
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeutral and cationic enantiopure group 13 iminophosphonamide complexes†
Bhupendra
Goswami
 ,
Ravi
Yadav
,
Christoph
Schoo
and
Peter W.
Roesky
*
,
Ravi
Yadav
,
Christoph
Schoo
and
Peter W.
Roesky
*
Institut für Anorganische Chemie, Karlsruher Institut für Technologie (KIT), Engesserstr. 15, Geb. 30.45, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 17th December 2019
Abstract
Synthesis and reactivity of enantiopure iminophosphonamide ligand L-H (L = [Ph2P{N(R)CH(CH3)Ph}2]) with group 13 metal compounds has been investigated. The reaction of L-H with LiAlH4 afforded the aluminium monohydride complex [L2AlH]. The monochloride complexes [L2MCl] (M = Al, Ga) were accessed by reacting corresponding MCl3 (M = Al, Ga) with L-Li. Furthermore, the tetracoordinated aluminium cation [L2Al]+[GaCl4]− and gallium cation [L2Ga]+[AlCl4]− were obtained by chloride abstraction from [L2MCl] (M = Al, Ga), respectively. The title complexes represent the first examples of enantiopure group 13 metal complexes coordinated by chiral iminophosphonamides. All complexes have been characterized by single crystal X-ray diffraction, multinuclear NMR, EA and IR studies.
Introduction
Over the past decades, electron rich chelating mono anionic amido-ligands have widely been used in coordination chemistry to stabilize metal complexes.1–6 The isoelectronic analogy of nitrogen-based species with their oxygen counterpart is the guiding principle for the preparation of these N-donor ligands. Amidinate anions of the general formula [RC(NR′)2]− and iminophosphonamide anions of the general formula [R2P(NR′)2]− can be considered as the nitrogen analogue of the carboxylate and phosphinate anions, respectively. To date, ligand design is an important aspect in coordination chemistry to fine-tune specific structural and reactivity properties in metal complexes.7 In this regard, NXN ligand scaffolds are very attractive due to the possibility of different substituents for R and many variants for X such as CR (amidinate),2–4 BR (boraamidinate),8 C(NR2) (guanidinates),9 N (triazenide),10–12 SR (sulfinamidinate),13–16 and PR2 (iminophosphonamide).17The coordination chemistry with the achiral version of NXN ligand systems is very well explored. However, chemistry dealing with the chiral analogues of NXN ligand systems is scarce.18–21 In 2011, our group has described the synthesis of chiral amidinate (NCN)22 and subsequently synthesized corresponding alkaline earth metal and lanthanide complexes, which were active catalysts for hydroamination, hydrophosphination reactions and ring opening polymerization of racemic lactide.23–28 In comparison to amidinate, iminophosphonamide ligands show different structural features such as X–N bond lengths and NXN bite angles.29 In addition, the iminophosphonamide ligand system features an active nucleus for 31P NMR spectroscopy in the ligand backbone, being a useful tool to monitor the reaction progress. Having this in mind, very recently, we have reported the novel enantiopure iminophosphonamide L-H (L = [Ph2P{N(R)CH(CH3)Ph}2]) with chiral substituents at both nitrogen atoms and the corresponding alkali metal complexes L-M (M = Li, Na, K, Rb, and Cs).29 Interestingly, the alkali metal complexes (L-M) show remarkable TADF (thermally activated delayed fluorescence).
Halide, alkyl, and hydride substituted group 13 metal complexes are widely used as catalysts in Lewis acid mediated reactions for example Friedel–Craft and Diels–Alder reaction,30,31 initiator for cationic polymerization,32 hydroboration,33–35 hydro functionalization,36 and epoxidation of alkenes.37 In this regard, the cationic complexes are also of great interest due to increased electrophilicity resulting from the cationic charge, possibly enhance the substrate coordination and activation.32,38
Although achiral iminophosphonamides have a rich coordination chemistry, the structurally characterized group 13 metal complexes are limited to a few NPN ligand backbones such as [Ph2P(NSiMe3)2]−,39 [Ph2P(DipN)(NtBu)]− (Dip = 2,6-iPr2C6H3),40,41 [tBuP(H)(DipN)2]−,42 [Ph2P(NDip)2]−,43 and [rac-[trans-1,2-C6H12{NP(Ph2)N(Ar)}2]− (where Ar = 2,4,6-Me3C6H2 or 2,6-Me2C6H3).44 To the best of our knowledge, the coordination chemistry of group 13 metal complexes with the chiral iminophosphonamide ligand was not investigated yet.
Herein, we report the synthesis and characterization of chiral group 13 (Al and Ga) complexes utilising the recently reported chiral iminophosphonamide ligand (L).29

Results and discussion
Reacting LiAlH4 with L-H in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, resulted in the formation of complex [L2AlH] (1) in 65% yield (Scheme 1). The 1H NMR spectrum of complex 1 shows a doublet corresponding to Ph(CH)CH3 protons at δ 1.90 ppm with a coupling constant of 3JHH = 6.45 Hz. However, the corresponding Ph(CH)CH3 resonances for the ligand (L-H) are observed at δ 1.66 and 1.13 ppm due to proton (NH) exchange (tautomerization).29 The Ph(CH)CH3 protons of complex 1 appear as a broad signal at δ 4.99 ppm (Δν1/2 ≈ 55 Hz) without fine resolution. A resonance for the Al–H could not be observed possibly due to quadrupole relaxation of 27Al nuclei.45 In the IR spectrum of complex 1, a peak at 1693 cm−1 was assigned for the vibrational stretching of the Al–H bond (Fig. S18, ESI†). However, this value is not in the literature reported range of 1780–1853 cm−1 for related Al–H valence modes.39,40,45,46 This unusual red shift in the Al–H frequency for complex 1 could be due to stronger donor ability of ligand L.
:2, resulted in the formation of complex [L2AlH] (1) in 65% yield (Scheme 1). The 1H NMR spectrum of complex 1 shows a doublet corresponding to Ph(CH)CH3 protons at δ 1.90 ppm with a coupling constant of 3JHH = 6.45 Hz. However, the corresponding Ph(CH)CH3 resonances for the ligand (L-H) are observed at δ 1.66 and 1.13 ppm due to proton (NH) exchange (tautomerization).29 The Ph(CH)CH3 protons of complex 1 appear as a broad signal at δ 4.99 ppm (Δν1/2 ≈ 55 Hz) without fine resolution. A resonance for the Al–H could not be observed possibly due to quadrupole relaxation of 27Al nuclei.45 In the IR spectrum of complex 1, a peak at 1693 cm−1 was assigned for the vibrational stretching of the Al–H bond (Fig. S18, ESI†). However, this value is not in the literature reported range of 1780–1853 cm−1 for related Al–H valence modes.39,40,45,46 This unusual red shift in the Al–H frequency for complex 1 could be due to stronger donor ability of ligand L.
 | ||
| Scheme 1 Synthesis of complex 1. | ||
To further confirm the existence of the Al–H bond, the L2AlD (1D) isotopomer of complex 1 was synthesized by reacting LiAlD4 with L-H in the appropriate stoichiometric ratio. Complex 1D showed exactly the same 1H NMR as shown for complex 1H. Moreover, in the IR spectrum of 1D no peak was observed at 1693 cm−1 (Fig. S20 and S21, ESI†). Theoretical study suggests that the Al-D stretching frequency should appear at 1198 cm−1 due to isotopic shift.47 However, this region of IR spectrum is obstructed due to C–N stretch. Therefore, to confirm further theoretical calculations of these complexes were conducted. According to a theoretical calculation, the experimental spectra fit nicely with the calculated ones for both complexes 1H and 1D (see ESI†). Another method to differentiate between Al–H (D) bonding in 1H and 1D is provided by the investigation of the two usually very intense Al–H(D) deformation modes in the IR spectrum. They are found at 636 and 606 cm−1 (1H) as well as close to 482 cm−1 (1D). A second Al-D-deformation mode is presumably overlapped by the signals of the AlN4 framework at 537 and 508 cm−1 (both 1H and 1D). These findings are also confirmed by the DFT calculation.
The 31P{1H} NMR spectrum of complex 1 shows a single resonance at δ 33.6 ppm, which is downfield shifted compared to L-H (δ 2.7 ppm).29 Absence of N–H stretch in the IR of complex 1 further indicates the deprotonation of the ligand by LiAlH4.
Single crystals suitable for X-ray analysis were obtained from a saturated solution of complex 1 in diethyl ether. Complex 1 crystallises in an orthorhombic chiral space group P21212 with half of the molecule in the asymmetric unit cell. The aluminium centre in complex 1 is pentacoordinated and form a distorted trigonal bipyramidal (tbp) polyhedron (Fig. 1). The hydride atom could be found and refined in the difference Fourier map.
 | ||
| Fig. 1 Molecular structure of complex 1 in the solid state. All hydrogen atoms except the hydride are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Al–N1 2.040(2), Al–N2 1.932(2), Al–H 1.48(4), P1–N1 1.621(2), P1–N2 1.622(2); N1–Al–N1′ 165.03(9), N2–Al–N1′ 97.68(7), N2–Al–N1 74.93(7), N2–Al–N2′ 121.99(11), N1–Al–H 97.49(5), N2–Al–H 119.01(5), P1–Al–P1′ 139.9(4), N2–P1–N1 96.46(9). | ||
Two nitrogen atoms (N2 and N2′) of the iminophosphonamide ligand and the hydride form the equatorial plane, the sum of bond angles involving Al in this plane is 360°. The remaining two nitrogen atoms of the ligand backbone (N1 and N1′) occupy the axial positions with a N1–Al–N1′ bond angle of 165.03(9)°, which is significantly wider than the equatorial N2–Al–N2′ bond angle (121.99(11)°).
The Al–N1 bond length of 2.040(2) Å is longer than 1.932(2) Å of Al–N2 equatorial bond length. Deviation in the bond angles from the ideal tbp geometry is clearly arising from the chelating effect of the bidentate ligand (N1–P1–N2 96.46(9)°). The N2–Al–N1 angle (74.93(7)°) in complex 1 is almost similar to the N–Al–N bite angle reported in [{Ph2P(NSiMe3)2}2AlH] (75.66(5)°).39
Using our previously reported ligand lithium salt (L-Li).29 and subsequent reaction with MCl3 (M = Al, Ga) resulted in a facile elimination of LiCl and formation of [L2MCl] (M = Al (2), Ga (3); Scheme 2). Both complexes 2 and 3 were fully characterized by multinuclear NMR, IR, elemental analysis as well as single crystal X-ray diffraction studies. Complexes 2 and 3 are stable in the solid state at room temperature for several months under an inert atmosphere and are soluble in organic solvents such as thf, Et2O and toluene while they are insoluble in n-hexane.
 | ||
| Scheme 2 Synthesis of complex 2 and 3. | ||
In the 1H NMR spectra of 2 and 3, the Ph(CH)CH3 protons of the ligand backbone show broad resonances at δ 5.17 (Δν1/2 ≈ 140 Hz) (2) and 5.10 ppm (Δν1/2 ≈ 40 Hz) (3). A broad resonance at δ 1.89 (Δν1/2 ≈ 40 Hz) ppm could be observed for Ph(CH)CH3 protons of 2 whereas, same resonance for complex 3 showed a doublet at δ 1.90 ppm with 3JHH = 6.52 Hz.
To investigate the dynamic behaviour of these complexes (2 and 3) a VT (variable temperature) 1H NMR of complex 2 in thf-d8 was recorded in the temperature range of 283–173 K with 10 K decrease in each spectrum (Fig. S6†). The broad resonance for Ph(CH)CH3 at room temperature in the area of 4.3–5.3 ppm splits into two different signals at low temperature with integral ratio of 1:1. While cooling down from 253 to 243 K the doublet at δ 1.59 ppm (3JHH = 6.58 Hz) of Ph(CH)CH3 splits into two different signals.
At 203 K one signal could be detected as a doublet at δ 1.47 ppm (3JHH = 6.40 Hz) whereas the other one is merged with the solvent peak at δ 1.73 ppm. Furthermore, the 31P{1H} NMR signals for 2 and 3 show downfield shift as compared to L-Li (δ 35.6 (2) and 36.5 (3) vs. 29.7 ppm (L-Li)). The solid-state structures of complexes 2 and 3 show that both are isostructural and crystallizes in orthorhombic chiral space group P21212 with half of the molecule in the asymmetric unit cell. Likewise, complex 1, complexes 2 and 3 adopt distorted trigonal bipyramidal geometry with the central metal atoms surrounded by one chlorine atom and four nitrogen atoms of the ligand backbone (Fig. 2 and 3). Since complex 2 and 3 are isostructural, only complex 2 is discussed in detail here. The average P–N bond length in complex 2 is 1.6195(3) Å, which is in the range of previously reported single and double P–N bonds.48
 | ||
| Fig. 2 Molecular structure of complex 2 in the solid state. All the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Al–N1 1.909(3), Al–N2 2.040(3), Cl–Al 2.174(2), P1–N1 1.620(3), P1–N2 1.619(3); N1–Al–N1′ 126.4(2), N2–Al–N2′ 166.6(2), N1–Al–N2 75.13(13), N1′–Al–N2 98.69(14), N1–Al–Cl 116.81(11), N2–Al–Cl 96.72(10), P1–Al–P1′ 142.76(8), N2–P1–N1 96.2(2). | ||
 | ||
| Fig. 3 Molecular structure of complex 3 in the solid state. All the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Ga–N1 1.950(6), Ga–N2 2.087(5), Ga–Cl 2.240(2), P1–N1 1.617(6), P1–N2 1.625(7); N1–Ga–Cl 116.2(2), N2–Ga–Cl 96.88(13), N2–Ga–N2′ 166.2(3), N1–Ga–N1′ 127.7(3), N1–Ga–N2 73.4(2), N1–Ga–N2′ 100.4(2), N1–P1–N2 96.4(3). | ||
The Al–Cl bond length in complex 2 (Al–Cl 2.174(2) Å) is within the range (2.141(2)–2.202(6) Å) of related bis-amidinate aluminium monochloride complexes reported before.49–52 The N1–P1–N2 bond angle of 96.2(2)° in complex 2 is slightly narrower than 96.46(9)° as seen in case of complex 1, however wider than 93.90(6)° as reported for [{Ph2P(NSiMe3)2}2AlH].39 In complex 2, the two individual four membered N2PAl plane are twisted to each other with a dihedral angle of 49.92°.
Attempted synthesis of mono and trisubstituted complexes failed, possibly the ligand is not bulky enough to stabilize the monosubstituted product and may be trisubstituted products suffers from steric crowding of the ligand. These results suggest that disubstituted product is thermodynamically more stable.
In order to access cationic analogues of complexes 2 and 3, GaCl3 and AlCl3 were used as halide abstracting agents. The stoichiometric reaction of complex 2 with GaCl3 and 3 with AlCl3 in thf resulted in the formation of the expected products [L2Al]+[GaCl4]− (4) and [L2Ga]+[AlCl4]− (5), respectively (Scheme 3). By using GaCl3 for the chloride abstraction of the aluminium complex 2 and AlCl3 for the same reaction of the gallium complex 3, we show that ligand scrambling does not take place. Complexes 4 and 5 are sparingly soluble in n-hexane and toluene. The symmetric nature of the ligands around the metal centre in both 4 and 5 could be seen by appearance of a single set of signals for Ph(CH)CH3 and Ph(CH)CH3 protons in the corresponding 1H NMR spectrum. In accordance, the 31P{1H} NMR spectrum shows singlets at δ 43.3 ppm for complex 4, and at δ 48.6 ppm for complex 5. Further, upon heating the NMR samples of complexes 4 and 5 (in CDCl3) at 50 °C for 16 h, no changes were observed in the 1H and 31P{1H} NMR, suggesting that the cationic complexes (4 and 5) are stable under these conditions. Both complexes 4 and 5 crystallizes in a chiral trigonal space group P3121 with two halves of the cationic part and one anionic [MCl4]− (M = Ga (4) and Al (5)) part in the asymmetric unit cell. The central metal atoms in both cationic and anionic parts of [L2Al]+[GaCl4]−4 and [L2Ga]+[AlCl4]−5 adopt distorted tetrahedral geometry (Fig. 4 and 5). The central metal atom forms two N2PM planes with NPN ligand backbones, which are twisted with the dihedral angle of 89.97(1)° (4) and 87.53(2)° (5). The average M–N bond distance 1.864(4) Å (M = Al, 4) is smaller than 1.931(6) Å (M = Ga, 5). Obviously, the larger M–N bond distance in case of complex 5 is due to the larger ionic radii of gallium compared to aluminium.
 | ||
| Scheme 3 Synthesis of complex 4 and 5. | ||
 | ||
| Fig. 4 Molecular structure of complex 4 in the solid state. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Al–N1 1.857(4), Al–N2 1.871(4), Ga–Cl1 2.166(2), Ga–Cl2 2.171(2), Ga–Cl3 2.158(2), Ga–Cl4 2.15(2), P1–N1 1.631(4), P2–N2 1.629(4); N1–Al–N1′ 79.2(2), N1–Al–N2 126.3(2), N1–Al–N2′ 126.3(2), N2–Al–N2′ 79.7(2), P2–Al–P1 180.0, Cl1–Ga–Cl2 110.89(6), Cl3–Ga–Cl1 109.87(7), Cl3–Ga–Cl2 106.93(7), Cl4–Ga–Cl1 108.97(7), Cl4–Ga–Cl2 108.19(9), Cl4–Ga–Cl3 112.0(9), N1–P1–N1′ 93.0(3), N2–P2–N2′ 94.7(3). | ||
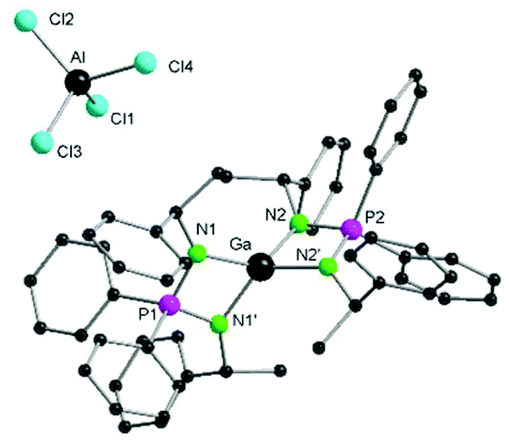 | ||
| Fig. 5 Molecular structure of 5 in the solid state. All the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Ga–N1 1.923(6), Ga–N2 1.938(6), Al–Cl1 2.129(3), Al–Cl2 2.119(4), Al–Cl3 2.134(4), Al–Cl4 2.109(4), P1–N1 1.629(6), P2–N2 1.626(6); N1–Ga–N1′ 76.9(4), N1–Ga–N2 126.5(3), N2–Ga–N2′ 77.2(4), N1–Ga–N2′ 129.0(3), Cl1–Al–Cl3 110.7(2), Cl2–Al–Cl1 109.8(2), Cl2–Al–Cl3 107.4(2), Cl4–Al–Cl1 109.2(2), Cl4–Al–Cl2 112.2(2), Cl4–Al–Cl3 107.5(2), N1–P1–N1′ 94.4(5), N2–P2–N2′ 96.1(5). | ||
Similarly, in case of the counter anion [MCl4]− the average M–Cl (M = Ga(4), Al(5)) bond distance 2.161(2) Å for complex 4 is larger compared to 2.123(4) Å for complex 5 and are in the expected range of literature reports.53,54
Complexes (1, 4 and 5) could be considered as a catalytically active species. Therefore, to check the catalytic activity of these complexes, an initial test reaction was conducted in the racemic lactide polymerization, however none of these complexes showed any catalytic activity. Further, in contrast to our previously reported alkali metal complexes of the ligand, observing these complexes (1–5) under UV light at room we did not observe any luminescence behavior of these complexes.
Conclusions
In conclusion, we have reacted the enantiopure ligand L-H (L = [Ph2P{N(R)CH(CH3)Ph}2]) and L-Li with group 13 metal compounds such as LiAlH4 and MCl3 (M = Al, Ga), respectively, to afford corresponding enantiopure monohydride (1) and monochloride complexes (2 and 3). Further the cationic complexes (4 and 5) were accessed by halide abstraction of complexes (2 and 3) using MCl3 (M = Al, Ga). To the best of our knowledge such enantiopure group 13 complexes based on chiral iminophosphonamide are not reported yet. All the complexes have been characterised thoroughly including their solid-state structure.Experimental section
All manipulations of air-sensitive materials were performed under the rigorous exclusion of oxygen and moisture in flame-dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−3 Torr) line, or in an argon-filled MBraun glove box. Hydrocarbon solvents (toluene, Et2O, n-pentane, n-heptane) were dried by using an MBraun solvent purification system (SPS-800), degassed and stored under vacuum over lithium aluminium hydride (LiAlH4). n-Hexane was pre-dried over CaCl2 before decantation and distillation from potassium and storage over 4 Å molecular sieves. Tetrahydrofuran was distilled under nitrogen from potassium benzophenoneketyl before storage over lithium aluminium hydride (LiAlH4). NMR spectra were recorded on a Bruker Avance II 300 MHz or Avance III 400 MHz. Elemental analyses were carried out with a Vario Micro Cube (Elementar Analysensysteme GmbH). IR spectra were obtained on a Bruker Tensor 37 FTIR spectrometer equipped with a room temperature DLaTGS detector, a diamond ATR (attenuated total reflection) unit, and a nitrogen flushed chamber In terms of their intensity, the signals were classified into the categories vs = very strong, s = strong, m = medium, w = weak and vw = very weak. (L-H)29 was prepared according to literature procedure.Synthesis of [L2AlH] (1)
LiAlH4 used in the following synthesis was purified by extracting with diethyl ether followed by filtration and removal of the solvent from the filtrate under vacuum to obtain LiAlH4 as white powder.To the mixture of L-H (425 mg, 1.0 mmol, 2.00 eq.) and LiAlH4 (19 mg, 0.5 mmol, 1.00 eq.) 40 mL of diethyl ether was added at room temperature. The reaction mixture was stirred overnight. After filtration and storing the concentrated filtrate at −30 °C for 2 days afforded colourless crystals suitable for X-ray analysis. The mother liquor was decanted-off and the crystals were washed with n-pentane (5 mL) and dried under vacuum.
Yield (based on crystals): 285 mg (65%). Elemental analysis calcd (%) for [C56H57N4P2Al] (875.03): C 76.87, H 6.57, N 6.40; found: C 76.93, H 6.97, N 6.20. 1H NMR (C6D6, 400 MHz): δ [ppm] = 7.63 (br, 8H, o-Arphos–H), 7.20–7.18 (m, 8H, Ar–H), 7.00–6.85 (m, 24H, o, m, p-Ar–H), 4.99 (br, 4H, Ph(CH)CH3), 1.90 (d, 3JHH = 6.45 Hz, 12H, Ph(CH)CH3). 13C{1H} NMR (C6D6, 100 MHz): δ [ppm] = 147.8 (Ar–Cq), 133.9 (d, 2JPC = 10.2 Hz, o-Arphos–CH), 131.7 (d, JPC = 91.0 Hz, Ar–CH), 130.8 (d, JPC = 2.7 Hz, Ar–CH), 128.0 (Ar–CH), 127.7 (Ar–CH), 127.6 (Ar–CH), 125.9 (Ar–CH), 54.1 (Ph(CH)CH3), 25.6 (d, 3JPC = 12.3 Hz, Ph(CH)CH3). 31P{1H} NMR (C6D6, 162 MHz): δ [ppm] = 33.6. IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] = 3081 (vw), 3054 (vw), 3024 (vw), 2969 (vw), 2958 (vw), 2929 (vw), 1693 (m), 1601 (w), 1590 (vw), 1493 (w), 1435 (w), 1369 (w), 1349 (w), 1273 (w), 1238 (w), 1220 (vs), 1202 (m), 1189 (w), 1136 (w), 1111 (m), 1103 (m), 1068 (w), 1044 (vw), 1021 (vw), 984 (w), 973 (w), 905 (vw), 856 (s), 826 (s), 783 (m), 771 (m), 753 (m), 740 (m), 719 (vs), 689 (w), 663 (m), 606 (m), 584 (w), 571 (s), 537 (s), 508 (w), 465 (vw), 418 (vw).
[cm−1] = 3081 (vw), 3054 (vw), 3024 (vw), 2969 (vw), 2958 (vw), 2929 (vw), 1693 (m), 1601 (w), 1590 (vw), 1493 (w), 1435 (w), 1369 (w), 1349 (w), 1273 (w), 1238 (w), 1220 (vs), 1202 (m), 1189 (w), 1136 (w), 1111 (m), 1103 (m), 1068 (w), 1044 (vw), 1021 (vw), 984 (w), 973 (w), 905 (vw), 856 (s), 826 (s), 783 (m), 771 (m), 753 (m), 740 (m), 719 (vs), 689 (w), 663 (m), 606 (m), 584 (w), 571 (s), 537 (s), 508 (w), 465 (vw), 418 (vw).
Synthesis of [L2AlCl] (2)
To the mixture of L-Li (1.00 g, 2.32 mmol, 2.00 eq.) and AlCl3 (155 mg, 1.16 mmol, 1.00 eq.), 60 mL of diethyl ether was added. The resulting solution was stirred overnight. After filtration and concentrating the filtrate afforded colourless crystals, suitable for X-ray structure analysis at room temperature. The mother liquor was decanted, and the crystals were washed with n-pentane (5 mL).
Yield (based on crystals): 750 mg (71%). Elemental analysis calcd (%) for [C56H56N4P2AlCl] (909.47): C 73.96, H 6.21, N 6.16; found: C 74.47, H 6.67, N 5.67. 1H NMR (C6D6, 400 MHz, 298 K): δ [ppm] = 7.68 (br, 8H, Ar–H), 7.09–6.84 (m, 32H, Ar–H), 5.17 (br, 4H, Ph(CH)CH3), 1.89 (br, 12H, Ph(CH)CH3). 1H NMR (thf-d8, 400 MHz, 298 K): δ [ppm] = 7.51 (br, 8H, Ar–H), 7.31 (br, 4H, Ar–H), 7.13 (br, 8H, Ar–H), 6.93–6.79 (br, 20H, Ar–H), 4.79 (br, 4H, Ph(CH)CH3), 1.59 (d, 3JHH = 6.58 Hz, 12H, Ph(CH)CH3). 13C{1H} NMR (C6D6, 100 MHz): δ [ppm] = 146.8 (Ar–Cq), 134.5–134.4 (Ar–CH), 131.1 (Ar–CH), 130.1 (Ar–CH), 128.2–127.9 (Ar–CH), 127.7 (Ar–CH), 127.5 (d, JPC = 11.9 Hz, Ar–CH), 126.0 (Ar–CH), 54.3 (Ph(CH)CH3), 26.4 (Ph(CH)CH3). 31P{1H} NMR (C6D6, 162 MHz): δ [ppm] = 35.6. IR (ATR): [cm−1] = 3057 (vw), 2989 (vw), 2960 (w), 2860 (vw), 1496 (w), 1484 (vw), 1452 (w), 1439 (m), 1434 (m), 1372 (w), 1364 (w), 1215 (w), 1205 (w), 1190 (w), 1185 (vs), 1128 (s), 1114 (s), 1094 (m), 1068 (m), 1045 (m), 1039 (m), 1025 (m), 998 (w), 974 (vs), 907 (m), 858 (m), 837 (m), 823 (m), 776 (w), 759 (m), 752 (m), 743 (m), 734 (w), 730 (w), 722 (w), 706 (vs), 663 (m), 641 (m), 617 (w), 590 (m), 579 (s), 539 (m), 516 (vs), 487 (m), 470 (m), 456 (m), 414 (m).
Synthesis of [L2GaCl] (3)
Following the procedure described above for 2 the reaction of L-Li (1.00 g, 2.32 mmol, 2.00 eq.) and GaCl3 (205 mg, 1.16 mmol, 1.00 eq.) in 60 mL of diethyl ether, afforded colourless crystals, suitable for X-ray structure analysis at room temperature. The solvent was decanted, and the product was washed with n-pentane (5 mL).
Yield (based on crystals): 650 mg (59%). Elemental analysis calcd (%) for [C56H56N4P2GaCl] (952.21): C 70.64, H 5.93, N 5.88; found: C 70.74, H 6.57, N 5.58. 1H NMR (C6D6, 400 MHz): δ [ppm] = 7.69–7.65 (m, 8H, Ar–H), 7.11–7.10 (m, 8H, Ar–H), 7.01–6.85 (m, 24H, o, m, p-Ar–H), 5.10 (br, 4H, Ph(CH)CH3), 1.90 (d, 3JHH = 6.52 Hz, 12H, Ph(CH)CH3). 13C{1H} NMR (C6D6, 100 MHz): δ [ppm] = 147.0 (Ar–Cq), 134.2 (d, JPC = 10.3 Hz, Ar–CH), 131.1 (Ar–CH), 130.1 (Ar–CH), 128.1 (Ar–CH), 127.7 (Ar–CH), 127.6 (d, JPC = 12.0 Hz, Ar–CH), 126.0 (Ar–CH), 54.5 (Ph(CH)CH3), 26.4 (Ph(CH)CH3). 31P{1H} NMR (C6D6, 162 MHz): δ [ppm] = 36.5. IR (ATR): [cm−1] = 3058 (vw), 3025 (vw), 2960 (vw), 2925 (vw), 1493 (w), 1451 (w), 1436 (m), 1372 (w), 1276 (w), 1238 (m), 1205 (s), 1182 (m), 1142 (vw), 1129 (vw), 1113 (w), 1104 (w), 1068 (vw), 1043 (w), 999 (w), 868 (m), 829 (m), 772 (m), 749 (s), 743 (m), 693 (vs), 658 (w), 633 (w), 570 (m), 530 (vs), 508 (m), 446 (w).
Synthesis of [L2Al]+[GaCl4]− (4)
To the mixture of 2 (150 mg, 0.33 mmol, 1.00 eq.) and GaCl3 (29 mg, 0.33 mmol, 1.00 eq.), 10 mL of thf was added at room temperature. All the volatiles were removed in vacuo after stirring for 2 hours to obtain white powder. Single crystals suitable for X-ray structure analysis were grown from concentrated benzene solution at room temperature.
Yield (based on crystals): 98 mg (55%). Elemental analysis calcd (%) for [C56H56N4P2AlGaCl4] (1085.54): C 61.96, H 5.20, N 5.16; found: C 62.69, H 5.25, N 5.08. 1H NMR (CDCl3, 400 MHz): δ [ppm] = 7.64–7.60 (m, 4H, Ar–H), 7.42–7.38 (m, 8H, Ar–H), 7.35–7.30 (m, 8H, Ar–H), 7.17–7.13 (m, 4H, Ar–H), 7.10–7.06 (m, 8H, Ar–H), 7.01–6.99 (m, 8H, Ar–H), 4.35–4.27 (m, 4H, Ph(CH)CH3), 1.56 (dd, 3JHH = 6.67 Hz, 4JPH = 1.44 Hz, 12H, Ph(CH)CH3). 13C{1H}-NMR (CDCl3, 100 MHz): δ [ppm] = 143.9 (d, JPC = 3.9 Hz, Ar–Cq), 134.0 (d, JPC = 2.9 Hz, Ar–CH), 133.0 (d, JPC = 11.2 Hz, Ar–CH), 129.2 (d, JPC = 12.8 Hz, Ar–CH), 128.8 (Ar–CH), 127.8 (Ar–CH), 126.7 (Ar–CH), 124.7 (d, JPC = 99.5 Hz, Ar–CH), 53.6 (Ph(CH)CH3), 27.8 (d, 3JPC = 12.8 Hz, Ph(CH)CH3). 31P{1H} NMR (THF-d8, 162 MHz): δ [ppm] = 43.3. IR (ATR): [cm−1] = 3027 (vw), 2976 (vw), 2923 (vw), 1589 (w), 1493 (w), 1455 (m), 1437 (w), 1371 (vw), 1313 (w), 1277 (m), 1208 (w), 1139 (s), 1124 (m), 1111 (w), 1069 (m), 1045 (w), 1000 (w), 974 (vw), 883 (w), 857 (vs), 766 (m), 752 (w), 747 (m), 693 (vs), 651 (vw), 617 (w), 599 (w), 518 (vs), 478 (m), 439 (vw).
Synthesis of [L2Ga]+[AlCl4]− (5)
Following the procedure described above for 4, the reaction of 3 (150 mg, 0.16 mmol, 1.00 eq.) and AlCl3 (7 mg, 0.16 mmol, 1.00 eq.) afforded white powder. Single crystals suitable for X-ray structure analysis were grown from concentrated benzene solution at room temperature.
Yield (based on crystals): 104 mg (60%). Elemental analysis calcd (%) for [C56H56N4P2AlGaCl4] (1085.54): C 61.96, H 5.20, N 5.16; found: C 61.98, H 5.17, N 5.22. 1H NMR (CDCl3, 400 MHz): δ [ppm] = 7.64–7.60 (m, 4H, Ar–H), 7.44–7.31 (m, 16H, Ar–H), 7.18–7.10 (m, 12H, Ar–H), 7.02–7.00 (m, 8H, Ar–H), 4.31 (dq, 2JPH = 9.42 Hz, 3JHH = 6.63 Hz, 4H, Ph(CH)CH3), 1.55 (dd, 3JHH = 6.71 Hz, 4JPH = 1.85 Hz, 12H, Ph(CH)CH3). 13C{1H} NMR (CDCl3, 100 MHz): δ [ppm] = 143.9 (d, JPC = 3.8 Hz, Ar–Cq), 133.9 (Ar–CH), 132.9 (d, JPC = 11.3 Hz, Ar–CH), 129.2 (d, JPC = 12.8 Hz, Ar–CH), 128.8 (Ar–CH), 127.9 (Ar–CH), 126.8 (Ar–CH), 125.1 (d, JPC = 99.6 Hz, Ar–CH), 54.4 (Ph(CH)CH3), 27.9 (d, 3JPC = 12.4 Hz, Ph(CH)CH3). 31P{1H} NMR (CDCl3, 162 MHz): δ [ppm] = 48.6. IR (ATR): [cm−1] = 3062 (vw), 3029 (vw), 2970 (w), 2931 (w), 2845 (vw), 1589 (vw), 1493 (w), 1452 (w), 1436 (m), 1367 (w), 1278 (w), 1242 (w), 1205 (s), 1191 (s), 1140 (w), 1110 (w), 1102 (w), 910 (w), 828 (s), 775 (m), 758 (s), 746 (m), 721 (m), 695 (vs), 664 (w), 633 (m), 616 (m), 568 (m), 558 (s), 518 (s), 506 (m), 449 (w).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Dahlen is acknowledged for her support in proof reading. Dr Ralf Köppe is acknowledged for his support in the interpretation of the IR spectra of 1 and the corresponding theoretical investigations.Notes and references
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- F. T. Edelmann, Coord. Chem. Rev., 1994, 137, 403–481 CrossRef.
- F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC.
- P. C. Junk and M. L. Cole, Chem. Commun., 2007, 1579–1590 RSC.
- P. W. Roesky, Chem. Soc. Rev., 2000, 29, 335–345 RSC.
- Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387–415 CrossRef CAS.
- M. Stradiotto and R. J. Lundgren, Ligand Design in Metal Chemistry: Reactivity and Catalysis, John Wiley & Sons, Ltd., 2016 Search PubMed.
- C. Fedorchuk, M. Copsey and T. Chivers, Coord. Chem. Rev., 2007, 251, 897–924 CrossRef CAS.
- A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS.
- S.-O. Hauber, F. Lissner, G. B. Deacon and M. Niemeyer, Angew. Chem., Int. Ed., 2005, 44, 5871–5875 CrossRef CAS PubMed.
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall, P. Takolpuckdee, A. K. Tomov, A. J. P. White, D. J. Williams, M. R. J. Elsegood and S. H. Dale, Inorg. Chem., 2007, 46, 9988–9997 CrossRef CAS PubMed.
- S.-O. Hauber and M. Niemeyer, Inorg. Chem., 2005, 44, 8644–8646 CrossRef CAS.
- F. Pauer, J. Rocha and D. Stalke, J. Chem. Soc., Chem. Commun., 1991, 1477–1479 RSC.
- F. Pauer and D. Stalke, J. Organomet. Chem., 1991, 418, 127–145 CrossRef CAS.
- R. Fleischer, B. Walfort, A. Gbureck, P. Scholz, W. Kiefer and D. Stalke, Chem. – Eur. J., 1998, 4, 2266–2274 CrossRef CAS.
- F. T. Edelmann, F. Knösel, F. Pauer, D. Stalke and W. Bauer, J. Organomet. Chem., 1992, 438, 1–10 CrossRef CAS.
- S. Collins, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS.
- C. Averbuj, E. Tish and M. S. Eisen, J. Am. Chem. Soc., 1998, 120, 8640–8646 CrossRef CAS.
- L. A. Koterwas, J. C. Fettinger and L. R. Sita, Organometallics, 1999, 18, 4183–4190 CrossRef CAS.
- H. Brunner, J. Lukassek and G. Agrifoglio, J. Organomet. Chem., 1980, 195, 63–76 CrossRef CAS.
- N. Li and B.-T. Guan, Adv. Synth. Catal., 2017, 359, 3526–3531 CrossRef CAS.
- P. Benndorf, C. Preuß and P. W. Roesky, J. Organomet. Chem., 2011, 696, 1150–1155 CrossRef CAS.
- T. S. Brunner, P. Benndorf, M. T. Gamer, N. Knöfel, K. Gugau and P. W. Roesky, Organometallics, 2016, 35, 3474–3487 CrossRef CAS.
- J. Kratsch, M. Kuzdrowska, M. Schmid, N. Kazeminejad, C. Kaub, P. Oña-Burgos, S. M. Guillaume and P. W. Roesky, Organometallics, 2013, 32, 1230–1238 CrossRef CAS.
- T. P. Seifert, T. S. Brunner, T. S. Fischer, C. Barner-Kowollik and P. W. Roesky, Organometallics, 2018, 37, 4481–4487 CrossRef CAS.
- P. Benndorf, J. Kratsch, L. Hartenstein, C. M. Preuss and P. W. Roesky, Chem. – Eur. J., 2012, 18, 14454–14463 CrossRef CAS PubMed.
- P. Benndorf, J. Jenter, L. Zielke and P. W. Roesky, Chem. Commun., 2011, 47, 2574–2576 RSC.
- M. He, M. T. Gamer and P. W. Roesky, Organometallics, 2016, 35, 2638–2644 CrossRef CAS.
- T. J. Feuerstein, B. Goswami, P. Rauthe, R. Köppe, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. Sci., 2019, 10, 4742–4749 RSC.
- Y. Hayashi, J. J. Rohde and E. J. Corey, J. Am. Chem. Soc., 1996, 118, 5502–5503 CrossRef CAS.
- K. B. Starowieyski, Chemistry of Aluminium, Gallium, Indium and Thallium, Springer Netherlands, London, UK, 1993 Search PubMed.
- M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 1997, 119, 8125–8126 CrossRef CAS.
- Z. Yang, M. Zhong, X. Ma, K. Nijesh, S. De, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 2548–2551 CrossRef CAS PubMed.
- Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225–10229 CrossRef CAS PubMed.
- A. Harinath, I. Banerjee, J. Bhattacharjee and T. K. Panda, New J. Chem., 2019, 43, 10531–10536 RSC.
- A. Ford and S. Woodward, Angew. Chem., Int. Ed., 1999, 38, 335–336 CrossRef CAS PubMed.
- L. A. Berben, Chem. – Eur. J., 2015, 21, 2734–2742 CrossRef CAS PubMed.
- S. Dagorne, I. A. Guzei, M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 2000, 122, 274–289 CrossRef CAS.
- B. Nekoueishahraki, H. W. Roesky, G. Schwab, D. Stern and D. Stalke, Inorg. Chem., 2009, 48, 9174–9179 CrossRef CAS PubMed.
- B. Prashanth and S. Singh, Dalton Trans., 2014, 43, 16880–16888 RSC.
- B. Prashanth, N. Srungavruksham and S. Singh, ChemistrySelect, 2016, 1, 3601–3606 CrossRef CAS.
- J. Vrána, R. Jambor, A. Růžička, M. Alonso, F. De Proft and L. Dostál, Eur. J. Inorg. Chem., 2014, 2014, 5193–5203 CrossRef.
- A. L. Hawley, C. A. Ohlin, L. Fohlmeister and A. Stasch, Chem. – Eur. J., 2017, 23, 447–455 CrossRef CAS PubMed.
- S. A. Ahmed, M. S. Hill, P. B. Hitchcock, S. M. Mansell and O. S. John, Organometallics, 2007, 26, 538–549 CrossRef CAS.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- C.-Y. Lin, C.-F. Tsai, H.-J. Chen, C.-H. Hung, R.-C. Yu, P.-C. Kuo, H. M. Lee and J.-H. Huang, Chem. – Eur. J., 2006, 12, 3067–3073 CrossRef CAS PubMed.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. A. Vyvyan, Introduction to Spectroscopy, Cengage Learning, 2008 Search PubMed.
- H. R. Allcock, in Phosphorus-nitrogen Compounds, Academic Press, 1972, pp. 385–395 Search PubMed.
- M. P. Coles, D. C. Swenson, R. F. Jordan and V. G. Young, Organometallics, 1997, 16, 5183–5194 CrossRef CAS.
- A. L. Brazeau, G. A. DiLabio, K. A. Kreisel, W. Monillas, G. P. A. Yap and S. T. Barry, Dalton Trans., 2007, 3297–3304 RSC.
- A. P. Kenney, G. P. A. Yap, D. S. Richeson and S. T. Barry, Inorg. Chem., 2005, 44, 2926–2933 CrossRef CAS PubMed.
- M. L. Cole, A. J. Davies, C. Jones, P. C. Junk, A. I. McKay and A. Stasch, Z. Anorg. Allg. Chem., 2015, 641, 2233–2244 CrossRef CAS.
- M.-A. Muñoz-Hernández, T. S. Keizer, S. Parkin, B. Patrick and D. A. Atwood, Organometallics, 2000, 19, 4416–4421 CrossRef.
- C. L. B. Macdonald, J. D. Gorden, A. Voigt, S. Filipponi and A. H. Cowley, Dalton Trans., 2008, 1161–1176 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR spectra and crystallographic studies. CCDC 1959904–1959908. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04082j |
| This journal is © The Royal Society of Chemistry 2020 |