
Improvement of the O2 storage/release rate of YMnO3 nanoparticles synthesized by the polymerized complex method†
Yusuke
Asakura,  *a
Shu
Yin
*a
*a
Shu
Yin
*a
*a
Shu
Yin
*a
Abstract
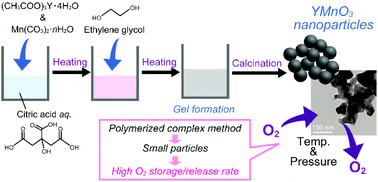
YMnO3 nanoparticles with a diameter of ca. 100 nm, which were synthesized by the polymerized complex method, exhibited a high O2 storage/release rate, because nanoparticle morphology increased the O2 accessible surface area of the material. This material with a high O2 storage/release rate is promising as a separator of O2 from air.
 Please wait while we load your content...
Please wait while we load your content...

