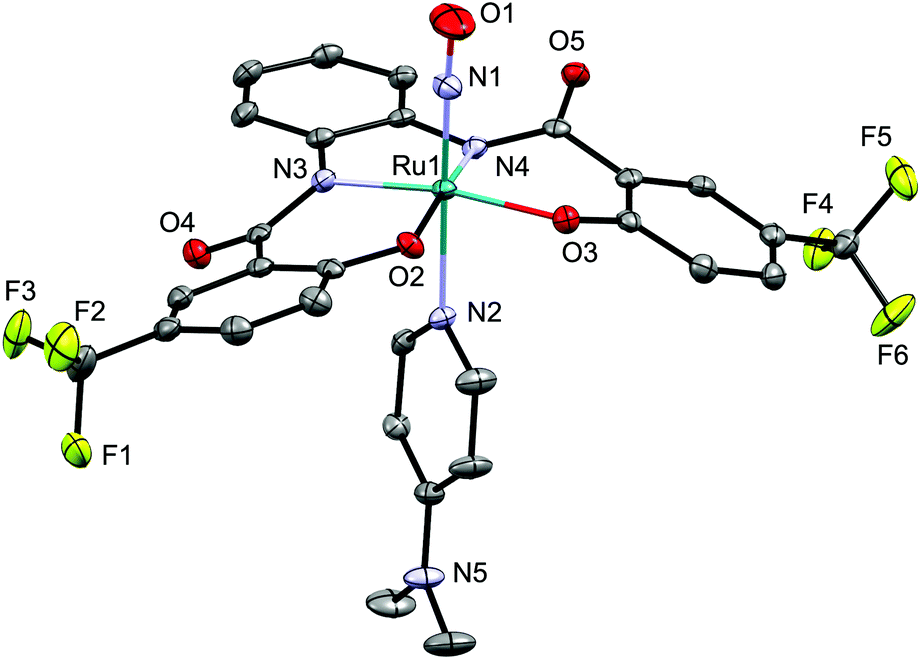 | ||
| Fig. 5 Molecular structure of the anionic part of NO complex 6. Only one of the two independent anions per unit cell is shown. Hydrogen atoms are omitted for clarity and only selected atoms are labelled. Selected bond lengths (Å) and angles (°): O1–N1 1.144(3); N1–Ru1 1.752(2); Ru1–N2 2.1325(19); O1–N1–Ru1 174.9(2); N1–Ru1–N2 176.57(8). | ||
Properties of a series of NE complexes
The selected metric parameters, Wiberg bond indices (WBI) and the natural population analysis (NPA) charge distributions for a series of the obtained NE complexes (E = O, 6; E = S, 4d; E = Se, 5d) are summarized in Table 3 together with those of nitrido complex 3d for comparison. The Ru–NE bond lengths in 6, 4d, and 5d are longer compared to the Ru–N bond length in 3d [Ru–N (3d): 1.6040(15) Å; Ru–NO (6): 1.75 Å (mean); Ru–NS (4d): 1.75 Å (mean); Ru–NSe (5d): 1.73 Å (mean)]. The WBIs of the Ru–NE bonds in 6, 4d, and 5d decrease compared to the Ru–N bond in 3d. Considering the previously reported WBI for nitrido complex [(Ir(N){N(CHCHPtBu2)2}][PF6]13 and iridium NE complexes [(Ir(NE){N(CHCHPtBu2)2}][PF6],7c,12 the Ru–N triple bond in 3d should turn into a double bond after the nitrido ligand bonds with chalcogen atoms to generate 4d and 5d.| E | Nothing (3d) | O (6) | S (4d) | Se (5d) |
|---|---|---|---|---|
| a Distances and bond angles of both molecules per unit cell are shown. b The calculation of WBIs and NPA charges were performed at the B3PW91/SBKJC(d) level of theory. | ||||
| Bond angles | ||||
| dmap–Ru–N | — | 176.57(8) | 173.53(19) | 173.79(16) |
| 176.82(8) | 173.78(19) | 172.78(17) | ||
| Ru–N–E | — | 174.9(2) | 171.0(3) | 171.2(3) |
| 177.27(19) | 172.2(3) | 169.9(3) | ||
| Bond lengths | ||||
| RuN–E | — | 1.144(3) | 1.511(4) | 1.670(4) |
| 1.149(3) | 1.516(4) | 1.671(4) | ||
| Ru–NE | 1.6040(15) | 1.752(2) | 1.746(4) | 1.734(4) |
| 1.744(2) | 1.746(4) | 1.732(4) | ||
| N(dmap)–Ru | — | 2.1325(19) | 2.155(4) | 2.173(4) |
| 2.1327(18) | 2.157(4) | 2.173(4) | ||
| Wiberg bond index | ||||
| RuN–E | — | 1.8352 | 1.6889 | 1.5792 |
| Ru–NE | 2.3049 | 1.4144 | 1.3629 | 1.4072 |
| N(dmap)–Ru | — | 0.3154 | 0.2881 | 0.2753 |
| NPA charges | ||||
| Ru | 1.111 | 0.883 | 0.980 | 1.006 |
| N (NE) | −0.065 | 0.401 | −0.348 | −0.409 |
| E | — | −0.224 | 0.370 | 0.390 |
The Ru–NE bond lengths are comparable to the sum of the covalent double-bond radii (Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N: 1.74 Å),27 and the WBIs corroborate double-bond character for the Ru–NE bonds in the NE complexes (Ru–NO: 1.4144; Ru–NS: 1.3629; Ru–NSe: 1.4072). The N–E bond lengths [N–O: 1.15 Å (mean); N–S: 1.51 Å (mean); N–Se: 1.67 Å (mean)] are longer than the calculated covalent triple-bond radii (N
N: 1.74 Å),27 and the WBIs corroborate double-bond character for the Ru–NE bonds in the NE complexes (Ru–NO: 1.4144; Ru–NS: 1.3629; Ru–NSe: 1.4072). The N–E bond lengths [N–O: 1.15 Å (mean); N–S: 1.51 Å (mean); N–Se: 1.67 Å (mean)] are longer than the calculated covalent triple-bond radii (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O: 1.07 Å; NS: 1.49 Å; NSe: 1.61 Å),27 but similar to the corresponding double-bond radii (NO: 1.17 Å; NS: 1.54 Å; NSe: 1.67 Å).27 Taking the WBIs into account (N–O: 1.8352; N–S: 1.6889; N–Se: 1.5792), the N–E bonds in 4d, 5d, and 6 exhibit double-bond character.
O: 1.07 Å; NS: 1.49 Å; NSe: 1.61 Å),27 but similar to the corresponding double-bond radii (NO: 1.17 Å; NS: 1.54 Å; NSe: 1.67 Å).27 Taking the WBIs into account (N–O: 1.8352; N–S: 1.6889; N–Se: 1.5792), the N–E bonds in 4d, 5d, and 6 exhibit double-bond character.
The obtained data revealed characteristic features of the NE ligands, i.e., the order of π-back donation and trans influence of the NE ligands. The length of the Ru–NSe bond in 5d slightly decreases compared to the corresponding Ru–NO and Ru–NS bonds in 6 and 4d [Ru–NO (6): 1.75 Å (mean); Ru–NS (4d): 1.75 Å (mean); Ru–NSe (5d): 1.73 Å (mean)]. This result could be explained in terms of an increased π-back donation from the ruthenium center to the NSe ligand, which appears to reflect a better π-accepting character of the NSe ligand relative to the lighter chalcogen homologues. This trend is consistent with previously reported estimations based on theoretical calculations.28 However, it should also be noted here that this trend contradicts previous studies on complexes such as [Cr(NS)(OH2)5]2+,29 [Os(NE)Cl2Tp]7a or [Ru(NE)Cl3(AsPh3)2],7d,12 in which the respective NO ligands appear to be better π-acceptors than the NS and NSe ligands, as well as the studies on [Ir(NE){N(CHCHPtBu2)2}][PF6],7c,13 where the π-acceptor ability of the NS ligand is lower than that of the NO and NSe ligands.30
The aforementioned π-accepting nature of the NSe ligand in 5d was corroborated by the NPA charge distribution on the RuNSe moiety. The positive charge on the ruthenium atom is greater in 5d than in 6 and 4d [RuNO (6): 0.883; RuNS (4d): 0.980; RuNSe (5d): 1.006], while the negative charge on the nitrogen atom of the NE ligand is greater in 5d than in 6 and 4d [RuNO (6): 0.401; RuNS (4d): −0.348; RuNSe (5d): −0.409]. Therefore, the NSe ligand appears to be a better π-acceptor than the NO and NS ligands.
The NSe ligand in 5d exhibits a stronger trans influence than the NO and NS ligands in 6 and 4d, respectively. The N(dmap)–RuNE bond length slightly increases in the order E = O < S < Se [N–RuNO (6): 2.13 Å (mean); N–RuNS (4d): 2.16 Å (mean); N–RuNSe (5d): 2.17 Å (mean)], and the corresponding WBIs of N(dmap)–Ru decrease in the same order [N–RuNO (6): 0.3154; N–RuNS (4d): 0.2880; N–RuNSe (5d): 0.2753]. Considering the order of trans influence, the NSe ligand appears to exhibit a better σ-donating ability than the lighter chalcogen homologues. This trend is consistent with the data obtained for [Ru(NE)Cl3(AsPh3)2] (E = O, S, Se)7d,12 wherein the NS and NSe ligands exert a stronger trans influence than the NO ligand.
Conclusions
Anionic ruthenium NS and NSe complexes were synthesized via the reaction of the corresponding nitrido complexes with elemental sulfur or selenium in the presence of DMAP, which provided the first anionic NSe complexes. The structural properties of these NE (E = O, S, Se) complexes were determined by single-crystal X-ray diffraction analyses and theoretical calculations, which revealed that in these complexes, the NSe ligand is the best π-acceptor with the strongest trans influence.Experimental
General considerations
Unless otherwise noted, all reactions were performed in oven-dried (110 °C) glassware under an atmosphere of dry argon (balloon). Reagents were obtained from common commercial sources and used as received. Anhydrous solvents for reactions were purchased from Kanto Chemical Co., Inc. and used as received. Solvents for workup, column chromatography, and recrystallization were of ‘reagent grade’. The following compounds were prepared as described in the literature: [Bu4N][Ru(N)Cl4],20 tetrahydro-2-[4-(trifluoromethyl)phenoxy]-2H-pyran,17 and [Ru(NO)Cl3].26 Commercially available sulfur powder was recrystallized from benzene prior to use. TLC was performed on Merck TLC silica gel 60 F254 plates. Silica gel used for flash column chromatography (silica gel 60N; spherical; neutral; 40–50 μm) was obtained from Kanto Chemical Co., Inc. Alumina used for column chromatography (Aluminium Oxide 90; active neutral; 63–200 μm) was obtained from Merck.Melting points are uncorrected. IR spectra were recorded using a diamond-attenuated total reflectance (ATR) unit and are corrected. NMR spectra were recorded at ambient temperature on a JEOL ECZ 400 spectrometer (1H: 400 MHz; 13C: 100 MHz; 19F: 375 MHz; 31P: 161 MHz). Chemical shifts are reported in δ, relative to residual 1H and 13C{1H} signals of CDCl3 (1H: δ 7.24; 13C{1H}: 77.16) and (CD3)2SO with 0.03% v/v TMS (1H: δ 2.50; 13C{1H}: δ 39.52) or to the external 19F signal of C6F6 (δ −164.9) or 31P{1H} signal of H3PO4 (δ 0.00). ESI-HRMS were obtained on an FT-ICR mass spectrometer. Elemental analyses were performed on a Perkin-Elmer 2400 series II CHN analyser. The number of solvent molecules in the products was determined by elemental analysis and 1H NMR spectroscopy.
General procedure for the syntheses of N,N′-(1,2-phenylene)bis(2-hydroxybenzamide) and derivatives (1a–c)
A previous reported procedure was slightly modified.15 Ph3PCl2 (4.8 equiv.) was added to a CHCl3 (40 mL) solution of phenylenediamine (1.0 equiv.) and salicylic acid (2.4 equiv.), and the mixture was stirred under reflux for 17 h. The resulting mixture was poured into iced water and extracted with EtOAc. The organic layer was consecutively treated with an aqueous solution of HCl (4 M), a saturated aqueous solution of NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was passed through silica gel (EtOAc) in order to remove Ph3PO, and the fraction Rf ≈ 0.8 was collected and concentrated in vacuo. The residual material was purified by column chromatography on silica gel (EtOAc/hexane) to afford the product as a white solid.:1 was used as the eluent after Ph3PO was removed. 1a·0.1EtOAc was isolated as a white solid (0.981 g, 2.75 mmol, 92% yield). The NMR data of synthesized 1a was consistent with reported data.31
:1 was used as the eluent after Ph3PO was removed. 1b·0.1EtOAc was isolated as a white solid (1.14 g, 2.68 mmol, 89% yield). The 1H NMR data matched those reported previously.3213C{1H} NMR ((CD3)2SO with 0.03% v/v TMS): δ 170.4 (EtOAc), 164.9, 157.0, 133.4, 131.0, 129.1, 126.0, 125.5, 123.1, 119.3, 118.7, 59.8 (EtOAc), 20.8 (EtOAc), 14.1 (EtOAc). HRMS (ESI−) m/z: [M − H]− calcd for C20H13N2O435Cl2: 415.0255; found: 415.0258. IR (ATR, cm−1): νNH = 3311, νCO = 1645. mp: 248.6–251.4 °C.
:3 was used as the eluent after Ph3PO was removed. 1c·0.1EtOAc was isolated as white solid (1.24 g, 2.51 mmol, 84% yield). 1H NMR ((CD3)2SO with 0.03% v/v TMS): δ 11.81 (s, 2H), 10.53 (s, 2H), 8.16 (s, 2H), 7.99 (d, J = 2.6 Hz, 2H), 7.49 (dd, J = 8.8, 2.7 Hz, 2H), 7.03 (d, J = 8.9 Hz, 2H), 4.02 (q, J = 7.1, 0.2H, EtOAc), 1.98 (s, 0.3H, EtOAc), 1.17 (t, J = 7.1 Hz, 0.3H, EtOAc). 13C{1H} NMR ((CD3)2SO with 0.03% v/v TMS): δ 164.6, 156.6, 133.6, 131.0, 129.3, 127.4, 126.2, 123.2, 119.2, 118.6. HRMS (ESI−) m/z: [M − H]− calcd for C20H11N2O435Cl4: 482.9477; found 482.9478. IR (ATR, cm−1): νNH = 3332, νCO = 1636. mp: 284.7–286.7 °C.
:10, v/v) to afford 2 as a colourless oil (0.251 g, 0.789 mmol, 71% yield). 1H NMR (CDCl3): δ 8.02 (d, J = 2.3 Hz, 1H), 7.63 (dd, J = 8.9, 2.5 Hz, 1H), 7.29 (d, J = 8.7 Hz, 1H), 5.58 (t, J = 2.7 Hz, 1H), 4.36 (qd, J = 7.1, 1.9 Hz, 2H), 3.83 (dd, J = 11.2, 3.0 Hz, 1H), 3.60 (d, J = 11.0 Hz, 1H), 1.61–2.04 (m, 6H), 1.37 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (CDCl3): δ 165.5, 158.7, 130.0 (dd, J = 3.9, 2.9 Hz), 128.8 (q, J = 3.9 Hz), 124.0 (q, J = 271.7 Hz), 123.2 (q, J = 33.7 Hz), 121.8, 116.2, 96.6, 61.9, 61.4, 30.1, 25.2, 18.1, 14.4. 19F NMR (CDCl3): δ −60.3. HRMS (ESI+) m/z: [M + Na]+ calcd for C15H17O4F3Na+: 341.0974; found 341.0971. IR (ATR, cm−1): νCO = 1732.
General procedure for the syntheses of N,N′-(1,2-phenylene)bis(5-trifluoromethyl-2-hydroxybenzamide) derivatives 1d and 1e
A previous reported procedure was slightly modified.18a NaHMDS (1.0 M solution in hexane, 2.0 equiv.) was added dropwise to a THF (2.0 mL) solution of 2 (1.0 equiv.) and phenylenediamine (0.45 equiv.) at 0 °C. The mixture was stirred for 5 min at 0 °C before it was allowed to warm to room temperature, where it was stirred for 16 h. The resulting mixture was added to a saturated aqueous solution of NH4Cl and extracted with EtOAc. The organic layer was treated with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to leave a red residue.This residue was dissolved in THF (1.0 mL) and 2 M HCl (1.9 mL, 2.2 equiv.) was added. The mixture was stirred at room temperature for 8 h. The resulting mixture was added to a saturated aqueous solution of NaHCO3 and extracted with EtOAc. The organic layer was treated with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (EtOAc/hexane) to afford the product.
:1 was used as the eluent. 1d·0.2EtOAc was isolated as a pale yellow solid (0.258 g, 0.514 mmol, 69% yield over 2 steps). 1H NMR ((CD3)2SO with 0.03% v/v TMS): δ 12.41 (s, 2H), 10.52 (s, 2H), 8.32 (s, 2H), 7.76–7.82 (m, 4H), 7.32 (q, J = 3.2, 2H), 7.16 (d, J = 8.7 Hz, 2H), 4.03 (q, J = 7.1, 0.4H, EtOAc), 1.99 (s, 0.6H, EtOAc), 1.17 (t, J = 7.1 Hz, 0.6H, EtOAc). 13C{1H} NMR ((CD3)2SO with 0.03% v/v TMS): δ 170.4 (EtOAc), 164.9, 161.1, 131.0, 130.4, 127.5, 126.1, 125.6, 124.3 (q, J = 271.7 Hz), 120.1 (q, J = 32.8 Hz), 118.3, 117.8, 59.8 (EtOAc), 20.7 (EtOAc), 14.1 (EtOAc). 19F NMR ((CD3)2SO with 0.03% v/v TMS): δ −58.4. HRMS (ESI−) m/z: [M − H]− calcd for C22H13N2O4F6: 483.0779, found 483.0785. IR (ATR, cm−1): νNH = 3324, νCO = 1615. mp: 234.2–237.0 °C.
:3 was used as the eluent. 1e·0.1EtOAc was isolated as pale yellow solid (0.284 g, 0.505 mmol, 66% yield over 2 steps). 1H NMR ((CD3)2SO with 0.03% v/v TMS): δ 12.33 (s, 2H), 10.59 (s, 2H), 8.29 (s, 2H) 8.16 (s, 2H), 7.79 (d, J = 8.7, 2.3 Hz, 2H), 7.17 (d, J = 8.2 Hz, 2H), 4.03 (q, J = 7.1 Hz, 0.2H, EtOAc), 1.99 (s, 0.3H, EtOAc), 1.17 (t, J = 7.1 Hz, 0.3H, EtOAc). 13C{1H} NMR ((CD3)2SO with 0.03% v/v TMS): δ 171.1 (EtOAc), 164.6, 160.7, 131.1, 130.6, 127.6, 127.5, 126.4, 124.3 (q, J = 270.7 Hz), 120.1 (q, J = 32.8 Hz), 118.3, 117.8, 59.8 (EtOAc), 20.8 (EtOAc), 14.1 (EtOAc). 19F NMR ((CD3)2SO with 0.03% v/v TMS): δ −58.4. HRMS (ESI−) m/z: [M − H]− calcd for C22H11N2O4F635Cl2: 551.0008, found 551.0006. IR (ATR, cm−1): νNH = 3357, νCO = 1647. mp: 274.6–277.5 °C.
General procedure for the syntheses of nitrido complexes [Ph4P][RuN(hybebR1,R2)] (3)
Nitrido complexes 3 were synthesized according to a modified literature procedure.19a 2,6-Lutidine (excess, 0.73 mL) was added to a MeOH/THF = 3:1 (5.1 mL:1.7 mL) solution of 1 (0.500 mmol, 1.00 equiv.) and [Bu4N][Ru(N)Cl4]19 (0.250 g, 0.500 mmol, 1.0 equiv.), and the mixture was stirred for 16 h at room temperature. The solvent was removed in vacuo, and the black/brown residue was purified by column chromatography on alumina (CHCl3). The orange band was collected and concentrated in vacuo, and the residue was recrystallized from CH2Cl2/hexane to afford [Bu4N][Ru(N)(hybebH,H)] as an orange solid. The orange complex (1.0 equiv.) was dissolved in CH2Cl2 (10.0 mL per mmol) and [Ph4P]Br (1.0 equiv.) was added. After stirring for 8 h, water was added, and the mixture was extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on alumina (CHCl3) and recrystallized (CH2Cl2/hexane) to afford the product.
General procedure for the syntheses of NS complexes [Ph4P][Ru(NS)(hybebR1,R2)(dmap)] (4)
1/8 S8 (0.160 g, 5.00 mmol, 10 equiv. as S) and DMAP (0.611 g, 5.00 mmol, 10 equiv.) were added to a MeCN solution (20 mL) of nitrido complex 3 (0.500 mmol, 1.0 equiv.). After the mixture was stirred at room temperature for the reaction time shown in Table 1, the solvent was removed in vacuo. The residue was dissolved in a small amount of acetone, and a large amount of water was added. The resulting brown solid was collected by filtration and recrystallized from acetone/MTBE (methyl tert-butyl ether) to afford 4 as black crystals.General procedure for the syntheses of NSe complexes [Ph4P][Ru(NSe)(hybebR1,R2)(dmap)] (5)
A 30 mL two-necked flask containing grey selenium powder (0.790 g, 10.0 mmol, 20 equiv.) was heated under vacuum, before argon gas was introduced. In a separate flask, nitrido complex 3 (0.500 mmol, 1.0 equiv.) and DMAP (0.611 g, 5.00 mmol, 10 equiv.) were dissolved in MTBE/CH2Cl2 (15 mL:1 mL) at room temperature. The resulting solution was added to the flask containing the dried selenium, and the mixture was stirred at room temperature for the reaction time shown in Table 2. The solvent was removed in vacuo to leave a black residue. During the subsequent manipulations, the temperature of the solution containing the product should be kept below 0 °C. To remove the selenium powder, the residue was suspended at 0 °C in CH2Cl2 and filtered through a pad of Celite into a glass tube that was kept at 0 °C. The filtrate was layered with MTBE and stored in a refrigerator (0 °C) for one day to afford 5 as black crystals, which were collected by suction filtration or decantation, and dried under vacuum.
During the measurements of the 13C{1H} NMR spectra, 5c–e decomposed to generate nitrido complexes 3c–e, DMAP, and selenium. For this reason, the 13C{1H} NMR spectra exhibit signals of 5c–e together with those of 3c and DMAP (Fig. S32, S34, and S36, ESI†).
Synthesis of NO complex [Ph4P][Ru(NO)(hybebH,CF3)(dmap)] (6)
Initially, a previously reported procedure was modified.25 A solution of 1d·0.1EtOAc (0.178 g, 0.360 mmol, 1.0 equiv.) in DMF (12 mL) was added to NaH, and the mixture was stirred for 0.5 h. The colour of the mixture changed from yellow to brown. The mixture was then transferred to a DMF (5 mL) solution of [RuNOCl3]25 (0.0855 g, 0.360 mmol, 1.0 equiv.) and DMAP (0.0440 g, 0.360 mmol, 1.0 equiv.). The mixture was stirred under reflux for 2 h, before the solvent was removed by distillation. The black residue was dissolved in acetonitrile and filtered, before the filtrate was concentrated in vacuo. The crude product was purified by recrystallization from acetone/MTBE to yield the sodium salt as a black solid.Next, a solution of [Ph4P]Br (0.0717 g, 0.171 mmol, 1.0 equiv.) in CH2Cl2 (3 mL) was added and the mixture was stirred overnight. The mixture was transferred into water and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on alumina (CHCl3/MeOH = 20:1, v/v) and the product was recrystallized from CH2Cl2/MTBE to afford 6 as black crystals (0.132 g, 0.123 mmol, 34% yield from 1d·0.1EtOAc). 1H NMR (CDCl3): δ 8.99 (td, J = 6.3, 2.7 Hz, 2H), 8.58 (d, J = 1.4 Hz, 2H), 7.83 (d, J = 7.3 Hz, 2H), 7.75 (dd, J = 7.9, 6.7 Hz, 4H), 7.59 (td, J = 7.8, 3.5 Hz, 8H), 7.42–7.47 (m, 8H), 7.21–7.24 (m, 2H), 7.08 (d, J = 8.7 Hz, 2H), 6.79 (q, J = 3.3 Hz, 2H), 6.00 (d, J = 6.8 Hz, 2H), 2.72 (s, 6H). 13C{1H} NMR (CDCl3): δ 170.9, 166.7, 154.7, 147.3, 145.3, 135.9 (d, J = 2.9 Hz), 134.3 (d, J = 10.6 Hz), 131.5 (d, J = 3.9 Hz), 130.8 (d, J = 13.5 Hz), 127.1 (d, J = 1.9 Hz), 125.7 (q, J = 270.7 Hz), 124.2, 122.7, 122.6, 121.7, 117.4 (d, J = 89.6 Hz), 116.3 (q, J = 32.8 Hz), 105.9, 39.0. 19F NMR (CDCl3): δ −58.6. 31P{1H} NMR (CDCl3): δ 23.5. HRMS (ESI−) m/z: [M − Ph4P − dmap]− calcd for C22H10F6N3O5Ru: 611.9568; found 611.9582. IR (ATR, cm−1): νNO = 1810.
X-ray diffraction studies
All diffraction data were collected at −173 °C on a Bruker Apex II Ultra X-ray diffractometer equipped with a Mo-Kα radiation (λ = 0.71073 Å) source. Intensity data were processed using the Apex3 software suite. The solution of the structures and the corresponding refinements were carried out using the Yadokari-XG33 graphical interface. The positions of the non-hydrogen atoms were determined by using the SHELXT34 program and refined on F2 by full-matrix least-squares techniques using the SHELXL-201835 program. All non-hydrogen atoms were refined with anisotropic thermal parameters, while all hydrogen atoms were placed using AFIX instructions. Details of the diffraction data are summarized in Tables S1–S4 (ESI†). For 4d and 5d, the structures were refined as an inversion twin. Although two residual peaks slightly above 1 e Å−3 were observed in the final difference map, these have no chemical meaning.Computational details
All density functional theory (DFT) calculations were performed using the Gaussian 09 package.36 The computers used in this study are part of the computer facilities of the Academic Center for Computing Media Studies (ACCMS) at Kyoto University (Japan). The geometries of the anionic part of 4d, 5d, and 6 were fully optimized using the B3PW9137 density functional with the effective core potential (ECP) proposed by Stevens et al.,38 SBKJC, with double-ζ polarization functions, which is denoted SBKJC(d) in this paper. Vibrational analyses based on force constant matrices (Hessians) were carried out at the stationary points in order to identify minima (all positive constants), transition states (one negative force constant), or higher-order saddle points. NAO-based Wiberg bond indices39 and natural population analysis (NPA) charge distributions40 were estimated using the NBO6.0 program41 based on the optimized structures. Optimized Cartesian coordinates of anionic part for 4d, 5d, and 6 are summarized in Tables S5–S7 (ESI†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI Grant Number 23550082. We would thank Prof. Youichi Ishii (Chuo University, Japan) for his generous support as well as Mr Wataru Abe and Mr Takashi Kurihara (Chuo University, Japan) for their assistance in the early stages of this work. We would like to thank members of the Ishii Research Group for elemental analysis measurements (Chuo University, Japan), and Prof. Mao Minoura for his help on the solution of the solid-state structures. Furthermore, we would like to thank ACCMS at Kyoto University and the Research Center for Computational Science Okazaki (Japan) for providing access to their computational facilities.Notes and references
- (a) F. Murad, Angew. Chem., Int. Ed., 1999, 38, 1856–1868 CrossRef CAS; (b) R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS; (c) L. J. Ignarro, Angew. Chem., Int. Ed., 1999, 38, 1882–1892 CrossRef CAS; (d) K. Ghimire, H. M. Altmann, A. C. Straub and J. S. Isenberg, Am. J. Physiol.: Cell Physiol., 2017, 312, C254–C262 CrossRef PubMed; (e) A. De Mel, F. Murad and A. M. Seifalian, Chem. Rev., 2011, 111, 5742–5767 CrossRef CAS PubMed.
- (a) T. W. Hayton, P. Legzdins and W. B. Sharp, Chem. Rev., 2002, 102, 935–991 CrossRef CAS PubMed; (b) B. Machura, Coord. Chem. Rev., 2005, 249, 2277–2307 CrossRef CAS; (c) M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093–2114 CrossRef CAS PubMed.
- For studies on NS, see: (a) W. M. Irvine, M. Senay and A. J. Lovell, Icarus, 2000, 143, 412–414 CrossRef CAS PubMed; (b) S. B. Yaghlane, S. Lahmar, Z. B. Lakhdar and M. Hochlaf, J. Phys. B: At., Mol. Opt. Phys., 2005, 38, 3395–3403 CrossRef; (c) S. B. Yaghlane and M. Hochlaf, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 015101 CrossRef. For studies on NSe, see: (d) J. M. Brown and H. Uehara, J. Chem. Phys., 1987, 87, 880–884 CrossRef CAS; (e) D. H. Shi, P. L. Li, J. F. Sun and Z. L. Zhu, Spectrochim. Acta, Part A, 2014, 117, 109–119 CrossRef CAS PubMed; (f) L. Andrews and P. Hassanzadeh, J. Chem. Soc., Chem. Commun., 1994, 1523–1524 RSC. For theoretical studies on NE molecules, see: (g) R. T. Boeré and T. L. Roemmele, in Comprehensive Inorganic Chemistry II, ed. J. Reedikj and K. Poeppelmeier, Elsevier, 2013, vol. 1, pp. 375–411. DOI:10.1016/B978-0-08-097774-4.00117-0.
- (a) W. Petz, Coord. Chem. Rev., 2008, 252, 1689–1733 CrossRef CAS; (b) L. Busetto and V. Zanotti, Inorg. Chim. Acta, 2008, 361, 3004–3011 CrossRef CAS; (c) Y. Mutoh, N. Kozono, K. Ikenaga and Y. Ishii, Coord. Chem. Rev., 2012, 256, 589–605 CrossRef CAS; (d) A. F. Hill and C. M. A. McQueen, Organometallics, 2012, 31, 2482–2485 CrossRef CAS; (e) A. Suzuki, T. Arai, K. Ikenaga, Y. Mutoh, N. Tsuchida, S. Saito and Y. Ishii, Dalton Trans., 2017, 46, 44–48 RSC; (f) I. A. Cade, A. F. Hill and C. M. A. McQueen, Dalton Trans., 2019, 48, 2000–2012 RSC; (g) B. J. Frogley, A. F. Hill and L. J. Watson, Dalton Trans., 2019, 48, 12598–12606 RSC.
- J. Chatt and J. R. Dilworth, J. Chem. Soc., Chem. Commun., 1974, 508 RSC.
- (a) J. F. Berry, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS; (b) X.-Y. Yi, Y. Liang and C. Li, RSC Adv., 2013, 3, 3477–3486 RSC; (c) J. M. Smith, in Progress of Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, 2014, ch. 6, vol. 58, pp. 417–470 Search PubMed; (d) W.-L. Man, W. W. Y. Lam and T.-C. Lau, Acc. Chem. Res., 2014, 47, 427–439 CrossRef CAS PubMed.
- (a) T. J. Crevier, S. Lovell and J. M. Mayer, J. Am. Chem. Soc., 1998, 120, 6607–6608 CrossRef CAS; (b) B. L. Tran, R. Thompson, S. Ghosh, X. Gao, C.-H. Chen, M.-H. Baik and D. J. Mindiola, Chem. Commun., 2013, 49, 2768–2770 RSC; (c) M. G. Scheibel, I. Klopsch, H. Wolf, P. Stollberg, D. Stalke and S. Schneider, Eur. J. Inorg. Chem., 2013, 3836–3839 CrossRef CAS; (d) H.-Y. Ng, W.-M. Cheung, E. K. Huang, K.-L. Wong, H. H.-Y. Sung, I. D. Williams and W. H. Leung, Dalton Trans., 2015, 44, 18459–18468 RSC; (e) H.-Y. Ng, N.-M. Lam, M. Yang, X.-Y. Yi, I. D. Williams and W.-H. Leung, Inorg. Chim. Acta, 2013, 394, 171–175 CrossRef CAS.
- For reactions with carbon disulphide, see: (a) E.-S. El-Samanody, K. D. Demadis, L. A. Gallagher, T. J. Meyer and P. S. White, Inorg. Chem., 1999, 38, 3329–3336 CrossRef CAS PubMed; (b) K. D. Demadis, E.-S. El-Samanody, T. J. Meyer and P. S. White, Inorg. Chem., 1998, 37, 838–839 CrossRef CAS.
- For reactions with thiosulfate salt, see: (a) A. Wu, A. Dehestani, E. Saganic, T. J. Crevier, W. Kaminsky, D. E. Cohen and J. M. Mayer, Inorg. Chim. Acta, 2006, 359, 2842–2849 CrossRef CAS; (b) X.-Y. Yi, T. C. H. Lam, Y.-K. Sau, Q.-F. Zhang, I. D. Williams and W.-H. Leung, Inorg. Chem., 2007, 46, 7193–7198 CrossRef CAS PubMed.
- (a) B. W. S. Kolthammer and P. Legzdins, J. Am. Chem. Soc., 1978, 100, 2247–2248 CrossRef CAS; (b) M. Herberhold and L. Haumaier, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1980, 35, 1277–1280 Search PubMed; (c) D. S. Bohle, C.-H. Hung, A. K. Powell, B. D. Smith and S. Wocadlo, Inorg. Chem., 1997, 36, 1992–1993 CrossRef CAS PubMed.
- For theoretical studies of NE complexes, see: (a) S. F. Vyboishchikov and G. Frenking, Theor. Chem. Acc., 1999, 102, 300–308 Search PubMed; (b) J. W. Dethlefsen, E. D. Hedegård, R. D. Rimmer, P. C. Ford and A. Døssing, Inorg. Chem., 2009, 48, 231–238 CrossRef CAS PubMed; (c) J. R. Dethlefsen, A. Døssing and E. D. Hedegård, Inorg. Chem., 2010, 49, 8769–8778 CrossRef CAS PubMed; (d) A. Døssing, Coord. Chem. Rev., 2016, 306, 544–557 CrossRef.
- D. H. F. Souza, G. Oliva, A. Teixeira and A. A. Batista, Polyhedron, 1995, 14, 1031–1034 CrossRef CAS.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- (a) J. W. Bats, K. K. Pandey and H. W. Roesky, J. Chem. Soc., Dalton Trans., 1984, 2081–2083 RSC; (b) K. K. Pandey, H. W. Roesky, M. Noltemeyer and G. M. Sheldrick, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1984, 39, 590–593 Search PubMed; (c) J. B. Raynor, T. J. Kemp and A. M. Thyer, Inorg. Chim. Acta, 1992, 193, 191–196 CrossRef CAS; (d) U. Abram, R. Hübener, R. Wollert, R. Krimse and W. Hiller, Inorg. Chim. Acta, 1993, 206, 9–14 CrossRef CAS; (e) M. Reinel, T. Höcher, U. Abram and R. Kirmse, Z. Anorg. Allg. Chem., 2003, 629, 853–861 CrossRef CAS.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- I. Azumaya, T. Okamoto, F. Imabeppu and H. Takayanagi, Tetrahedron, 2003, 59, 2325–2331 CrossRef CAS.
- H. Geneste and B. Schäfer, Synthesis, 2001, 2259–2262 CrossRef CAS.
- (a) J. Wang, M. Rosingana, R. P. Discordia, N. Soundararajan and R. Polniaszek, Synlett, 2001, 1485–1487 CrossRef CAS; (b) G. Li, C.-L. Ji, X. Hong and M. Szostak, J. Am. Chem. Soc., 2019, 141, 11161–11172 CrossRef CAS PubMed.
- (a) P.-M. Chan, W. Y. Yu, C.-M. Che and K.-K. Cheung, J. Chem. Soc., Dalton Trans., 1998, 3183–3190 RSC; (b) K.-L. Yip, W.-Y. Yu, P.-M. Chan, N.-Y. Zhu and C.-M. Che, Dalton Trans., 2003, 3556–3566 RSC.
- W. P. Griffith and D. Pawson, J. Chem. Soc., Dalton Trans., 1973, 1315–1320 RSC.
- L. Bonomo, E. Solari, R. Scopelliti and C. Floriani, Angew. Chem., Int. Ed., 2001, 40, 2529–2531 CrossRef CAS.
- G.-S. Fang, J.-S. Huang, N. Zhu and C.-M. Che, Eur. J. Inorg. Chem., 2004, 1341–1348 CrossRef CAS.
- D. Sellmann, M. W. Wemple, W. Donaubauer and F. Heinemann, Inorg. Chem., 1997, 36, 1397–1402 CrossRef CAS PubMed.
- R. M. Clarke and T. Storr, J. Am. Chem. Soc., 2016, 138, 15299–15302 CrossRef CAS PubMed.
- V. Panwar, A. Bansal, S. S. Ray and S. L. Jain, RSC Adv., 2016, 6, 71550–71556 RSC.
- C. A. Johnson, S. Sharma, B. Subramaniam and A. S. Borovik, J. Am. Chem. Soc., 2005, 127, 9698–9699 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- (a) K. K. Pandey, M. Massoudipour and V. Paleria, Indian J. Chem., 1990, 29A, 260–262 CAS; (b) K. K. Pandey, R. B. Sharma and P. K. Pandit, Inorg. Chim. Acta, 1990, 169, 207–210 CrossRef CAS; (c) K. K. Pandey, J. Coord. Chem., 1990, 22, 307–313 CrossRef; (d) S. F. Vyboishchikov and G. Frenking, Theor. Chem. Acc., 1999, 102, 300–308 Search PubMed; (e) C. M. Teague, T. A. O'Brien and J. F. O'Brien, J. Coord. Chem., 2002, 55, 627–631 CrossRef CAS.
- J. W. Dethlefsen and A. Døssing, Inorg. Chim. Acta, 2009, 362, 259–262 CrossRef CAS.
- K. K. Pandey and P. Patidar, Polyhedron, 2014, 68, 87–91 CrossRef CAS.
- A. D. Keramidas, A. B. Papaioannou, A. Vlahos, T. A. Kabanos, G. Bonas, A. Makriyannis, C. P. Rapropoulou and A. Terzis, Inorg. Chem., 1996, 35, 357–367 CrossRef CAS PubMed.
- Y. A. Ibrahim and A. H. M. Elwahy, Synthesis, 1993, 503–508 CrossRef CAS.
- (a) K. Wakita, Yadokari-XG, Software for Crystal Structure Analysis, 2001 Search PubMed; (b) C. Kabuto, S. Akine and E. Kwon, J. Crystallogr. Soc. Jpn., 2009, 51, 218–224 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (c) J. Lübben, C. M. Wandtke, C. B. Hübschle, M. Ruf, G. M. Sheldrick and B. Dittrich, Acta Crystallogr., Sect. A: Found. Adv., 2019, 75, 50–62 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, K. Ehara, R. Toyota, J. Fukuda, M. Hasegawa, T. Ishida, Y. Nakajima, M. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1996, 98, 5648–5652 CrossRef; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- (a) W. J. Stevens, H. Basch and M. Krauss, J. Chem. Phys., 1984, 81, 6026–6033 CrossRef; (b) W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Can. J. Chem., 1992, 70, 612–630 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- (a) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1998, 88, 899–926 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra, X-ray structures and computational data of 3d, 4d, 5d and 6. CCDC 1946037–1946040. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04219a |
| This journal is © The Royal Society of Chemistry 2020 |