
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning a modular system – synthesis and characterisation of a boron-rich s-triazine-based carboxylic acid and amine bearing a galactopyranosyl moiety†
Martin
Kellert
 a,
Paul
Hoppenz
b,
Peter
Lönnecke
a,
Dennis J.
Worm
b,
Bernd
Riedl
c,
Johannes
Koebberling
c,
Annette G.
Beck-Sickinger
b and
Evamarie
Hey-Hawkins
*a
a,
Paul
Hoppenz
b,
Peter
Lönnecke
a,
Dennis J.
Worm
b,
Bernd
Riedl
c,
Johannes
Koebberling
c,
Annette G.
Beck-Sickinger
b and
Evamarie
Hey-Hawkins
*a
aFaculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bFaculty of Life Sciences, Institute of Biochemistry, Leipzig University, Brüderstrasse 34, 04103 Leipzig, Germany
cBayer AG, Aprather Weg 18A, 42113 Wuppertal, Germany
First published on 26th November 2019
Abstract
Introduction of a bis(isopropylidene)-protected galactopyranosyl moiety in s-triazine-based boron-rich carboxylic acids and amines results in soluble and suitable coupling partners for tumour-selective biomolecules with applications as selective agents for boron neutron capture therapy (BNCT). Bearing either a carboxylic acid or primary amine as a functional group, these compounds are highly versatile and thus largely extend the possible coupling strategies with suitable biomolecules. Modification of the gastrin-releasing peptide receptor (GRPR) selective agonist [D-Phe6, β-Ala11, Ala13, Nle14]Bn(6–14) with the carboxylic acid derivative yielded a bioconjugate with an optimal receptor activation and internalisation profile. This demonstrates the great potential of this approach for the development of novel boron delivery agents.
Introduction
Since the first report by Locher in 1936, boron neutron capture therapy (BNCT) has been developed as a very promising approach for cancer treatment.1 It combines two non-toxic components to produce cytotoxic species, which are able to destroy malignant tissue. Boron-10 containing drugs bearing a tumour-selective moiety to address the infected site are highly advantageous.2–8 Ideally, these bioconjugates accumulate selectively in the malignant tissues in a required amount of 10–30 μg g−1 tumour tissue and can then be irradiated with thermal or epithermal neutrons.2,9 Depending on the biomolecule, the BNCT agent is either just accumulated in the tumour tissue or internalised into the cancer cells, which of course increases the efficacy of this treatment.10,11,12–20 The particles which are generated upon neutron capture are lithium and helium nuclei (α particles).2,21,22 These particles with a high linear energy transfer (LET) have a mean free path of about 5 to 10 μm, and thus, a limited radius of destructive action.2,5,6,21,22 The combination of suitable boron-rich molecules with tumour-selective biomolecules opens up a very selective tumour therapy which only affects malignant tissue and spares normal tissue.10,12,14–17,23 However, there are still some major challenges, including the selectivity of the chosen biomolecules for a specific type of tumour, the required high concentration of boron-10 in the cancer cell, the water solubility of the final bioconjugate, and the neutron beam quality,24 which are also the focus of current research.13,17–19,25,26Recently, we reported the synthesis of s-triazine-based boron-rich carboxylic acids.27 Preliminary studies showed that the incorporation of more than one s-triazine-based bis(carboranyl) derivative into the breast tumour-selective peptide [F7,P34]-neuropeptide Y leads to a decrease or even total loss of the hY1 receptor activation potency. It was assumed that this was caused by the strong hydrophobic character of the carborane clusters attached to the peptide producing a hydrophobic collapse of the bioconjugate.28 Here, we describe the incorporation of an α-D-galactopyranosyl-substituted glycine derivative to reduce the hydrophobicity and improve the water solubility, which are important features for successful BNCT.5–7,29 We also demonstrate the expansion of potential receptor targets for tumour addressing by utilising the recently developed gastrin-releasing peptide receptor (GRPR, BB2) selective peptide agonist [D-Phe6, β-Ala11, Ala13, Nle14]Bn(6–14) (sBB2L).30 The GRPR is commonly used as a drug shuttle system because of its frequent overexpression in breast and prostate cancer and its ability to repetitively internalise together with the peptide drug conjugate leading to an intracellular drug accumulation.31 Furthermore, inclusion of either a carboxylic acid or an amine moiety as functional group further extends the scope for potential coupling partners (Fig. 1).
 | ||
| Fig. 1 A modular approach using 9-mercapto-1,7-dicarba-closo-dodecaborane(12) (pale red), glycine or ethylene diamine (blue), 1,3,5-triazine (green) and an α-D-galactopyranose derivative (yellow) to produce less hydrophobic boron-rich s-triazine derivatives (R = COOH, CH2NH2). | ||
The only comparable compound using an s-triazine scaffold in combination with carboranes, monosaccharides and carboxylic acids was reported by Panza and co-workers.32 In this case, ortho-carborane is used, which is a potential problem as this isomer is prone to undergo deboronation reactions if good nucleophiles associated with basic or even neutral conditions are present.33 This may cause problems with bioavailability or side effects when employed in therapy.17,34 Using meta-carborane assures the integrity of the carborane cluster.
Results and discussion
Based on the synthetic procedures described in our previous work, [4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]glycine (3) was prepared from glycine and 2-chloro-4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine (2), which was synthesised from 9-mercapto-(1,7-dicarba-closo-dodecaborane)(12) (1) and cyanuric chloride.27,35 The following step involved the base-assisted introduction of the monosaccharide moiety as 1,2:3,4-di-O-isopropylidene-6-deoxy-α-D-galactopyranosyl-6-triflate (4), prepared from 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (Scheme 1, step c).36 A protection strategy is required also to retain the monosaccharide as the α anomer.37 Although, acetyl protecting groups are well known and widely used,38 we favour isopropylidene (acetonide) protecting groups, because the corresponding α-D-galactopyranose is commercially available, whereas the respective 1,2,3,4-tetraacetyl derivative has to be synthesised. Furthermore, there is no interference of the acetonide groups with the cluster signals in 1H NMR spectroscopy.39 In addition, acetonides are only cleavable under strongly acidic (and aqueous) conditions, whereas acetyl protecting groups are also cleaved by strong acids and bases and nucleophiles like enolates, organolithium compounds and Grignard reagents.38 | ||
Scheme 1 Synthetic strategy for the desired product 5 resulting in compound 6 instead. (a) 1, diisopropylethylamine (DIPEA), MeCN, reflux, 5 h, quant.; (b) glycine, NaOH, MeCN/H2O/ethyl acetate (EtOAc) (3.33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1), 95 °C, 6 h, 60%; (c) trifluoromethanesulfonic anhydride, 2,4,6-collidine, CH2Cl2, rt, 4 h, 93%; (d) 4, K2CO3, THF, rt, 2 d, 63%. :2:1), 95 °C, 6 h, 60%; (c) trifluoromethanesulfonic anhydride, 2,4,6-collidine, CH2Cl2, rt, 4 h, 93%; (d) 4, K2CO3, THF, rt, 2 d, 63%. | ||
Unexpectedly, the reaction of 3 with 4 did not give N-[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]-N-(1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)glycine (5); instead the carboxylic acid ester (1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]glycinate (6) was obtained with N,N-diisopropylethylamine (DIPEA) or potassium carbonate as base (Scheme 1, step d). A similar reaction was observed by Hughes et al. when reacting α-trifluoromethyl benzyl triflates with octanoic acid.40
These observations led to a different synthetic approach (Scheme 2). Starting from cyanuric chloride and tert-butyl glycinate hydrochloride the dichloro species tert-butyl(4,6-dichloro-1,3,5-triazin-2-yl)glycinate (7) was prepared to introduce a protected carboxylic acid group and thus avoid the side reaction leading to 6 (Scheme 1, step d). Subsequently, the carborane clusters were introduced under mild basic conditions (b in Scheme 2). However, only a mixture of the mono- (8) and disubstituted derivative (9) could be obtained, which could not be separated by column chromatography (details are given in the ESI†). Attempts to introduce a galactopyranosyl moiety via the reaction of the mixture of 8 and 9 with the triflate 4 failed irrespective of the used base (DIPEA or K2CO3). This indicates that the secondary amine group in 8 and 9 is a weak nucleophile and not able to undergo a substitution reaction. A possible reason for this may be the resonance stabilisation of the adjacent s-triazine ring which might even be enhanced by the electron-withdrawing effect of the chloro and carborane substituents.
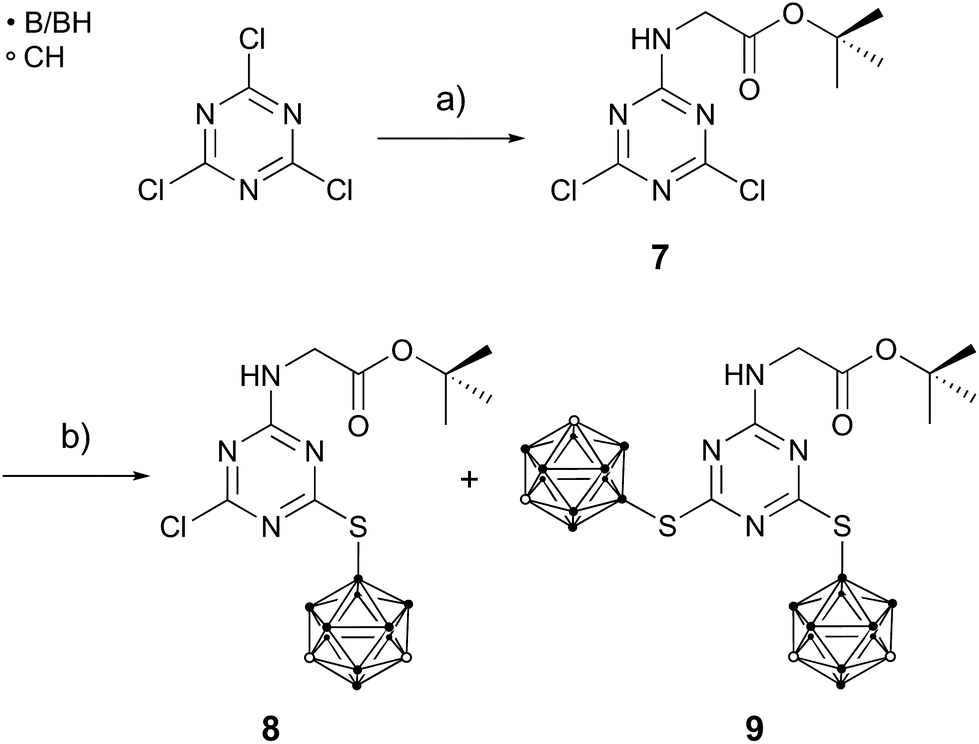 | ||
| Scheme 2 Proposed synthetic strategy for 9 as a precursor for 5. (a) tert-Butyl glycinate hydrochloride, DIPEA, THF, rt, 2 d, 82%; (b) 1, DIPEA, MeCN, 80 °C, 1 d. | ||
Since the carboxylate could act as a nucleophile towards triflates (Scheme 1, step d) and the directly attached secondary amine at the s-triazine backbone is a weak nucleophile and undergoes no substitution reaction (Scheme 2), these observations led to a third approach in the synthesis of compound 5 (Scheme 3).
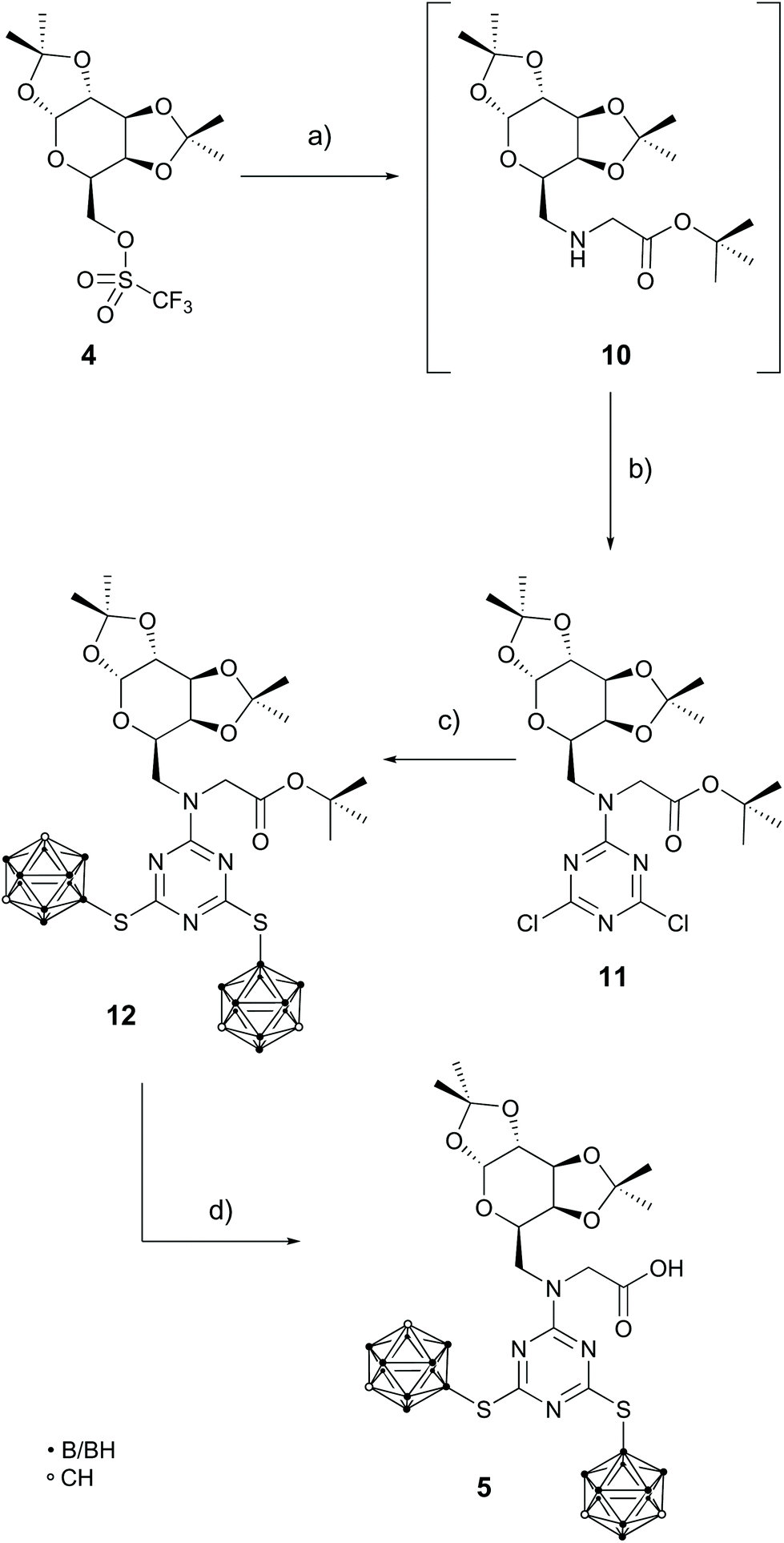 | ||
| Scheme 3 Successful synthetic route to the glycine derivative 5: (a) tert-butyl glycinate hydrochloride, DIPEA, MeCN, 45 °C, 2 d, not isolated; (b) cyanuric chloride, DIPEA, MeCN, 35 °C, 2 d, 77%*; (c) 1, K2CO3, MeCN, reflux, 2 d, 98%; (d) trifluoroacetic acid, DCM, rt, 4 h, 83%; * = yield over two steps. | ||
Starting from 1,2:3,4-di-O-isopropylidene-6-deoxy-α-D-galactopyranosyl-6-triflate (4) and tert-butyl glycinate hydrochloride the monosubstituted s-triazine derivative tert-butyl-N-(4,6-dichloro-1,3,5-triazin-2-yl)-N-(1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)glycinate (11) was prepared in a one-pot reaction via tert-butyl-N-(1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)glycinate (10) as intermediate (a and b in Scheme 3). Subsequently, the introduction of the two 9-mercapto-meta-carborane clusters (1) was achieved with K2CO3 as base resulting in almost quantitative formation of tert-butyl-N-[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]-N-(1′,2′:3′,4′-di-O-iso-propylidene-6′-deoxy-α-D-galactopyranos-6′-yl)glycinate (12) from 11.
Successful reproduction of the mentioned synthetic approach (Scheme 3, steps a to c) was already reported using a three carborane-bearing mercapto derivative as a nucleophile.26
Attempts to prepare 12 or 5 directly in one-pot syntheses, using DIPEA as a base, failed and only produced the mono-substituted derivative tert-butyl-N-[4-chloro-6-(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]-N-(1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)glycinate (SP1) in 15% yield or the side product N-ethyl-N-isopropyl-4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine-2-amine (SP2) in 10% yield (details are given in the ESI†). Selective cleavage of the ester group of 12 (under maintenance of the isopropylidene protecting groups of the galactopyranosyl moiety) with trifluoroacetic acid in anhydrous toluene or dichloromethane gave the final product 5.38,41
The synthetic approach described in Scheme 3 was adapted for ethylene diamine (Scheme 4) to introduce an additional functional group for incorporation of various other biomolecules. In this case, tert-butyl N-(2-aminoethyl)carbamate and triflate 4 were converted to tert-butyl-N-{2-[(4,6-dichloro-1,3,5-triazin-2-yl)-2-(1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)-amino]ethyl}carbamate (14), with compound 13 as intermediate, in 68% yield over two steps. Introduction of 9-mercapto-dicarba-closo-dodecaborane (1), in the same manner as described for compound 12, gave tert-butyl-N-{2-[(4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl)-2-(1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)-amino]ethyl}carbamate (15) in almost quantitative yield. The observed high yields in the reaction of 1 with 11 or 14 show the pronounced nucleophilicity of the 9-mercapto-dicarba-closo-dodecaborane (1) under mild basic conditions. Cleavage of the tert-butoxycarbonyl protecting group in 15 was achieved as described for 5 (Scheme 3). The primary amine 16, N1-[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]-N1-(1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)ethane-1,2-diamine, was obtained in 91% yield.
 | ||
| Scheme 4 Adapted synthetic strategy to prepare compound 16: (a) tert-butyl N-(2-aminoethyl)carbamate, DIPEA, MeCN, 40 °C, 2 d, not isolated; (b) cyanuric chloride, DIPEA, MeCN, 35 °C, 2 d, 68%*; (c) 1, K2CO3, MeCN, reflux, 2 d, 99%; (d) trifluoroacetic acid, DCM, rt, 4 h, 91%; * = yield over two steps. | ||
All isolated compounds were fully characterised by NMR and IR spectroscopy, mass spectrometry and melting point determination. Additionally, compounds 6, 11 and 12 were characterised by single crystal X-ray diffraction.
The final product 5 was characterised by high-resolution mass spectrometry and NMR spectroscopy. A very broad signal in the 1H NMR spectrum at 6.34 ppm was assigned to a protonated tertiary amine indicating the presence of a zwitterion (NHR3+/CO2−). In comparison, typical carboxylic acid protons have chemical shifts around 9 to 13 ppm.39
The characterisation of 6 was very challenging, as the obtained spectroscopic data were not unambiguous for the proposed structure. However, colourless crystals suitable for X-ray diffraction were obtained from an acetone solution confirming the formation of (1′,2′:3′,4′-di-O-isopropylidene-6′-deoxy-α-D-galactopyranos-6′-yl)[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]glycinate (6). Two independent molecules of 6 are linked by hydrogen bonds between the hydrogen atom of the secondary amine group and one nitrogen atom of the s-triazine ring (Fig. 2). The observation of hydrogen bonds was already described for compounds of this substance class.27
 | ||
| Fig. 2 Molecular structure of 6. Hydrogen atoms are omitted for clarity, except the hydrogen atoms attached to the secondary amine groups drawn with a fixed atom radius of 13.5 pm. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths, distances (pm) and bond angles (°): N4–H1N4 86(4), H1N4⋯N7 213(4), N4⋯N7 299.4(4), N8–H1N8 85(4), H1N8⋯N3 217(4), N8⋯N3 302.5(4), N4–C5 133.7(4), S1–B10 186.9(4), S1–C3 174.8(4), S2–B20 186.4(4), S2–C4 175.7(3), N8–C26 133.5(4), S3–B30 186.7(4), S3–C24 176.1(3), S4–B40 187.0(4), S4–C25 175.8(4); N4–H1N4⋯N7 173(4), N8–H1N8⋯N3 176(3), H1N4–N4–C5 121(3), C5–N4–C6 120.5(3), C6–N4–H1N4 114(3), C3–S1–B10 107.2(2), C4–S2–B20 106.9(2), H1N8–N8–C26 116(3), C26–N8–C27 122.4(3), C27–N8–H1N8 122(3), C24–S3–B30 108.3(2), C25–S4–B40 106.4(2). | ||
Compound 11 crystallised from chloroform solution with two molecules in the asymmetric unit (Fig. 3).
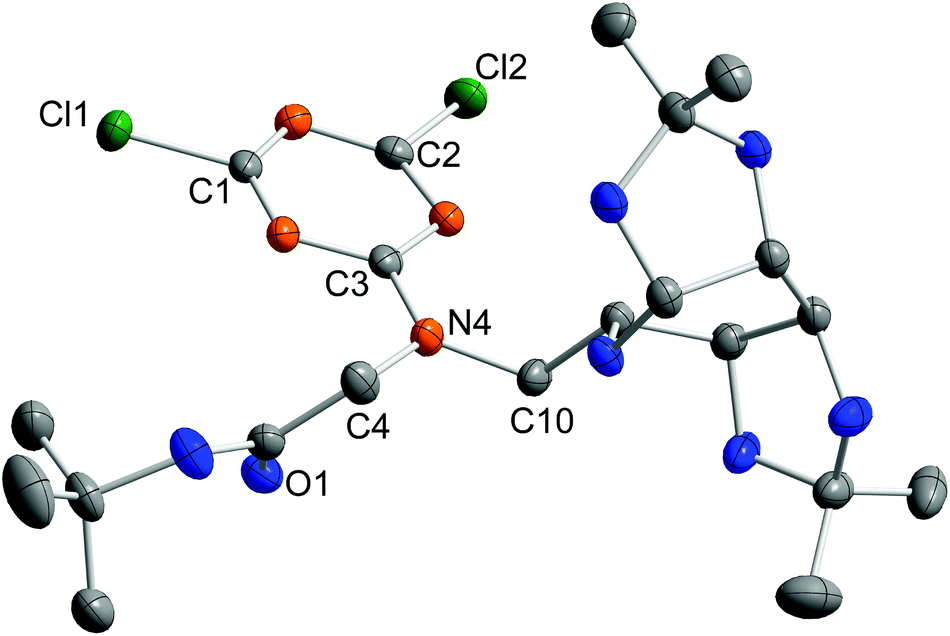 | ||
| Fig. 3 Molecular structure of 11. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Only one of the two symmetry-independent molecules is shown. Selected bond length (pm) and bond angles (°): N4–C3 133.4(3) [133.7(3)], N4–C4 145.5(3) [145.1(3)], N4–C10 146.3(3) [146.4(3)], C1–Cl1 172.7(2) [172.3(2)], C2–Cl2 172.9(2) [172.4(2)]; C3–N4–C4 118.2(2) [118.2(2)], C4–N4–C10 120.8(2) [119.1(1)], C10–N4–C3 120.9(2) [122.2(2)] (values for the second molecule are given in brackets [ ]). | ||
In the case of compound 12, mass spectrometry was a very informative characterisation method, as the characteristic isotopic pattern for molecules with 20 boron atoms was observed.42,43 Single crystals of 12 could be obtained from n-hexane/ethyl acetate solution. The molecular structure is depicted in Fig. 4.
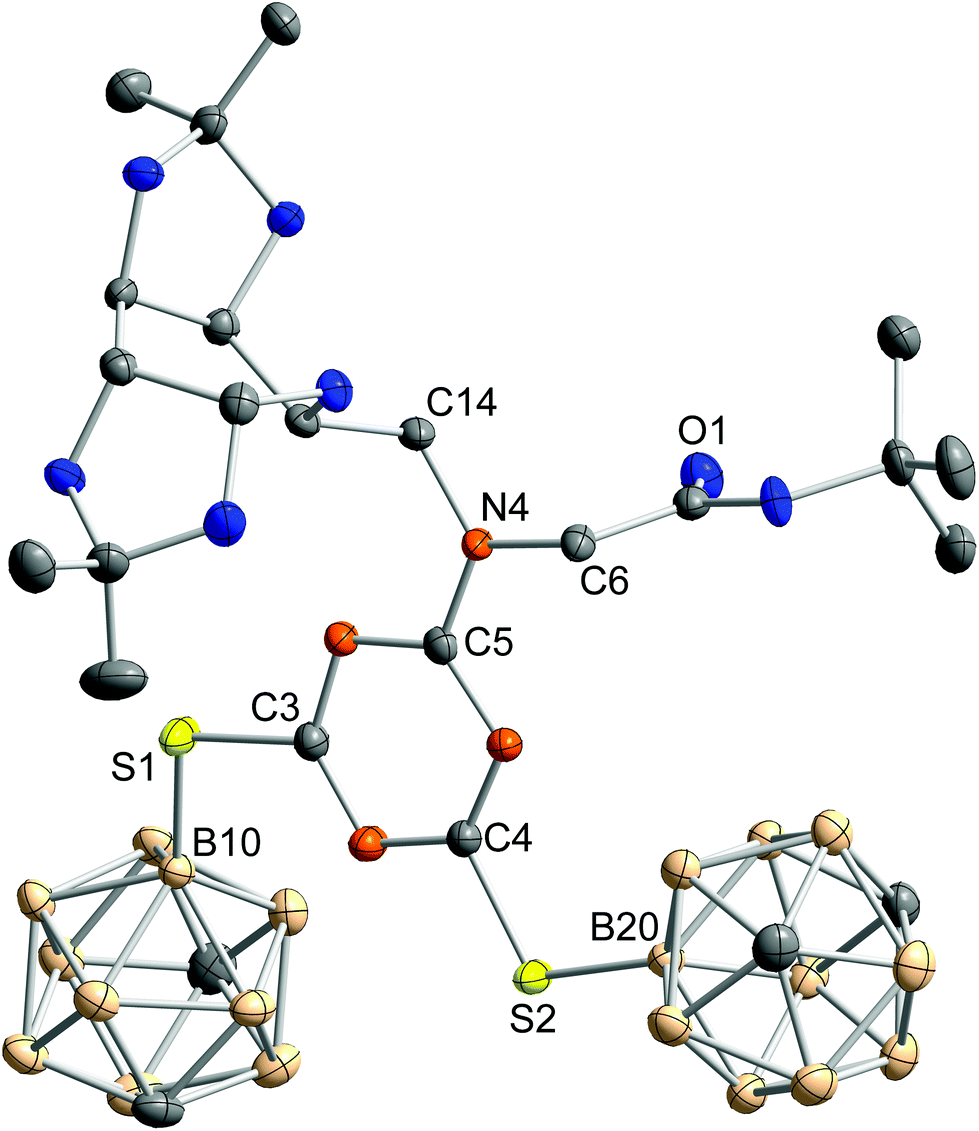 | ||
| Fig. 4 Molecular structure of 12. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond length (pm) and bond angles (°): N4–C5 135.2(4), S1–C3 175.6(3), S1–B10 186.5(3), S2–C4 175.0(3), S2–B20 186.0(3); C5–N4–C6 121.2(2), C6–N4–C14 119.1(2), C14–N4–C5 119.7(2), C3–S1–B10 106.7(2), C4–S2–B20 107.6(1). | ||
The carbamate 14 and the following products 15 and 16 were characterised by NMR and mass spectrometry confirming the successful synthesis. For compound 15, the mass spectrum was very characteristic, because the isotopic pattern clearly showed the replacement of two chloro substituents by two carborane clusters.42,43
In summary, the two target molecules 5 and 16 were obtained in good to excellent yield. Due to their different functional groups (R–COOH in 5 and R–NH2 in 16), coupling with a large variety of different biomolecules as tumour-selective carriers can be envisaged for application in BNCT.4,8,12,15,17,20
Biological studies
The galactopyranosyl derivative 5 was incorporated into the recently developed GRPR selective peptide [D-Phe6, β-Ala11, Ala13, Nle14]Bn(6–14) (sBB2L)30 to investigate, whether the ratio of one galactopyranosyl unit per two carborane clusters is sufficient to counterbalance the hydrophobicity of the carborane clusters yielding biologically active bioconjugates. The necessity for increased hydrophilicity was concluded from previous studies concerning the introduction of meta-carboranes as single clusters17 or s-triazine-based derivatives bearing no monosaccharide group.27 Three different peptide conjugates were synthesised (Fig. 5A) by a combination of automated and manual solid phase peptide synthesis (SPPS).30,44 The s-triazine derivative 5 as well as the previously published derivative without any galactopyranosyl moiety (compound #4 in Kellert et al., ref. 27) were coupled manually in three-fold molar excess to an N-terminally introduced three ethylene glycol-unit spacer (EG3). The reaction was carried out overnight at room temperature in dimethylformamide (DMF) containing three equivalents 1-hydroxybenzotriazole (HOBt) and N,N′-diisopropylcarbodiimide (DIC), yielding conjugates 17 and 18. To enhance the boron loading per molecule a branching step was introduced using (2S)-2,3-diaminopropanoic acid (Dap) allowing the incorporation of two carborane building blocks 5 as deprotected moieties 5* (Fig. 5A). This strategy facilitated the generation of conjugate 19 bearing 40 boron atoms. | ||
| Fig. 5 Chemical structure and exemplary analytical data of conjugate 18. (A) Schematic structures of the GRPR selective conjugates 17, 18 and 19 modified with the glycine derivative 5* and the previously published derivative #4 without a galactopyranosyl moiety.27 Peptide sequence is presented as one letter amino acid code with special amino acids indicated (f = D-Phe; b = β-Ala, X = L-norleucine). (B) Reversed phase (RP)-HPLC analysis of conjugate 18 using an Aeris PEPTIDE 3.6 μm XB-C18 column (phenomenex) and a linear gradient of 20–70% (v/v) eluent B (MeCN + 0.08% TFA (v/v)) in eluent A (H2O + 0.1% TFA (v/v)) over 40 min with UV detection at λ = 220 nm. (C) ESI-HCT MS showing the multiply charged ion species of 18. MWcal/mono: 1891.24 Da. | ||
Analyses of all three conjugates were performed with analytical reversed-phase high performance liquid chromatography (RP-HPLC) and electrospray ionisation mass spectrometry (ESI-MS) as well as with MALDI-TOF-MS (Fig. 5B and C). The exemplary RP-HPLC chromatogram of 18 displayed the main peak at 17.2 min retention time and a small shoulder on the right side. Since this shoulder had the same mass as the desired product and conjugate 17 did not exhibit any shoulders, the deoxygalactosyl moiety is suggested to cause this shoulder by mutarotation (Fig. 5B).
The three bioconjugates were tested in receptor activation and internalisation studies. For this purpose, the human GRPR was selected due to its remarkable overexpression on various tumour tissues like small cell lung,45 breast46 and prostate cancer.47 The neuromedin B receptor (NMBR) and the bombesin receptor subtype 3 (BRS-3), which are also part of the bombesin receptor family are only occasionally expressed in these tumours.48
The use of the recently published GRPR selective ligand (sBB2L) allows the specific targeting of cancer cells while the accumulation in healthy tissues is kept to a minimum.30 Thus, the risk of side effects is reduced. Compared to the unmodified ligand sBB2L, which displayed an EC50 value of 0.12 nM at the GRPR, conjugate 17 exhibited a ca. 20-fold reduced potency (EC50 2.2 nM; Fig. 6A). This can be explained by the insufficient solubility of derivate #4 which was recently demonstrated by the incorporation into the even longer and more hydrophilic peptide [F7,P34]-NPY.27 However, conjugate 18 bearing an additional deoxygalactopyranosyl moiety regained the activity at the GRPR and, with an EC50 value of 0.17 nM, demonstrated wild type like potency. In addition, fluorescence microscopy studies revealed that 17 and 18 induce internalisation of the GRPR at a peptide concentration of 100 nM. After one hour of stimulation, the membrane bound receptor was completely translocated into intracellular vesicles, as was observed for the unmodified sBB2L (Fig. 6B).
 | ||
| Fig. 6 Biological evaluation of carborane-modified GRPR selective conjugates. (A) GRPR receptor activation data of synthesised peptide conjugates. Sigmoidal concentration–response curves were determined from at least two independent Ca2+ mobilisation experiments, each performed in duplicate. All curves were normalised to the control. (B) Receptor internalisation studies of HEK293 cells stably expressing the GRPR fused to tGFP (green). Cells were stimulated with 100 nM peptide for 1 h at 37 °C and cell nuclei were stained with Hoechst 33342 (blue). Scale bar: 10 μm. | ||
Due to the improved receptor activation of conjugate 18, Dap was introduced to allow the incorporation of more than one carborane-based building block per peptide molecule. The resulting conjugate 19, bearing two deprotected molecules 5* (40 boron atoms), displayed a strongly reduced potency at the GRPR and showed nearly no internalisation after 1 h of stimulation with 100 nM peptide. This indicates that building block 5, featuring a ratio of one deoxygalactopyranosyl unit per two carborane clusters, is not optimal for the generation of highly carborane-loaded peptide conjugates. Nevertheless, the incorporation of the deoxygalactopyranosyl unit improved the hydrophilicity, which allowed the synthesis of a double modified conjugate (19). This was previously not possible with building block #4 using standard peptide purification methods (data not shown). These observations demonstrate the necessity of hydrophilicity providing moieties to generate highly boron-loaded bioconjugates for tumour delivery. Therefore, conjugate 18 can be considered as a promising selective boron delivery agent in BNCT.
Conclusions
By modifying s-triazine derivatives with a galactopyranosyl moiety the modular system based on readily available building blocks like 9-mercapto-1,7-dicarba-closo-dodecaborane (1) and cyanuric chloride was further extended to compounds with reduced hydrophobicity. The final products, carboxylic acid 5 and amine 16, are highly suitable for the synthesis of bioconjugates, shown here exemplarily for 5 being introduced to the GRPR selective peptide [D-Phe6, β-Ala11, Ala13, Nle14]Bn(6–14) (18 and 19). Receptor activation and internalisation studies revealed an improved performance in comparison to the conjugate without monosaccharide moiety (17). This study corroborates the advantageous influence of galactopyranosyl moieties for the development of highly carborane-loaded biomolecules.Experimental section
General experimental part
All reactions were carried out under nitrogen atmosphere using Schlenk techniques, if not reported otherwise. Anhydrous dichloromethane, toluene and tetrahydrofuran were obtained with an MBRAUN solvent purification system MB SPS-800 and stored over molecular sieves (3 Å) under nitrogen atmosphere. Acetonitrile and 2,4,6-collidine were dried over CaH2, distilled and stored over molecular sieves (3 Å) under nitrogen atmosphere. 9-Mercapto-1,7-dicarba-closo-dodecaborane(12) (1), 2-chloro-4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine (2), [4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]glycine (3) and 1,2:3,4-di-O-isopropylidene-6-deoxy-α-D-galactopyranosyl-6-triflate (4) were prepared and characterised according to the literature procedure.27,35,36 All other solvents and chemicals were purchased and used as received. Thin-layer chromatography (TLC) with silica gel 60 F254 on glass available from Merck KGaA was used for monitoring the reactions. Carborane-containing spots were visualised with a 5–10% solution of PdCl2 in methanol. For chromatography, silica gel (60 Å) with a particle diameter in the range of 0.035 to 0.070 mm, the Biotage® Isolera 1 or the Biotage® Isolera 4 automatic purification system with SNAP (particle diameter: 0.040 to 0.065 mm) and SNAP Ultra (spherical particle, diameter: 0.025 mm) cartridges were used. The triazine and carborane species were detected by an integrated UV/Vis detector (Isolera 1, Biotage) or evaporative light scattering detector (ELSD) A-120 (Isolera 4, Biotage). For chromatography, solvents were distilled before use. NMR measurements were carried out on a Bruker AVANCE III HD spectrometer with an Ascend™ 400 magnet at room temperature. Tetramethylsilane was used as internal standard for 1H and 13C{1H} NMR spectra, and 11B and 11B{1H} NMR spectra were referenced to the Ξ scale.49 NMR spectra were recorded at the following frequencies: 1H: 400.16 MHz, 13C: 100.63 MHz, 11B: 128.38 MHz; chemical shifts are reported in ppm. Assignment of the 1H and 13C signals was based on 2D NMR spectra (H,H-COSY, HSQC, HMQC, HMBC). Identification of the boron atom attached to sulfur was possible by comparison of the proton-coupled and -decoupled 11B NMR spectra. NMR data were interpreted with MestReNova.50 NMR signals that appear as broad overlapping signals with the shape of a multiplet in either 1H, 11B{1H} or 11B NMR spectra are described as ‘br’ (broad). In this case, the superscript a is added (bra). The numbering schemes of the entire assignments of all chemical shifts from selected synthesised compounds are given in the ESI.† IR data were obtained with a PerkinElmer FT-IR spectrometer Spectrum 2000 as KBr pellets and on a Thermo Scientific Nicolet iS5 with an ATR unit in the range from 4000 to 400 cm−1. Electrospray ionisation mass spectrometry was performed with an ESI ESQUIRE 3000 PLUS spectrometer with an IonTrap analyser from Bruker Daltonics or on a MicroTOF spectrometer from Bruker Daltonics with a ToF analyser in negative or positive mode. Dichloromethane, acetonitrile, methanol or mixtures of these solvents were used for the measurements. Melting points were determined with a Gallenkamp MPD350·BM2.5 melting point device. Melting points are not corrected.X-ray diffraction experiments
Measurements were performed with a Gemini diffractometer (Rigaku Oxford Diffraction) with Mo-Kα radiation (λ = 71.073 pm), ω-scan rotation. Data reduction was performed with CrysAlis Pro51 including the program SCALE3 ABSPACK52 for empirical absorption correction. The structures were solved by dual space methods (SHELXT-2014)53 and the refinement of all non-hydrogen atoms was performed with SHELXL-2018.54 Hydrogen atoms were mostly located on difference Fourier maps calculated at the final stage of the structure refinement, and only for disordered regions or poor diffracting samples they were calculated on idealised positions using the riding model. Structure figures were generated with Diamond.55 CCDC 1958031 (6), 1958032 (8), 1958033 (9), 1958034 (11), 1958035 (12), 1958036 (SP1), 1958037 (SP2) and 1958038 (SP3)† contain supplementary crystallographic data for this paper.:1 to 100% ethyl acetate, v/v) 84 mg (0.11 mmol, 63%, Rf = 0.49, n-hexane/ethyl acetate, 1:1, v/v) of compound 6 was obtained as a pure white solid. Colourless crystals of 6 suitable for X-ray structure determination were obtained from acetone at room temperature. Crystallographic data are given in Table S1,† and the molecular structure is depicted in Fig. 2. Tm: 167–168 °C (ethyl acetate). IR (KBr): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3436 (m), 3056 (w), 2988 (w), 2936 (w), 2608 (s), 1749 (m), 1563 (s), 1518 (s), 1497 (s), 1455 (w), 1412 (m), 1383 (m), 1298 (m), 1252 (m), 1211 (m), 1174 (m), 1116 (w), 1070 (s), 1001 (m), 955 (w), 921 (w), 892 (w), 864 (m), 850 (m), 804 (w), 759 (w), 730 (w), 677 (w), 627 (w), 585 (w), 512 (w) cm−1. 1H NMR (acetone-d6): δ = 1.316 (s, 3H, C14 or 14′H3), 1.322 (s, 3H, C15 or 15′H3), 1.38 (s, 3H, C14 or 14′H3), 1.47 (s, 3H, C15 or 15′H3), 1.50 to 3.50 (bra, 18H, 2 × B10H9), 3.69 (br, s, 2H, 2 × C1 or 1′H), 3.74 (br, s, 2H, 2 × C1 or 1′H), 4.06 (m, 1H, C7H), 4.19 (m, 2H, C6H2), 4.29 (m, 1H, C9H), 4.33 (m, 2H, C5H2), 4.37 (m, 1H, C10H), 4.64 (dd, 3JHH = 7.9 Hz, 3JHH = 2.5 Hz, 1H, C8H), 5.47 (d, 3JHH = 5.0 Hz, 1H, C11H), 7.05 ppm (t, 3JHH = 6.6 Hz, 1H, N4H). 13C{1H} NMR (acetone-d6): δ = 24.7, 25.2, 26.3 and 26.4 (s, CH3, C14H3, C14′H3, C15H3 and C15′H3), 42.9 (s, CH2, C5H2), 55.6 and 55.9 (br, s, CH, 2 × C1H and 2 × C1′H), 64.7 (s, CH2, C6H2), 66.6 (s, CH, C7H), 71.3 (s, CH, C10H), 71.5 (s, CH, C8H), 71.8 (s, CH, C9H), 97.1 (s, CH, C11H), 109.2 (s, Cq, Cq13), 109.9 (s, Cq, Cq12), 164.7 (s, Cq, Cq3), 170.7 (s, Cq, 2 × Cq2), 179.8 ppm (s, Cq, Cq, C16O). 11B{1H} NMR (acetone-d6): δ = −18.5 (s, 2B), −17.0 (s, 2B), −14.1 (s, 4B), −12.8 (s, 4B), −10.4 (s, 2B), −5.9 (s, 4B), −3.4 ppm (s, 2B, 2 × BS). 11B NMR (acetone-d6): δ = −20.4 to −9.2 (bra, 14B), −5.9 (d, 1JBH = 149 Hz, 4B), −3.4 ppm (s, 2B, 2 × BS). HRMS (ESI+): C21H44B20N4O7S2, m/z calcd: 768.45326 ([M + Na]+); found: 768.45314 (100%); m/z calcd: 746.47131 ([M + H]+); found 746.47144 (17%).
:3 to 1:2, v/v) 2.50 g (8.96 mmol, 82%, Rf = 0.61, ethyl acetate/n-hexane, 1:2, v/v) of compound 7 was obtained as a slightly yellow solid. Tm: 140–145 °C (ethyl acetate/n-hexane, decomposition). IR (KBr): = 3430 (m), 3273 (m), 3169 (m), 2981 (m), 2937 (w), 1739 (s), 1678 (m), 1612 (s), 1552 (s), 1524 (m), 1422 (m), 1393 (m), 1370 (m), 1323 (m), 1253 (s), 1233 (s), 1161 (s), 1130 (m), 1067 (w), 1029 (w), 978 (w), 934 (w), 896 (w), 846 (m), 801 (m), 756 (w), 691 (w), 657 (w), 552 (w), 424 (w) cm−1. 1H NMR (acetone-d6): δ = 1.46 (s, 9H, C(CH3)3), 4.13 (d, 3JHH = 6.2 Hz, 2H, CH2), 8.15 ppm (vbr, t, 3JHH = 7.0 Hz, 1H, NH). 13C{1H} NMR (acetone-d6): δ = 28.1 (s, CH3, C(CH3)3), 44.2 (s, CH2), 82.3 (s, Cq, Cq(CH3)3), 167.5 (s, Cq, CqN), 168.4 (s, Cq, CqO), 170.6, 171.1 ppm (s, Cq, 2 × CqCl). HRMS (ESI+): C9H12Cl2N4O2, m/z calcd: 301.02352 ([M + Na]+); found: 301.02354 (100%).
:3,4-di-O-isopropylidene-6-deoxy-α-D-galactopyranosyl-6-triflate (4) and 0.80 g (4.77 mmol, 1.21 eq.) tert-butyl glycinate hydrochloride, evacuated, purged with nitrogen and 40 mL MeCN were added. Subsequently, 2.00 mL (1.52 g, 11.8 mmol, 2.47 eq.) DIPEA were slowly added and the mixture was stirred for 2 d at 45 °C. A solution of 1.83 g (9.92 mmol, 2.51 eq.) cyanuric chloride and 0.87 mL (0.66 g, 5.12 mmol, 1.30 eq.) DIPEA in 10 mL MeCN was slowly added at 0 °C and the reaction mixture was stirred for 2 d at 35 °C. The reaction was stopped by adding 50 mL of a saturated NaCl solution; the resulting layers were separated. The aqueous layer was extracted three times with 50 mL ethyl acetate. The combined organic layers were dried over MgSO4, filtered and the solvent was removed under reduced pressure. After column chromatography (ethyl acetate/n-hexane, 1:5, v/v) 1.59 g (3.05 mmol, 77%, Rf = 0.37) of the title compound was obtained as a slightly yellow solid. Colourless crystals of 11 suitable for X-ray structure determination were obtained from CHCl3 at room temperature. Crystallographic data are given in Table S2,† and the molecular structure is depicted in Fig. 3. Tm: 122–124 °C (ethyl acetate). IR (KBr): = 3441 (s), 2984 (m), 2937 (w), 1741 (s), 1568 (s), 1489 (s), 1458 (w), 1417 (w), 1372 (m), 1328 (m), 1289 (w), 1228 (s), 1172 (s), 1114 (m), 1071 (s), 1003 (m), 980 (m), 906 (w), 883 (w), 848 (m), 800 (m), 773 (w), 746 (w), 651 (w), 599 (w), 551 (w), 512 (w) cm−1. 1H NMR (CDCl3): δ = 1.31 (s, 3H, C15 or 15′H3), 1.34 (s, 3H, C16 or 16′H3), 1.44 (s, 3H, C15 or 15′H3), 1.47 (s, 9H, C(C1H3)3), 1.50 (s, 3H, C16 or 16′H3), 3.68 (dd, 2JHH = 14.3 Hz, 3JHH = 8.1 Hz, 1H, C7HH), 3.88 (dd, 2JHH = 14.3 Hz, 3JHH = 5.1 Hz, 1H, C7HH), 4.19 (d, 2JHH = 17.5 Hz, 1H, C4HH), 4.21 (m, 1H, C8H), 4.26 (dd, 3JHH = 7.9 Hz, 3JHH = 1.8 Hz, 1H, C10H), 4.30 (dd, 3JHH = 5.0 Hz, 3JHH = 2.5 Hz, 1H, C11H), 4.44 (d, 2JHH = 17.5 Hz, 1H, C4HH), 4.62 (dd, 3JHH = 7.9 Hz, 3JHH = 2.5 Hz, 1H, C9H), 5.49 ppm (d, 3JHH = 4.9 Hz, 1H, C12H). 13C{1H} NMR (CDCl3): δ = 24.4, 25.0, 25.9 and 26.0 (s, CH3, C15H3, C15′H3, C16H3 and C16′H3), 28.0 (s, CH3, C(C1H3)3), 50.1 (s, CH2, C7H2), 52.2 (s, CH2, C4H2), 65.3 (s, CH, C8H), 70.5 (s, CH, C11H), 70.7 (s, CH, C9H), 71.1 (s, CH, C10H), 82.4 (s, Cq, Cq2), 96.3 (s, CH, C12H), 108.9 (s, Cq, Cq14), 109.5 (s, Cq, Cq13), 165.5 (s, Cq, Cq5N), 167.5 (s, Cq, 2 × Cq6Cl), 169.9 ppm (s, Cq, Cq3O). LRMS (ESI+): C21H30Cl2N4O2, m/z calcd: 521.2 ([M + H]+); found: 521.2 (21%); m/z calcd: 543.1 ([M + Na]+); found: 543.1 (55%).
:3 to 100% ethyl acetate, v/v) 3.01 g (3.75 mmol, 98%, Rf = 0.25, ethyl acetate/n-hexane, 1:3, v/v) of compound 12 were obtained as a colourless solid. Colourless crystals of 12 suitable for X-ray structure determination were obtained from ethyl acetate layered with n-hexane at room temperature. Crystallographic data are given in Table S2,† and the molecular structure is depicted in Fig. 5. Tm: 240–242 °C (ethyl acetate, decomposition). IR (KBr): = 3253 (w), 3134 (w), 3064 (m), 3038 (m), 2983 (m), 2952 (m), 2602 (s), 2562 (m), 1967 (w), 1741 (s), 1614 (m), 1534 (s), 1510 (s), 1486 (s), 1421 (m), 1401 (w), 1383 (m), 1370 (s), 1331 (m), 1316 (m), 1301 (m), 1261 (s), 1250 (s), 1222 (s), 1182 (s), 1153 (s), 1102 (s), 1070 (s), 1044 (m), 1007 (s), 976 (m), 952 (s), 919 (w), 901 (m), 879 (w), 864 (m), 848 (s), 803 (m), 772 (w), 757 (w), 731 (m) cm−1. 1H NMR (CDCl3): δ = 1.29 (s, 3H, C16 or 16′H3), 1.34 (s, 3H, C17 or 17′H3), 1.44 (s, 12H, C16 or 16′H3 and C(C7H3)3), 1.46 (s, 3H, C17 or 17′H3), 1.66 to 3.56 (bra, 18H, 2 × B10H9), 2.96 (br, s, 4H, 4xC1H), 3.65 (dd, 2JHH = 14.4 Hz, 3JHH = 8.2 Hz, 1H, C8HH), 3.99 (dd, 2JHH = 14.4 Hz, 3JHH = 4.6 Hz, 1H, C8HH), 4.19 (m, 1H, C9H), 4.26 (m, 2H, C11H and C12H), 4.37 (d, 2JHH = 17.8 Hz, 1H, C4HH), 4.58 (dd, 3JHH = 8.0 Hz, 3JHH = 2.4 Hz, 1H, C10H), 4.68 (d, 2JHH = 17.8 Hz, 1H, C4HH), 5.49 ppm (d, 3JHH = 5.0 Hz, 1H, C13H). 13C{1H} NMR (CDCl3): δ = 24.4, 25.1, 26.0 and 26.1 (s, CH3, C16H3, C16′H3, C17H3 and C17′H3), 28.1 (s, CH3, C(C7H3)3), 47.9 (s, CH2, C8H2), 50.2 (s, CH2, C4H2), 53.8 (br, s, CH, 4xC1H), 66.4 (s, CH, C9H), 70.5 (s, CH, C12H), 70.8 (s, CH, C10H), 71.5 (s, CH, C11H), 81.4 (s, Cq, Cq6), 96.3 (s, CH, C13H), 108.8 (s, Cq, Cq15), 109.2 (s, Cq, Cq14), 163.2 (s, Cq, Cq3), 169.3 (s, Cq, Cq2), 178.5 ppm (s, Cq, Cq5). 11B{1H} NMR (CDCl3): δ = −18.7 (s, 2B), −17.4 (s, 2B), −14.0 (s, 4B), −12.8 (s, 4B), −10.0 (s, 2B), −5.6 (s, 4B), −3.1 ppm (s, 2B, 2 × BS). 11B NMR (CDCl3): δ = −20.5 to −15.8 (bra, 4B), −15.4 to −11.4 (bra, 8B), −10.0 (d, 1JBH = 150 Hz, 2B), −5.6 (d, 1JBH = 158 Hz, 4B), −3.1 ppm (s, 2B, 2 × BS). HRMS (ESI+): C25H52B20N4O7S2, m/z calcd: 802.53420 ([M + H]+); found: 802.53393 (100%).
:3 to 100% ethyl acetate to ethyl acetate/methanol, 10:1, v/v) and 775 mg (1.04 mmol, 83%) of compound 5 were isolated as an off-white solid. Tm: 167–169 °C (ethyl acetate). IR (KBr): = 3435 (s), 3056 (m), 2984 (m), 2935 (m), 2608 (s), 1682 (m), 1533 (s), 1513 (s), 1483 (s), 1432 (w), 1407 (w), 1383 (m), 1316 (m), 1247 (m), 1211 (s), 1176 (s), 1070 (s), 997 (m), 977 (w), 954 (m), 919 (w), 904 (w), 864 (s), 849 (s), 802 (w), 759 (w), 727 (w), 677 (w), 624 (w), 513 (w) cm−1. 1H NMR (CDCl3): δ = 1.28 (s, 3H, C14 or 14′H3), 1.34 (s, 3H, C15 or 15′H3), 1.43 (s, 3H, C14 or 14′H3), 1.46 (s, 3H, C15 or 15′H3), 1.55 to 3.30 (bra, 18H, 2 × B10H9), 2.98 (br, s, 4H, 2 × C1H and 2 × C1′H), 3.54 (dd, 2JHH = 14.4 Hz, 3JHH = 8.4 Hz, 1H, C6HH), 4.10 (m, 1H, C6HH), 4.19 (m, 1H, C7H), 4.27 (m, 2H, C9H and C10H), 4.58 (m, 1H, C8H), 4.64 (br, m, 2H, C4H2), 5.48 (d, 3JHH = 5.0 Hz, 1H, C11H), 6.34 ppm (vbr s, 1H, C5OOH or NR3H+). 13C{1H} NMR (CDCl3): δ = 24.3, 25.1, 26.0 and 26.1 (s, CH3, C14H3, C14′H3, C15H3 and C15′H3), 48.6 (s, CH2, C6H2), 49.9 (s, CH2, C4H2), 53.9 and 54.2 (br, s, CH, 2 × C1H and 2 × C1′H), 66.5 (s, CH, C7H), 70.5 (s, CH, C10H), 70.8 (s, CH, C8H), 71.5 (s, CH, C9H), 96.2 (s, CH, C11H), 108.9 (s, Cq, Cq13), 109.3 (s, Cq, Cq12), 163.3 (s, Cq, Cq3), 175.3 (s, Cq, Cq5), 178.6 ppm (br, s, Cq, 2 × Cq2). 11B{1H} NMR (CDCl3): δ = −19.8 to −4.3 (bra, 18B), −3.4 (s, 1B, BS), −3.1 ppm (s, 1B, BS). 11B NMR (CDCl3): δ = −21.0 to −4.1 (bra, 18B), −3.4 (s, 1B, BS), −3.1 ppm (s, 1B, BS). HRMS (ESI+): C21H44B20N4O7S2, m/z calcd: 746.47131 ([M + H]+); found: 746.47182 (100%).
:3, v/v) 1.90 g (3.46 mmol, 68%, Rf = 0.31) of compound 14 were obtained as a colourless solid. Tm: 107–109 °C (ethyl acetate/n-hexane, decomposition). IR (KBr): = 3412 (s), 2984 (s), 2937 (m), 1715 (s), 1574 (s), 1513 (w), 1481 (s), 1440 (m), 1384 (m), 1329 (m), 1237 (s), 1213 (m), 1165 (s), 1110 (w), 1070 (s), 1003 (m), 903 (w), 882 (w), 847 (m), 798 (m), 649 (w), 585 (w), 513 (w) cm−1. 1H NMR (acetone-d6): δ = 1.29 (s, 3 H, C17 or 17′H3), 1.35 (s, 3 H, C18 or 18′H3), 1.38 (s, 12 H, C17 or 17′H3 and C(C1H3)3), 1.43 (s, 3 H, C18 or 18′H3), 3.39 (m, 2 H, C5H2), 3.76 (m, br, 3 H, C6HH and C9HH and C9HH), 3.92 (m, 1 H, C6HH), 4.25 (m, 1 H, C10H), 4.28 (m, 1 H, C11H), 4.36 (dd, 3JHH = 5.1 Hz, 3JHH = 2.5 Hz, 1 H, C13H), 4.65 (dd, 3JHH = 7.9 Hz, 3JHH = 2.4 Hz, 1 H, C12H), 5.48 (d, 3JHH = 5.0 Hz, 1 H, C14H), 6.12 ppm (t, 3JHH = 6.3 Hz, 1 H, N4H). 13C{1H} NMR (acetone-d6): δ = 24.7, 25.2, 26.2 and 26.4 (s, CH3, C17H3, C17′H3, C18H3 and C18′H3), 28.6 (s, CH3, C(C1H3)3), 38.5 (s, CH2, C5H2), 50.1 (s, CH2, C6H2), 50.3 (s, CH2, C9H2), 65.7 (s, CH, C10H), 71.3 (s, CH, C13H), 71.7 (s, CH, C12H), 72.0 (s, CH, C11H), 78.8 (s, Cq, Cq2(CH3)3), 97.3 (s, CH, C14H), 109.3 (s, Cq, Cq15), 110.0 (s, Cq, Cq16), 156.7 (s, Cq, Cq3), 166.3 (s, Cq, Cq8), 170.27 and 170.32 ppm (s, Cq, 2 × Cq7). HRMS (ESI+): C22H33Cl2N5O7, m/z calcd: 572.1655 ([M + Na]+); found: 572.1651 (100%); m/z calcd: 1123.3383 ([2M + Na]+); found: 1123.3383 (28%); m/z calcd: 550.1836 ([M + H]+); found: 550.1833 (20%).
:2, v/v) 1.50 g (1.80 mmol, 99%, Rf = 0.21) of compound 15 was obtained as a colourless solid. IR (KBr): = 3455 (m), 3048 (m), 2981 (m), 2935 (m), 2608 (s), 1715 (s), 1625 (w), 1543 (s), 1511 (s), 1478 (s), 1434 (m), 1383 (m), 1369 (m), 1335 (m), 1315 (m), 1243 (s), 1214 (m), 1171 (s), 1104 (w), 1069 (s), 995 (m), 954 (m), 918 (w), 903 (w), 864 (m), 847 (m), 802 (w), 759 (w), 731 (w), 678 (w), 627 (w), 585 (w), 512 (w) cm−1. 1H NMR (CDCl3): δ = 1.28 (s, 3 H, C18 or 18′H3), 1.35 (s, 3 H, C19 or 19′H3), 1.41 (s, 12 H, C18 or 18′H3 and C(C9H3)3), 1.48 (s, 3 H, C19 or 19′H3), 1.55 to 3.55 (bra, 18 H, 2 × B10H9), 2.98 (br, s, 4 H, 2 × C1H and 2 × C1′H), 3.40 (m, 2 H, C5H2), 3.45 (m, 1 H, C10HH), 3.64 (dt, 3JHH = 13.3 Hz, 3JHH = 5.6 Hz, 1 H, C4HH), 4.03 (dd, 3JHH = 14.4 Hz, 3JHH = 3.2 Hz, 1 H, C10HH), 4.14 (m, 1 H, C4HH), 4.28 (m, 3 H, C11H, C12H and C14H), 4.59 (dd, 3JHH = 7.9 Hz, 3JHH = 2.3 Hz, 1 H, C13H), 5.33 (m, 1 H, N6H), 5.50 ppm (d, 3JHH = 4.9 Hz, 1 H, C15H). 13C{1H} NMR (CDCl3): δ = 24.4, 25.1, 26.06 and 26.08 (s, CH3, C18H3, C18′H3, C19H3 and C19′H3), 28.4 (s, CH3, C(C9H3)3), 39.7 (s, CH2, C5H2), 48.7 (s, CH2, C4H2), 49.0 (s, CH2, C10H2), 53.9 and 54.0 (s, CH, 2 × C1H and 2 × C1′H), 66.2 (s, CH, C11H), 70.5 (s, CH, C14H); 70.9 (s, CH, C13H), 71.7 (s, CH, C12H), 78.8 (s, Cq, Cq8), 96.3 (s, CH, C15H), 109.0 (s, Cq, Cq16), 109.3 (s, Cq, Cq17), 156.1 (s, Cq, Cq7), 163.3 (s, Cq, Cq3), 178.1 ppm (s, Cq, Cq2). 11B{1H} NMR (CDCl3): δ = −18.8 (s, 2B), −17.3 (s, 2B), −14.0 (s, 4B), −12.9 (s, 4B), −10.1 (s, 2B), −5.6 (s, 4B), −3.1 ppm (s, 2B, 2 × BS). 11B NMR (CDCl3): δ = −20.9 to −11.4 (bra, 12B), −10.1 (d, 1JBH = 151 Hz, 2B), −5.6 (d, 1JBH = 166 Hz, 4B), −3.1 ppm (s, 2B, 2 × BS). HRMS (ESI+): C26H55B20N5O7S2, m/z calcd: 831.55915 ([M + H]+); found: 831.56120 (100%).
:1 (v/v) to ethyl acetate, 100%) and 283 mg (388 μmol, 91%, Rf = 0.05 (100% ethyl acetate)) of compound 16 were obtained as an off-white solid. Tm: 147–150 °C (ethyl acetate, decomposition). IR (KBr): = 3442 (s), 3053 (m), 2989 (m), 2935 (m), 2609 (s), 2095 (w), 1680 (s), 1637 (m), 1531 (s), 1508 (s), 1483 (s), 1431 (m), 1383 (m), 1338 (w), 1316 (m), 1281 (w), 1250 (m), 1207 (s), 1175 (s), 1139 (m), 1109 (w), 1069 (s), 998 (m), 954 (m), 902 (w), 864 (m), 849 (m), 802 (m), 760 (w), 724 (m), 700 (w), 626 (w), 588 (w), 513 (w) cm−1. 1H NMR (acetone-d6): δ = 1.28 (s, 3 H, C15 or 15′H3), 1.34 (s, 3 H, C16 or 16′H3), 1.35 (s, 3 H, C15 or 15′H3), 1.44 (s, 3 H, C16 or 16′H3), 1.53 to 3.40 (bra, 18 H, 2 × B10H9), 3.56 (dd, 3JHH = 15.4 Hz, 3JHH = 10.2 Hz, 1 H, C7HH), 3.80 and 3.82 (s, CH, 2 × C1H and 2 × C1′H), 3.95 to 4.25 (br, m, 6 H, 2 × C4HH, 2 × C5HH, C7HH and C8H), 4.35 (m, 2 H, C9H and C11H), 4.62 (dd, 3JHH = 7.9 Hz, 3JHH = 2.5 Hz, 1 H, C10H), 5.51 ppm (d, 3JHH = 5.0 Hz, 1 H, C12H). 13C{1H} NMR (acetone-d6): δ = 24.7, 25.2, 26.3 and 26.4 (s, CH3, C15H3, C15′H3, C16H3 and C16′H3), 45.8 (s, CH2, C5H2), 47.9 (s, CH2, C4H2), 49.9 (s, CH2, C7H2), 56.0 and 56.1 (s, CH, 2 × C1H and 2 × C1′H), 67.2 (s, CH, C8H), 71.3 (s, CH, C11H), 71.8 (s, CH, C10H), 72.4 (s, CH, C9H), 97.3 (s, CH, C12H), 109.3 (s, Cq, Cq13), 110.0 (s, Cq, Cq14), 163.9 (s, Cq, Cq3), 179.0 and 179.5 ppm (s, Cq, 2 × Cq2). 11B{1H} NMR (acetone-d6): δ = −19.6 (s, 2B), −17.9 (s, 2B), −14.9 (s, 4B), −13.7 (s, 4B), −11.3 (s, 2B), −6.9 (s, 4B), −4.4 ppm (s, 2B, 2 × BS). 11B NMR (acetone-d6): δ = −21.3 to −12.3 (bra, 12B), −11.3 (d, 1JBH = 149 Hz, 2B), −6.9 (d, 1JBH = 165 Hz, 4B), −4.4 ppm (s, 2B, 2 × BS). HRMS (ESI+): C21H47B20N5O5S2, m/z calcd: 774.5604 ([M + C2H7N]+); found: 774.5620 (100%); m/z calcd: 731.5067 ([M + H]+); found: 731.5082 (54%).
= 3436 (m), 3056 (w), 2988 (w), 2936 (w), 2608 (s), 1749 (m), 1563 (s), 1518 (s), 1497 (s), 1455 (w), 1412 (m), 1383 (m), 1298 (m), 1252 (m), 1211 (m), 1174 (m), 1116 (w), 1070 (s), 1001 (m), 955 (w), 921 (w), 892 (w), 864 (m), 850 (m), 804 (w), 759 (w), 730 (w), 677 (w), 627 (w), 585 (w), 512 (w) cm−1. 1H NMR (acetone-d6): δ = 1.316 (s, 3H, C14 or 14′H3), 1.322 (s, 3H, C15 or 15′H3), 1.38 (s, 3H, C14 or 14′H3), 1.47 (s, 3H, C15 or 15′H3), 1.50 to 3.50 (bra, 18H, 2 × B10H9), 3.69 (br, s, 2H, 2 × C1 or 1′H), 3.74 (br, s, 2H, 2 × C1 or 1′H), 4.06 (m, 1H, C7H), 4.19 (m, 2H, C6H2), 4.29 (m, 1H, C9H), 4.33 (m, 2H, C5H2), 4.37 (m, 1H, C10H), 4.64 (dd, 3JHH = 7.9 Hz, 3JHH = 2.5 Hz, 1H, C8H), 5.47 (d, 3JHH = 5.0 Hz, 1H, C11H), 7.05 ppm (t, 3JHH = 6.6 Hz, 1H, N4H). 13C{1H} NMR (acetone-d6): δ = 24.7, 25.2, 26.3 and 26.4 (s, CH3, C14H3, C14′H3, C15H3 and C15′H3), 42.9 (s, CH2, C5H2), 55.6 and 55.9 (br, s, CH, 2 × C1H and 2 × C1′H), 64.7 (s, CH2, C6H2), 66.6 (s, CH, C7H), 71.3 (s, CH, C10H), 71.5 (s, CH, C8H), 71.8 (s, CH, C9H), 97.1 (s, CH, C11H), 109.2 (s, Cq, Cq13), 109.9 (s, Cq, Cq12), 164.7 (s, Cq, Cq3), 170.7 (s, Cq, 2 × Cq2), 179.8 ppm (s, Cq, Cq, C16O). 11B{1H} NMR (acetone-d6): δ = −18.5 (s, 2B), −17.0 (s, 2B), −14.1 (s, 4B), −12.8 (s, 4B), −10.4 (s, 2B), −5.9 (s, 4B), −3.4 ppm (s, 2B, 2 × BS). 11B NMR (acetone-d6): δ = −20.4 to −9.2 (bra, 14B), −5.9 (d, 1JBH = 149 Hz, 4B), −3.4 ppm (s, 2B, 2 × BS). HRMS (ESI+): C21H44B20N4O7S2, m/z calcd: 768.45326 ([M + Na]+); found: 768.45314 (100%); m/z calcd: 746.47131 ([M + H]+); found 746.47144 (17%).
:3 to 1:2, v/v) 2.50 g (8.96 mmol, 82%, Rf = 0.61, ethyl acetate/n-hexane, 1:2, v/v) of compound 7 was obtained as a slightly yellow solid. Tm: 140–145 °C (ethyl acetate/n-hexane, decomposition). IR (KBr): = 3430 (m), 3273 (m), 3169 (m), 2981 (m), 2937 (w), 1739 (s), 1678 (m), 1612 (s), 1552 (s), 1524 (m), 1422 (m), 1393 (m), 1370 (m), 1323 (m), 1253 (s), 1233 (s), 1161 (s), 1130 (m), 1067 (w), 1029 (w), 978 (w), 934 (w), 896 (w), 846 (m), 801 (m), 756 (w), 691 (w), 657 (w), 552 (w), 424 (w) cm−1. 1H NMR (acetone-d6): δ = 1.46 (s, 9H, C(CH3)3), 4.13 (d, 3JHH = 6.2 Hz, 2H, CH2), 8.15 ppm (vbr, t, 3JHH = 7.0 Hz, 1H, NH). 13C{1H} NMR (acetone-d6): δ = 28.1 (s, CH3, C(CH3)3), 44.2 (s, CH2), 82.3 (s, Cq, Cq(CH3)3), 167.5 (s, Cq, CqN), 168.4 (s, Cq, CqO), 170.6, 171.1 ppm (s, Cq, 2 × CqCl). HRMS (ESI+): C9H12Cl2N4O2, m/z calcd: 301.02352 ([M + Na]+); found: 301.02354 (100%).
:3,4-di-O-isopropylidene-6-deoxy-α-D-galactopyranosyl-6-triflate (4) and 0.80 g (4.77 mmol, 1.21 eq.) tert-butyl glycinate hydrochloride, evacuated, purged with nitrogen and 40 mL MeCN were added. Subsequently, 2.00 mL (1.52 g, 11.8 mmol, 2.47 eq.) DIPEA were slowly added and the mixture was stirred for 2 d at 45 °C. A solution of 1.83 g (9.92 mmol, 2.51 eq.) cyanuric chloride and 0.87 mL (0.66 g, 5.12 mmol, 1.30 eq.) DIPEA in 10 mL MeCN was slowly added at 0 °C and the reaction mixture was stirred for 2 d at 35 °C. The reaction was stopped by adding 50 mL of a saturated NaCl solution; the resulting layers were separated. The aqueous layer was extracted three times with 50 mL ethyl acetate. The combined organic layers were dried over MgSO4, filtered and the solvent was removed under reduced pressure. After column chromatography (ethyl acetate/n-hexane, 1:5, v/v) 1.59 g (3.05 mmol, 77%, Rf = 0.37) of the title compound was obtained as a slightly yellow solid. Colourless crystals of 11 suitable for X-ray structure determination were obtained from CHCl3 at room temperature. Crystallographic data are given in Table S2,† and the molecular structure is depicted in Fig. 3. Tm: 122–124 °C (ethyl acetate). IR (KBr): = 3441 (s), 2984 (m), 2937 (w), 1741 (s), 1568 (s), 1489 (s), 1458 (w), 1417 (w), 1372 (m), 1328 (m), 1289 (w), 1228 (s), 1172 (s), 1114 (m), 1071 (s), 1003 (m), 980 (m), 906 (w), 883 (w), 848 (m), 800 (m), 773 (w), 746 (w), 651 (w), 599 (w), 551 (w), 512 (w) cm−1. 1H NMR (CDCl3): δ = 1.31 (s, 3H, C15 or 15′H3), 1.34 (s, 3H, C16 or 16′H3), 1.44 (s, 3H, C15 or 15′H3), 1.47 (s, 9H, C(C1H3)3), 1.50 (s, 3H, C16 or 16′H3), 3.68 (dd, 2JHH = 14.3 Hz, 3JHH = 8.1 Hz, 1H, C7HH), 3.88 (dd, 2JHH = 14.3 Hz, 3JHH = 5.1 Hz, 1H, C7HH), 4.19 (d, 2JHH = 17.5 Hz, 1H, C4HH), 4.21 (m, 1H, C8H), 4.26 (dd, 3JHH = 7.9 Hz, 3JHH = 1.8 Hz, 1H, C10H), 4.30 (dd, 3JHH = 5.0 Hz, 3JHH = 2.5 Hz, 1H, C11H), 4.44 (d, 2JHH = 17.5 Hz, 1H, C4HH), 4.62 (dd, 3JHH = 7.9 Hz, 3JHH = 2.5 Hz, 1H, C9H), 5.49 ppm (d, 3JHH = 4.9 Hz, 1H, C12H). 13C{1H} NMR (CDCl3): δ = 24.4, 25.0, 25.9 and 26.0 (s, CH3, C15H3, C15′H3, C16H3 and C16′H3), 28.0 (s, CH3, C(C1H3)3), 50.1 (s, CH2, C7H2), 52.2 (s, CH2, C4H2), 65.3 (s, CH, C8H), 70.5 (s, CH, C11H), 70.7 (s, CH, C9H), 71.1 (s, CH, C10H), 82.4 (s, Cq, Cq2), 96.3 (s, CH, C12H), 108.9 (s, Cq, Cq14), 109.5 (s, Cq, Cq13), 165.5 (s, Cq, Cq5N), 167.5 (s, Cq, 2 × Cq6Cl), 169.9 ppm (s, Cq, Cq3O). LRMS (ESI+): C21H30Cl2N4O2, m/z calcd: 521.2 ([M + H]+); found: 521.2 (21%); m/z calcd: 543.1 ([M + Na]+); found: 543.1 (55%).
:3 to 100% ethyl acetate, v/v) 3.01 g (3.75 mmol, 98%, Rf = 0.25, ethyl acetate/n-hexane, 1:3, v/v) of compound 12 were obtained as a colourless solid. Colourless crystals of 12 suitable for X-ray structure determination were obtained from ethyl acetate layered with n-hexane at room temperature. Crystallographic data are given in Table S2,† and the molecular structure is depicted in Fig. 5. Tm: 240–242 °C (ethyl acetate, decomposition). IR (KBr): = 3253 (w), 3134 (w), 3064 (m), 3038 (m), 2983 (m), 2952 (m), 2602 (s), 2562 (m), 1967 (w), 1741 (s), 1614 (m), 1534 (s), 1510 (s), 1486 (s), 1421 (m), 1401 (w), 1383 (m), 1370 (s), 1331 (m), 1316 (m), 1301 (m), 1261 (s), 1250 (s), 1222 (s), 1182 (s), 1153 (s), 1102 (s), 1070 (s), 1044 (m), 1007 (s), 976 (m), 952 (s), 919 (w), 901 (m), 879 (w), 864 (m), 848 (s), 803 (m), 772 (w), 757 (w), 731 (m) cm−1. 1H NMR (CDCl3): δ = 1.29 (s, 3H, C16 or 16′H3), 1.34 (s, 3H, C17 or 17′H3), 1.44 (s, 12H, C16 or 16′H3 and C(C7H3)3), 1.46 (s, 3H, C17 or 17′H3), 1.66 to 3.56 (bra, 18H, 2 × B10H9), 2.96 (br, s, 4H, 4xC1H), 3.65 (dd, 2JHH = 14.4 Hz, 3JHH = 8.2 Hz, 1H, C8HH), 3.99 (dd, 2JHH = 14.4 Hz, 3JHH = 4.6 Hz, 1H, C8HH), 4.19 (m, 1H, C9H), 4.26 (m, 2H, C11H and C12H), 4.37 (d, 2JHH = 17.8 Hz, 1H, C4HH), 4.58 (dd, 3JHH = 8.0 Hz, 3JHH = 2.4 Hz, 1H, C10H), 4.68 (d, 2JHH = 17.8 Hz, 1H, C4HH), 5.49 ppm (d, 3JHH = 5.0 Hz, 1H, C13H). 13C{1H} NMR (CDCl3): δ = 24.4, 25.1, 26.0 and 26.1 (s, CH3, C16H3, C16′H3, C17H3 and C17′H3), 28.1 (s, CH3, C(C7H3)3), 47.9 (s, CH2, C8H2), 50.2 (s, CH2, C4H2), 53.8 (br, s, CH, 4xC1H), 66.4 (s, CH, C9H), 70.5 (s, CH, C12H), 70.8 (s, CH, C10H), 71.5 (s, CH, C11H), 81.4 (s, Cq, Cq6), 96.3 (s, CH, C13H), 108.8 (s, Cq, Cq15), 109.2 (s, Cq, Cq14), 163.2 (s, Cq, Cq3), 169.3 (s, Cq, Cq2), 178.5 ppm (s, Cq, Cq5). 11B{1H} NMR (CDCl3): δ = −18.7 (s, 2B), −17.4 (s, 2B), −14.0 (s, 4B), −12.8 (s, 4B), −10.0 (s, 2B), −5.6 (s, 4B), −3.1 ppm (s, 2B, 2 × BS). 11B NMR (CDCl3): δ = −20.5 to −15.8 (bra, 4B), −15.4 to −11.4 (bra, 8B), −10.0 (d, 1JBH = 150 Hz, 2B), −5.6 (d, 1JBH = 158 Hz, 4B), −3.1 ppm (s, 2B, 2 × BS). HRMS (ESI+): C25H52B20N4O7S2, m/z calcd: 802.53420 ([M + H]+); found: 802.53393 (100%).
:3 to 100% ethyl acetate to ethyl acetate/methanol, 10:1, v/v) and 775 mg (1.04 mmol, 83%) of compound 5 were isolated as an off-white solid. Tm: 167–169 °C (ethyl acetate). IR (KBr): = 3435 (s), 3056 (m), 2984 (m), 2935 (m), 2608 (s), 1682 (m), 1533 (s), 1513 (s), 1483 (s), 1432 (w), 1407 (w), 1383 (m), 1316 (m), 1247 (m), 1211 (s), 1176 (s), 1070 (s), 997 (m), 977 (w), 954 (m), 919 (w), 904 (w), 864 (s), 849 (s), 802 (w), 759 (w), 727 (w), 677 (w), 624 (w), 513 (w) cm−1. 1H NMR (CDCl3): δ = 1.28 (s, 3H, C14 or 14′H3), 1.34 (s, 3H, C15 or 15′H3), 1.43 (s, 3H, C14 or 14′H3), 1.46 (s, 3H, C15 or 15′H3), 1.55 to 3.30 (bra, 18H, 2 × B10H9), 2.98 (br, s, 4H, 2 × C1H and 2 × C1′H), 3.54 (dd, 2JHH = 14.4 Hz, 3JHH = 8.4 Hz, 1H, C6HH), 4.10 (m, 1H, C6HH), 4.19 (m, 1H, C7H), 4.27 (m, 2H, C9H and C10H), 4.58 (m, 1H, C8H), 4.64 (br, m, 2H, C4H2), 5.48 (d, 3JHH = 5.0 Hz, 1H, C11H), 6.34 ppm (vbr s, 1H, C5OOH or NR3H+). 13C{1H} NMR (CDCl3): δ = 24.3, 25.1, 26.0 and 26.1 (s, CH3, C14H3, C14′H3, C15H3 and C15′H3), 48.6 (s, CH2, C6H2), 49.9 (s, CH2, C4H2), 53.9 and 54.2 (br, s, CH, 2 × C1H and 2 × C1′H), 66.5 (s, CH, C7H), 70.5 (s, CH, C10H), 70.8 (s, CH, C8H), 71.5 (s, CH, C9H), 96.2 (s, CH, C11H), 108.9 (s, Cq, Cq13), 109.3 (s, Cq, Cq12), 163.3 (s, Cq, Cq3), 175.3 (s, Cq, Cq5), 178.6 ppm (br, s, Cq, 2 × Cq2). 11B{1H} NMR (CDCl3): δ = −19.8 to −4.3 (bra, 18B), −3.4 (s, 1B, BS), −3.1 ppm (s, 1B, BS). 11B NMR (CDCl3): δ = −21.0 to −4.1 (bra, 18B), −3.4 (s, 1B, BS), −3.1 ppm (s, 1B, BS). HRMS (ESI+): C21H44B20N4O7S2, m/z calcd: 746.47131 ([M + H]+); found: 746.47182 (100%).
:3, v/v) 1.90 g (3.46 mmol, 68%, Rf = 0.31) of compound 14 were obtained as a colourless solid. Tm: 107–109 °C (ethyl acetate/n-hexane, decomposition). IR (KBr): = 3412 (s), 2984 (s), 2937 (m), 1715 (s), 1574 (s), 1513 (w), 1481 (s), 1440 (m), 1384 (m), 1329 (m), 1237 (s), 1213 (m), 1165 (s), 1110 (w), 1070 (s), 1003 (m), 903 (w), 882 (w), 847 (m), 798 (m), 649 (w), 585 (w), 513 (w) cm−1. 1H NMR (acetone-d6): δ = 1.29 (s, 3 H, C17 or 17′H3), 1.35 (s, 3 H, C18 or 18′H3), 1.38 (s, 12 H, C17 or 17′H3 and C(C1H3)3), 1.43 (s, 3 H, C18 or 18′H3), 3.39 (m, 2 H, C5H2), 3.76 (m, br, 3 H, C6HH and C9HH and C9HH), 3.92 (m, 1 H, C6HH), 4.25 (m, 1 H, C10H), 4.28 (m, 1 H, C11H), 4.36 (dd, 3JHH = 5.1 Hz, 3JHH = 2.5 Hz, 1 H, C13H), 4.65 (dd, 3JHH = 7.9 Hz, 3JHH = 2.4 Hz, 1 H, C12H), 5.48 (d, 3JHH = 5.0 Hz, 1 H, C14H), 6.12 ppm (t, 3JHH = 6.3 Hz, 1 H, N4H). 13C{1H} NMR (acetone-d6): δ = 24.7, 25.2, 26.2 and 26.4 (s, CH3, C17H3, C17′H3, C18H3 and C18′H3), 28.6 (s, CH3, C(C1H3)3), 38.5 (s, CH2, C5H2), 50.1 (s, CH2, C6H2), 50.3 (s, CH2, C9H2), 65.7 (s, CH, C10H), 71.3 (s, CH, C13H), 71.7 (s, CH, C12H), 72.0 (s, CH, C11H), 78.8 (s, Cq, Cq2(CH3)3), 97.3 (s, CH, C14H), 109.3 (s, Cq, Cq15), 110.0 (s, Cq, Cq16), 156.7 (s, Cq, Cq3), 166.3 (s, Cq, Cq8), 170.27 and 170.32 ppm (s, Cq, 2 × Cq7). HRMS (ESI+): C22H33Cl2N5O7, m/z calcd: 572.1655 ([M + Na]+); found: 572.1651 (100%); m/z calcd: 1123.3383 ([2M + Na]+); found: 1123.3383 (28%); m/z calcd: 550.1836 ([M + H]+); found: 550.1833 (20%).
:2, v/v) 1.50 g (1.80 mmol, 99%, Rf = 0.21) of compound 15 was obtained as a colourless solid. IR (KBr): = 3455 (m), 3048 (m), 2981 (m), 2935 (m), 2608 (s), 1715 (s), 1625 (w), 1543 (s), 1511 (s), 1478 (s), 1434 (m), 1383 (m), 1369 (m), 1335 (m), 1315 (m), 1243 (s), 1214 (m), 1171 (s), 1104 (w), 1069 (s), 995 (m), 954 (m), 918 (w), 903 (w), 864 (m), 847 (m), 802 (w), 759 (w), 731 (w), 678 (w), 627 (w), 585 (w), 512 (w) cm−1. 1H NMR (CDCl3): δ = 1.28 (s, 3 H, C18 or 18′H3), 1.35 (s, 3 H, C19 or 19′H3), 1.41 (s, 12 H, C18 or 18′H3 and C(C9H3)3), 1.48 (s, 3 H, C19 or 19′H3), 1.55 to 3.55 (bra, 18 H, 2 × B10H9), 2.98 (br, s, 4 H, 2 × C1H and 2 × C1′H), 3.40 (m, 2 H, C5H2), 3.45 (m, 1 H, C10HH), 3.64 (dt, 3JHH = 13.3 Hz, 3JHH = 5.6 Hz, 1 H, C4HH), 4.03 (dd, 3JHH = 14.4 Hz, 3JHH = 3.2 Hz, 1 H, C10HH), 4.14 (m, 1 H, C4HH), 4.28 (m, 3 H, C11H, C12H and C14H), 4.59 (dd, 3JHH = 7.9 Hz, 3JHH = 2.3 Hz, 1 H, C13H), 5.33 (m, 1 H, N6H), 5.50 ppm (d, 3JHH = 4.9 Hz, 1 H, C15H). 13C{1H} NMR (CDCl3): δ = 24.4, 25.1, 26.06 and 26.08 (s, CH3, C18H3, C18′H3, C19H3 and C19′H3), 28.4 (s, CH3, C(C9H3)3), 39.7 (s, CH2, C5H2), 48.7 (s, CH2, C4H2), 49.0 (s, CH2, C10H2), 53.9 and 54.0 (s, CH, 2 × C1H and 2 × C1′H), 66.2 (s, CH, C11H), 70.5 (s, CH, C14H); 70.9 (s, CH, C13H), 71.7 (s, CH, C12H), 78.8 (s, Cq, Cq8), 96.3 (s, CH, C15H), 109.0 (s, Cq, Cq16), 109.3 (s, Cq, Cq17), 156.1 (s, Cq, Cq7), 163.3 (s, Cq, Cq3), 178.1 ppm (s, Cq, Cq2). 11B{1H} NMR (CDCl3): δ = −18.8 (s, 2B), −17.3 (s, 2B), −14.0 (s, 4B), −12.9 (s, 4B), −10.1 (s, 2B), −5.6 (s, 4B), −3.1 ppm (s, 2B, 2 × BS). 11B NMR (CDCl3): δ = −20.9 to −11.4 (bra, 12B), −10.1 (d, 1JBH = 151 Hz, 2B), −5.6 (d, 1JBH = 166 Hz, 4B), −3.1 ppm (s, 2B, 2 × BS). HRMS (ESI+): C26H55B20N5O7S2, m/z calcd: 831.55915 ([M + H]+); found: 831.56120 (100%).
:1 (v/v) to ethyl acetate, 100%) and 283 mg (388 μmol, 91%, Rf = 0.05 (100% ethyl acetate)) of compound 16 were obtained as an off-white solid. Tm: 147–150 °C (ethyl acetate, decomposition). IR (KBr): = 3442 (s), 3053 (m), 2989 (m), 2935 (m), 2609 (s), 2095 (w), 1680 (s), 1637 (m), 1531 (s), 1508 (s), 1483 (s), 1431 (m), 1383 (m), 1338 (w), 1316 (m), 1281 (w), 1250 (m), 1207 (s), 1175 (s), 1139 (m), 1109 (w), 1069 (s), 998 (m), 954 (m), 902 (w), 864 (m), 849 (m), 802 (m), 760 (w), 724 (m), 700 (w), 626 (w), 588 (w), 513 (w) cm−1. 1H NMR (acetone-d6): δ = 1.28 (s, 3 H, C15 or 15′H3), 1.34 (s, 3 H, C16 or 16′H3), 1.35 (s, 3 H, C15 or 15′H3), 1.44 (s, 3 H, C16 or 16′H3), 1.53 to 3.40 (bra, 18 H, 2 × B10H9), 3.56 (dd, 3JHH = 15.4 Hz, 3JHH = 10.2 Hz, 1 H, C7HH), 3.80 and 3.82 (s, CH, 2 × C1H and 2 × C1′H), 3.95 to 4.25 (br, m, 6 H, 2 × C4HH, 2 × C5HH, C7HH and C8H), 4.35 (m, 2 H, C9H and C11H), 4.62 (dd, 3JHH = 7.9 Hz, 3JHH = 2.5 Hz, 1 H, C10H), 5.51 ppm (d, 3JHH = 5.0 Hz, 1 H, C12H). 13C{1H} NMR (acetone-d6): δ = 24.7, 25.2, 26.3 and 26.4 (s, CH3, C15H3, C15′H3, C16H3 and C16′H3), 45.8 (s, CH2, C5H2), 47.9 (s, CH2, C4H2), 49.9 (s, CH2, C7H2), 56.0 and 56.1 (s, CH, 2 × C1H and 2 × C1′H), 67.2 (s, CH, C8H), 71.3 (s, CH, C11H), 71.8 (s, CH, C10H), 72.4 (s, CH, C9H), 97.3 (s, CH, C12H), 109.3 (s, Cq, Cq13), 110.0 (s, Cq, Cq14), 163.9 (s, Cq, Cq3), 179.0 and 179.5 ppm (s, Cq, 2 × Cq2). 11B{1H} NMR (acetone-d6): δ = −19.6 (s, 2B), −17.9 (s, 2B), −14.9 (s, 4B), −13.7 (s, 4B), −11.3 (s, 2B), −6.9 (s, 4B), −4.4 ppm (s, 2B, 2 × BS). 11B NMR (acetone-d6): δ = −21.3 to −12.3 (bra, 12B), −11.3 (d, 1JBH = 149 Hz, 2B), −6.9 (d, 1JBH = 165 Hz, 4B), −4.4 ppm (s, 2B, 2 × BS). HRMS (ESI+): C21H47B20N5O5S2, m/z calcd: 774.5604 ([M + C2H7N]+); found: 774.5620 (100%); m/z calcd: 731.5067 ([M + H]+); found: 731.5082 (54%).
Peptide synthesis
All bioconjugates were synthesised on a NovaSyn® TGR R resin from Merck (Darmstadt, Germany) with automated peptide synthesiser (SYRO I, MultiSynTech) on solid support in a 15 μmol scale. All reactions were carried out in DMF and all amino acids (AA) were N-α-Fmoc-protected and standard side chain protecting groups were used. Each AA and the reagents oxyma and DIC were added in 8-fold molar excess (equiv.) and each coupling step was performed twice with a reaction time of 40 min. Fmoc protecting group cleavage was achieved by using 40% piperidine for 3 min followed by incubation with 20% piperidine for 10 min. The cycle of coupling and Fmoc cleavage was repeated until the desired peptide length was achieved. After the peptide backbone was finished on solid support, a three-unit ethylene glycol spacer (EG3) was coupled manually to the N-terminus with 2 equiv. of Fmoc-NH-PEG3-COOH and 1.9 equiv. of HATU (O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium-hexafluorophosphate). DIPEA was added in 2-fold molar excess and the reaction was performed for approximately 18 h. Subsequently, Fmoc was cleaved using 30% piperidine for 10 min twice. The carborane building blocks #4 or 5 were coupled manually with 3 equiv. HOBt and DIC overnight to yield conjugates 17 and 18. A branching moiety was introduced to obtain conjugate 19. Fmoc-L-Dap(Fmoc)-OH was coupled manually with 3 equiv. HOBt and DIC overnight. Manual Fmoc-cleavage was performed as described above, and building block 5 was coupled as described above using 3 equiv. of 5 per free amino group. After completed synthesis the conjugates were cleaved from the resin and the side chain protecting groups were removed by incubation with TFA/triisopropylsilane (TIS)/H2O (90:5:5) for 2.5 h. Cleaved conjugates were precipitated in ice-cold Et2O/n-hexane (1:4) and subsequently lyophilised. Purification was performed with preparative RP-HPLC by applying a linear binary gradient system of eluent A (0.1% TFA in water) and eluent B (0.08% TFA in MeCN). UV detection was measured at λ = 220 nm. To confirm correct identity and purity of the synthesised conjugates, analytical RP-HPLC, MALDI-ToF-MS (Ultraflex III, Bruker) and ESI-MS (HCT, Bruker) were performed.
Stable transfection
The plasmid (pCMV6_GRPR-tGFP) was amplified using E. coli DH5α and purified (PureYield™, Promega, Madison, WI, USA). Correct constructs could be confirmed by sequencing. HEK293 cells were stably transfected with the plasmids using Lipofectamine™ 2000 and selected with G-418.Cell culture
The generation and cultivation of stably transfected HEK293_GRPR-tGFP cells was described before.30 Briefly, cells were cultivated in T75 cell culture flasks and were grown in DMEM/HAM's F12 (1:1, v/v) containing 15% FBS (v/v) and 1.0 mg mL−1 G-418 under standard conditions in an incubator (37 °C, 5% CO2, 95% humidity). After cells reached full confluency, they were split in desired ratios from 1:2 to 1:12 into new cell culture flasks, filled with fresh medium for further cultivation or seeded into cell culture vessels for assays.
Ca2+ mobilisation assay
HEK293_GRPR-tGFP cells were seeded into black poly-D-lysine coated 96-well plates (μCLEAR®, CELLSTAR®, Greiner Bio One) at a density of 180000 cells per well and were incubated under standard conditions overnight. At the following day, the medium was aspirated, and the cells were incubated for 40 min under standard conditions with Fluo-2-AM solution (2.3 μM Fluo-2-AM (abcam, Cambridge, UK), 0.06% (v/v) Pluronic® F-127 (Sigma-Aldrich, St Louis, MO, USA) in assay buffer). The fluorescence dye solution was replaced by assay buffer (20 mM HEPES, 2.5 mM Probenecid (Sigma-Aldrich, St Louis, MO, USA) in HBSS, pH 7.5) and basal Ca2+ signal was measured for 20 s with a Flexstation 3 (Molecular Devices, Sunnyvale, CA, USA) before ligand was added and Ca2+ response was determined for another 40 s (λEx = 485 nm, λEm = 525 nm). The x-over basal values were calculated, and data were normalised with GraphPad Prism 5.0 to the universal ligand [D-Phe6, β-Ala11, Phe13, Nle14]Bn(6–14).
Live cell microscopy
Receptor internalisation was investigated by using stably transfected HEK293 cells, which were seeded into 8-well μ-slides (ibiTreat, ibidi, Martinsried, Germany) and incubated for two days at standard conditions. At the assay day, cells were starved for 30 min with 200 μL OptiMEM® under standard conditions and OptiMEM® was subsequently replaced by 200 μL OptiMEM® containing 10−7 M peptide. Stimulation was carried out for 1 h and nuclei visualisation was achieved by addition of 1 μL Hoechst 33342 (Sigma-Aldrich, 0.5 mg mL−1) 30 min before image recording. Subsequently, cells were washed twice with OptiMEM® to remove non-internalised conjugates. Images were taken directly after washing while cells were maintained in OptiMEM®, using an Axio Observer·Z1 microscope equipped with an ApoTome Imaging System and a Heating Insert P Lab-Tek S1 unit (Zeiss, Oberkochen, Germany). Image processing was performed with AxioVision 3.1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ramona Oehme, Dr Susann Billig and Dr Claudia Birkemeyer for measuring the mass spectra, Stefanie Märcker-Recklies and Ines Rein for recording the infrared spectra, Dr Menyhárt B. Sárosi for the DFT calculations. We thank Ronny Müller, Kristin Löbner and Regina Reppich-Sacher for expert technical assistance. Funding by the Europäischer Fonds for regionale Entwicklung (EFRE), the European Union, the Free State of Saxony (ESF) and the Graduate School “SFB 1052 – Obesity Mechanisms” are gratefully acknowledged.Notes and references
- (a) G. L. Locher, Am. J. Roentgenol. Radium Ther., 1936, 36, 1–18 CAS; (b) W. H. Sweet, N. Engl. J. Med., 1951, 245, 875–878 CrossRef CAS PubMed.
- M. F. Hawthorne, Angew. Chem., Int. Ed. Engl., 1993, 32, 950–984 ( Angew. Chem. , 1993 , 105 , 997–1033 ) CrossRef.
- G. W. Kabalka, Expert Opin. Ther. Pat., 1998, 8, 545–551 CrossRef CAS.
- R. F. Barth, P. Mi and W. Yang, Cancer Commun., 2018, 38, 35 CrossRef PubMed.
- R. F. Barth, A. H. Soloway and R. G. Fairchild, Cancer Res., 1990, 50, 1061–1070 CAS.
- A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed.
- G. Calabrese, A. Daou, E. Barbu and J. Tsibouklis, Drug Discovery Today, 2018, 23, 63–75 CrossRef CAS PubMed.
- I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 1433–1450 CrossRef CAS.
- (a) V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef; (b) W. Petry and J. Neuhaus, Phys. J., 2007, 6, 31–37 CAS.
- X. Q. Pan, H. Wang, S. Shukla, M. Sekido, D. M. Adams, W. Tjarks, R. F. Barth and R. J. Lee, Bioconjugate Chem., 2002, 13, 435–442 CrossRef CAS.
- (a) S. Shukla, G. Wu, M. Chatterjee, W. Yang, M. Sekido, L. A. Diop, R. Müller, J. J. Sudimack, R. J. Lee, R. F. Barth and W. Tjarks, Bioconjugate Chem., 2003, 14, 158–167 CrossRef CAS PubMed; (b) Y. Iguchi, H. Michiue, M. Kitamatsu, Y. Hayashi, F. Takenaka, T.-i. Nishiki and H. Matsui, Biomaterials, 2015, 56, 10–17 CrossRef CAS PubMed; (c) A. Doi, S. Kawabata, K. Iida, K. Yokoyama, Y. Kajimoto, T. Kuroiwa, T. Shirakawa, M. Kirihata, S. Kasaoka, K. Maruyama, H. Kumada, Y. Sakurai, S.-i. Masunaga, K. Ono and S.-I. Miyatake, J. Neurooncol., 2008, 87, 287–294 CrossRef CAS PubMed; (d) I. Romero-Canelón, B. Phoenix, A. Pitto-Barry, J. Tran, J. J. Soldevila-Barreda, N. Kirby, S. Green, P. J. Sadler and N. P. E. Barry, J. Organomet. Chem., 2015, 796, 17–25 CrossRef; (e) W. Mier, D. Gabel, U. Haberkorn and M. Eisenhut, Z. Anorg. Allg. Chem., 2004, 630, 1258–1262 CrossRef CAS.
- R. Dubey, S. Kushal, A. Mollard, L. Vojtovich, P. Oh, M. D. Levin, J. E. Schnitzer, I. Zharov and B. Z. Olenyuk, Bioconjugate Chem., 2015, 26, 78–89 CrossRef CAS PubMed.
- B. Feng, K. Tomizawa, H. Michiue, S.-I. Miyatake, X.-J. Han, A. Fujimura, M. Seno, M. Kirihata and H. Matsui, Biomaterials, 2009, 30, 1746–1755 CrossRef CAS PubMed.
- H. Michiue, Y. Sakurai, N. Kondo, M. Kitamatsu, F. Bin, K. Nakajima, Y. Hirota, S. Kawabata, T.-i. Nishiki, I. Ohmori, K. Tomizawa, S.-I. Miyatake, K. Ono and H. Matsui, Biomaterials, 2014, 35, 3396–3405 CrossRef CAS PubMed.
- C.-H. Lai, Y.-C. Lin, F.-I. Chou, C.-F. Liang, E.-W. Lin, Y.-J. Chuang and C.-C. Lin, Chem. Commun., 2012, 48, 612–614 RSC.
- R. Otero, S. Seoane, R. Sigüeiro, A. Y. Belorusova, M. A. Maestro, R. Pérez-Fernández, N. Rochel and A. Mouriño, Chem. Sci., 2016, 7, 1033–1037 RSC.
- D. J. Worm, S. Els-Heindl, M. Kellert, R. Kuhnert, S. Saretz, J. Koebberling, B. Riedl, E. Hey-Hawkins and A. G. Beck-Sickinger, J. Pept. Sci., 2018, 32, e3119 CrossRef PubMed.
- G. Ciofani, V. Raffa, A. Menciassi and A. Cuschieri, Nanoscale Res. Lett., 2008, 4, 113–121 CrossRef PubMed.
- P. J. Kueffer, C. A. Maitz, A. A. Khan, S. A. Schuster, N. I. Shlyakhtina, S. S. Jalisatgi, J. D. Brockman, D. W. Nigg and M. F. Hawthorne, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6512–6517 CrossRef CAS PubMed.
- K. Kettenbach, H. Schieferstein, C. Grunewald, D. Iffland, L. M. Reffert, G. Hampel, C. L. Schütz, N. H. Bings and T. L. Ross, Radiochim. Acta, 2015, 103, 799–809 CAS.
- J. Chadwick and M. Goldhaber, Nature, 1935, 135, 65 CrossRef CAS.
- H. J. Taylor and M. Goldhaber, Nature, 1935, 135, 341 CrossRef CAS.
- Y. Hattori, S. Kusaka, M. Mukumoto, K. Uehara, T. Asano, M. Suzuki, S.-i. Masunaga, K. Ono, S. Tanimori and M. Kirihata, J. Med. Chem., 2012, 55, 6980–6984 CrossRef CAS PubMed.
- T. Hartman and J. Carlsson, Radiother. Oncol., 1994, 31, 61–75 CrossRef CAS.
- (a) V. M. Ahrens, R. Frank, S. Stadlbauer, A. G. Beck-Sickinger and E. Hey-Hawkins, J. Med. Chem., 2011, 54, 2368–2377 CrossRef CAS PubMed; (b) K. Y. Choi, G. Saravanakumar, J. H. Park and K. Park, Colloids Surf., B, 2012, 99, 82–94 CrossRef CAS PubMed; (c) S. Agosteo, P. Colautti, J. Esposito, A. Fazzi, M. V. Introini and A. Pola, Appl. Radiat. Isot., 2011, 69, 1664–1667 CrossRef CAS PubMed; (d) H. Kumada, A. Matsumura, H. Sakurai, T. Sakae, M. Yoshioka, H. Kobayashi, H. Matsumoto, Y. Kiyanagi, T. Shibata and H. Nakashima, Appl. Radiat. Isot., 2014, 88, 211–215 CrossRef CAS PubMed; (e) E. Durisi, K. Alikaniotis, O. Borla, F. Bragato, M. Costa, G. Giannini, V. Monti, L. Visca, G. Vivaldo and A. Zanini, Appl. Radiat. Isot., 2015, 106, 63–67 CrossRef CAS; (f) T. Onishi, H. Kumada, K. Takada, F. Naito, T. Kurihara and T. Sakae, Appl. Radiat. Isot., 2018, 140, 5–11 CrossRef CAS; (g) D. J. Worm, P. Hoppenz, S. Els-Heindl, M. Kellert, R. Kuhnert, S. Saretz, J. Köbberling, B. Riedl, E. Hey-Hawkins and A. G. Beck-Sickinger, J. Med. Chem., 2019 DOI:10.1021/acs.jmedchem.9b01136 , accepted.
- M. Kellert, P. Lönnecke, B. Riedl, J. Koebberling and E. Hey-Hawkins, Molecules, 2019, 24, 3288 CrossRef CAS PubMed.
- M. Kellert, D. J. Worm, P. Hoppenz, M. B. Sárosi, P. Lönnecke, B. Riedl, J. Koebberling, A. G. Beck-Sickinger and E. Hey-Hawkins, Dalton Trans., 2019, 48, 10834–10844 RSC.
- (a) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (b) Z. J. Lesnikowski, Collect. Czech. Chem. Commun., 2007, 72, 1646–1658 CrossRef CAS.
- R. L. Sneath, Jr., A. H. Soloway and A. S. Dey, J. Med. Chem., 1974, 17, 796–799 CrossRef PubMed.
- P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, J. Pept. Sci., 2019 DOI:10.1002/psc.3224.
- V. Sancho, A. Di Florio, T. W. Moody and R. T. Jensen, Curr. Drug Delivery, 2011, 8, 79–134 CrossRef CAS PubMed.
- S. Ronchi, D. Prosperi, F. Compostella and L. Panza, Synlett, 2004, 1007–1010 CAS.
- (a) J. Yoo, J.-W. Hwang and Y. Do, Inorg. Chem., 2001, 40, 568–570 CrossRef CAS; (b) R. A. Wiesboeck and M. F. Hawthorne, J. Am. Chem. Soc., 1964, 86, 1642–1643 CrossRef CAS.
- (a) R. Frank, V. M. Ahrens, S. Boehnke, A. G. Beck-Sickinger and E. Hey-Hawkins, ChemBioChem, 2016, 17, 308–317 CrossRef CAS PubMed; (b) S. A. Anufriev, S. A. Erokhina, I. B. Sivaev and V. I. Bregadze, Russ. Chem. Bull. Int. Ed., 2016, 65, 2704–2707 CrossRef CAS; Published in Russian in Izvestiya Akademii Nauk. Seriya Khimicheskaya, 2016, 11, 2704–2707; (c) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (d) M. J. Manning, C. B. Knobler, R. Khattar and M. F. Hawthorne, Inorg. Chem., 1991, 30, 2009–2017 CrossRef CAS.
- L. I. Zakharkin and I. V. Pisareva, Phosphorus, Sulfur, Silicon Relat. Elem., 1984, 20, 357–370 CrossRef CAS.
- M. Brackhagen, H. Boye and C. Vogel, J. Carbohydr. Chem., 2001, 20, 31–43 CrossRef CAS.
- P. W. Wertz, J. C. Garver and L. Anderson, J. Am. Chem. Soc., 1981, 103, 3916–3922 CrossRef CAS.
- P. G. M. Wuts and T. W. Greene, Greene's protective groups in organic synthesis, Wiley-Interscience, Hoboken, N.J, 4th edn, 2007 Search PubMed.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Thieme, Stuttgart, New York, 7th edn, 2005 Search PubMed.
- G. Hughes, P. O'Shea, J. Goll, D. Gauvreau and J. Steele, Tetrahedron, 2009, 65, 3189–3196 CrossRef CAS.
- D. Pappo, T. Mejuch, O. Reany, E. Solel, M. Gurram and E. Keinan, Org. Lett., 2009, 11, 1063–1066 CrossRef CAS.
- F. H. Scholes, R. J. S. Morrison, C. L. Raston and G. H. Kruppa, Int. J. Mass Spectrom., 2000, 202, 37–45 CrossRef CAS.
- J. F. Ditter, F. J. Gerhart and R. E. Williams, Analysis of Boranes and Carboranes by Mass Spectrometry, in Mass Spectrometry in Inorganic Chemistry, ed. J. L. Margrave, 1968, ch. 14, vol. 72, pp. 191–210 Search PubMed.
- V. Mäde, S. Els-Heindl and A. G. Beck-Sickinger, Beilstein J. Org. Chem., 2014, 10, 1197–1212 CrossRef.
- J. Mattei, R. D. Achcar, C. H. Cano, B. R. Macedo, L. Meurer, B. S. Batlle, S. D. Groshong, J. M. Kulczynski, R. Roesler, L. Dal Lago, A. T. Brunetto and G. Schwartsmann, Arch. Pathol. Lab. Med., 2014, 138, 98–104 CrossRef CAS.
- C. Morgat, G. MacGrogan, V. Brouste, V. Vélasco, N. Sévenet, H. Bonnefoi, P. Fernandez, M. Debled and E. Hindié, J. Nucl. Med., 2017, 58, 1401–1407 CrossRef CAS PubMed.
- J.-C. Reubi, S. Wenger, J. Schmuckli-Maurer, J.-C. Schaer and M. Gugger, Clin. Cancer Res., 2002, 8, 1139–1146 CAS.
- R. T. Jensen, J. F. Battey, E. R. Spindel and R. V. Benya, Pharmacol. Rev., 2008, 60, 1–42 CrossRef CAS PubMed.
- (a) R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CAS; (b) R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS.
- MestReNova, v12.00-20080, Mestrelab Research S.L., 2017 Search PubMed.
- CrysAlis Pro: Data Collection and Data Reduction Software Package, Rigaku Oxford Diffraction Search PubMed.
- SCALE3 ABSPACK: Empirical Absorption Correction using Sperical Harmonics Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Brandenburg, Diamond, v4.5.3, Crystal Impact GbR, Bonn, Germany Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full crystallographic data of 6, 8, 9, 11, 12 and SP1, SP2, and SP3, and synthetic procedures and discussion of side products 8, 9, SP1, SP2, and SP3. CCDC 1958031–1958038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04031e |
| This journal is © The Royal Society of Chemistry 2020 |