
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards dual SPECT/optical bioimaging with a mitochondrial targeting, 99mTc(I) radiolabelled 1,8-naphthalimide conjugate†
Adam H.
Day
a,
Juozas
Domarkas
b,
Shubhanchi
Nigam
b,
Isaline
Renard
 b,
Christopher
Cawthorne
b,
Benjamin P.
Burke
b,
Gurmit S.
Bahra
c,
Petra C. F.
Oyston
c,
Ian A.
Fallis
*a,
Stephen J.
Archibald
*b and
Simon J. A.
Pope
*a
b,
Christopher
Cawthorne
b,
Benjamin P.
Burke
b,
Gurmit S.
Bahra
c,
Petra C. F.
Oyston
c,
Ian A.
Fallis
*a,
Stephen J.
Archibald
*b and
Simon J. A.
Pope
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, Cymru/Wales, UK. E-mail: popesj@cardiff.ac.uk; s.j.archibald@hull.ac.uk
bPositron Emission Tomography Research Centre and Department of Biomedical Sciences, University of Hull, Cottingham Road, Hull, HU6 7RX UK
cDefence Science and Technology Laboratory, Porton Down, Salisbury, Wiltshire, UK
First published on 17th December 2019
Abstract
A series of six different 1,8-naphthalimide conjugated dipicolylamine ligands (L1–6) have been synthesised and characterised. The ligands possess a range of different linker units between the napthalimide fluorophore and dipcolylamine chelator which allow the overall lipophilicity to be tuned. A corresponding series of Re(I) complexes have been synthesised of the form fac-[Re(CO)3(L1–6)]BF4. The absorption and luminescence properties of the ligands and Re(I) complexes were dominated by the intramolecular charge transfer character of the substituted fluorophore (typically absorption ca. 425 nm and emission ca. 520 nm). Photophysical assessments show that some of the variants are moderately bright. Radiolabelling experiments using a water soluble ligand variant (L5) were successfully undertaken and optimised with fac-[99mTc(CO)3(H2O)3]+. Confocal fluorescence microscopy showed that fac-[Re(CO)3(L5)]+ localises in the mitochondria of MCF-7 cells. SPECT/CT imaging experiments on naïve mice showed that fac-[99mTc(CO)3(L5)]+ has a relatively high stability in vivo but did not show any cardiac uptake, demonstrating rapid clearance, predominantly via the biliary system along with a moderate amount cleared renally.
Introduction
The different bioimaging modalities each have intrinsic strengths and weaknesses, with no single clinically used technique providing comprehensive information. Prospective multimodal imaging agents are designed to combine the strengths of individual modalities, while offering complementary or additional clinically useful information not provided by a single technique.1,2 Single photon emission computed tomography (SPECT) is a high sensitivity nuclear imaging technique using short half-life gamma emitting radioisotopes that can image the whole body. In comparison, optical fluorescence imaging can utilise agents that emit visible or near-IR photons and can be applied to both pre-clinical in vivo and cellular microscopy studies. However, it lacks the excellent tissue penetration offered by nuclear imaging modalities.The combination of radioimaging and fluorescence imaging presents significant opportunities in the design of useful imaging agents.3 Combined single molecule SPECT/fluorescence agents can provide early phase high sensitivity imaging of in vivo biodistribution using SPECT, followed by longitudinal optical imaging.4 Additionally, PET or SPECT imaging can be used to identify diseased tissue non-invasively, which can then be accurately biopsied or surgically resected based upon the fluorescence imaging signal.5–7
The clinical use of 99mTc in SPECT imaging has been well established for many decades. 99mTc (t1/2 = 6.01 h, γ = 142.7 keV) is a gamma emitting radioactive isotope which is used in >80% of clinical nuclear diagnostic imaging scans, with over 40 million SPECT scans carried out per year worldwide.8 What is less well understood is the behaviour of Tc agents at the sub-cellular level, since the image resolution of SPECT does not allow such determinations. Therefore intrinsically luminescent or fluorescently tagged 99mTc SPECT agents9 are potentially helpful in correlating in vitro and in vivo imaging properties,10 and in determining their cellular uptake, trafficking and localisation.11 From an inorganic chemistry perspective, non-radioactive rhenium is the obvious choice for developing structurally analogous and chemically related complexes.12 Such an approach can be used to validate the development of appropriate chelates13 for 99mTc and develop experimental protocols for synthesis and purification.14 Indeed, rhenium complexes are now attracting considerable attention; in part for their potential as cancer therapy agents15 as well as the existence of accessible radioisotopes appropriate for radionuclide therapies/theranostic applications.16 Binuclear Re(I) carbonyl complexes have shown accumulation in mitochondria leading to cell death.17 Indeed a bimetallic Re(I)/99mTc(I) complex has also been described where the emissive nature of the Re(I) fragment can be exploited in optical studies.18 In the case of fluorescently tagged rhenium complexes, these species can be assessed with respect to cellular uptake using conventional confocal fluorescence microscopy techniques.19
Whilst substituted 1,8-naphthalimides have been previously investigated in biological assays for their therapeutic applications,20 more recent studies have shown that such species can also be very useful fluorophores for fluorescence bioimaging of cells.21–23 The relative ease of functionalisation together with advantageous absorption and emission properties in the visible region have led to a large number of reports on their application to cell imaging. For example, recent studies have shown that the 1,8-naphthalimide structure can dictate intracellular localisation, including effective mitochondrial targeting.24
Since mitochondria have essential roles in energy production, cell signalling, cell-cycle control and cell death, and mitochondrial function can be disrupted in both ischaemic heart cells and cancer cells, they are attractive targets. Thus the development of a SPECT/fluorescence multimodal imaging agents for targeting mitochondria would therefore allow for in vitro, in vivo and ex vivo examination of myocardial function and tumour growth.25 In this context, this current work describes the development of 1,8-naphthalimide functionalised chelates suitable for labelling with 99mTc(I) for mitochondrial targeted multimodal imaging. During the course of these studies, work by Turnbull et al. has shown that a series of related 4-amino-1,8-naphthalimide fluorophores can be complexed with Re(I)/99mTc(I). The overall charge of the complex was shown to strongly influence the biological attributes of the species.26
Synthesis and characterisation of ligands and complexes
Scheme 1 shows the different chelator structures that were synthesised in this study. The compounds were designed to meet the coordination chemistry requirements of M(I) (where M = Re, Tc) with a face-capping tridentate donor set, fulfilled by a dipicolylamine (DPA)27 chelating moiety. This is linked to the naphthalimide fluorophore for optical imaging via different length spacers of variable hydro- and lipophilicity. Thus, a series of six ligands were synthesised in three or four steps from 4-chloro-1,8-naphthalic anhydride (Scheme 2). Substitution at the 4-chloro position of the naphthalimide ring was achieved in DMSO at elevated temperature using an excess of diamine (often as the mono-Boc protected version). Following cleavage of the Boc group (trifluoroacetic acid in dichloromethane), treatment of N1–6 with 2-pyridinecarboxaldehyde in a one-pot reductive amination procedure gave the dipicolylamine derived target ligands, L1–6, in each case. All experimental details and spectroscopic characterisation data for the synthesis of intermediates and ligands are included in the Experimental section. | ||
| Scheme 1 Design aspects of the compounds synthesised in this study to provide a fluorophore functionalised chelate to the {M(CO)3}+ unit (M = 99mTc, Re). Six derivatives (L1–6) were synthesised with three different linker units. | ||
 | ||
| Scheme 2 Synthesis of the precursors and ligands. Reagents and conditions: (i) N-Boc-ethylenediamine, DMSO, heat; then TFA/DCM; (ii) 1,6-diaminohexane, DMSO, heat; (iii) 2-pyridinecarboxaldehyde, NaBH(OAc)3, 1,2-dichloroethane; (iv) N-Boc(2,2′-(ethylenedioxy)diethylamine, DMSO, heat; then TFA/DCM. | ||
All isotopes of technetium are radioactive, and so natural rhenium is commonly used as a chemical analogue28 to allow the synthetic and purification protocols to be optimised prior to the use of a radionuclide. The corresponding tricarbonyl Re(I) complexes were isolated by reacting29 the precursor compounds with fac-[Re(CO)3(MeCN)3]BF4 to yield fac-[Re(CO)3(L)]BF4 as air stable solids. Characterisation of the complexes was achieved using a range of spectroscopic and analytical techniques. The expected facial geometry with respect to the three carbonyl ligands was confirmed using IR spectroscopy. In all cases two C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretches were observed between 2040–1900 cm−1 which are attributed to the pseudo C3v symmetry of the complexes. In addition to this, the naphthalimide carbonyl stretches were observed at 1700–1625 cm−1 and the [BF4]− counter anion at ca. 1055 cm−1. High resolution mass spectrometry data were obtained for each complex identifying the cationic fragment with the appropriate isotopic distribution for rhenium. 1H NMR spectroscopy was able to clearly establish the formation of the complex in each case. Firstly, the methylene linkers of the dipicolylamine unit shift upon coordination and become diastereotopic with an observed coupling around 16 Hz.28 The aliphatic protons of the linker unit are also mildly shifted in the complex versus the ligand. The general pattern amongst the aromatic proton resonances is retained upon formation of the complex. Principally, data from 13C NMR studies allowed identification, in all cases, of the rhenium-bound carbonyls (ca. 195 ppm) and the naphthalimide carbonyls (ca. 165 ppm).
O stretches were observed between 2040–1900 cm−1 which are attributed to the pseudo C3v symmetry of the complexes. In addition to this, the naphthalimide carbonyl stretches were observed at 1700–1625 cm−1 and the [BF4]− counter anion at ca. 1055 cm−1. High resolution mass spectrometry data were obtained for each complex identifying the cationic fragment with the appropriate isotopic distribution for rhenium. 1H NMR spectroscopy was able to clearly establish the formation of the complex in each case. Firstly, the methylene linkers of the dipicolylamine unit shift upon coordination and become diastereotopic with an observed coupling around 16 Hz.28 The aliphatic protons of the linker unit are also mildly shifted in the complex versus the ligand. The general pattern amongst the aromatic proton resonances is retained upon formation of the complex. Principally, data from 13C NMR studies allowed identification, in all cases, of the rhenium-bound carbonyls (ca. 195 ppm) and the naphthalimide carbonyls (ca. 165 ppm).
Luminescence properties
Prior to radiolabelling and imaging studies, the electronic and optical properties of these species were investigated (Table 1). The UV-vis. spectra of all ligands were recorded in MeCN or chloroform solutions. Because of the relative hydrophilicity of the L5/L6 systems, additional data in aqueous solution was also obtained on these ligands and their complexes. The absorption spectra showed some common features, with higher energy absorptions associated with the naphthalimide and pyridyl based π–π* transitions, and a lower energy, broad peak around 440 nm which was assigned to an intramolecular charge transfer (ICT) transition from the amine-functionalised naphthalimide chromophore. The position of the ICT peak showed a slight shift that was dependent upon the amine substituent and was shown to be sensitive to the polarity of solvent, as expected for a charge transfer transition.30 The absorption spectra of the corresponding Re(I) complexes (for example, Fig. 1) were comparable, with only minor shifts of the ICT band upon formation of the cationic Re(I) complex that are likely to be a result of the cationic charge perturbing the ICT transition.31 Typically the molar absorption coefficients (Table 1) of the ICT bands in the complexes were in the range of 7000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.
000 M−1 cm−1.
 | ||
| Fig. 1 UV-vis. absorption spectrum for fac-[Re(CO)3(L5)]BF4 recorded in water. | ||
| Compound | λ abs (ε/M−1 cm−1)/nm | λ em/nma | τ obs/nsb | ϕ/%c |
|---|---|---|---|---|
| a Measurements obtained in aerated solutions; λex = 405 nm. b λ ex = 295 nm. c Using aerated MeCN solution of [Ru(bipy)3](PF6)2 as a reference. d In acetonitrile. e In chloroform. f In water. | ||||
| L1 | 434 (9310)d | 518d | 11.1d | 15d |
| 428 (7800)e | 508e | 9.8e | 19e | |
| L2 | 436 (10420)d |
521d | 10.5d | 22d |
| 427 (8160)e | 510e | 8.5e | 17e | |
| L3 | 433 (7300)d | 518d | 9.7d | 28d |
| 435 (8240)e | 508e | 1.4, 8.8 (94%)e | 28e | |
| L4 | 429 (8000)d | 517d | 9.5d | 31d |
| 430 (18600)e |
505e | 8.1e | 35e | |
| L5 | 448 (10220)f |
542f | 3.4, 6.0 (64%)f | 39f |
| 429 (8910)d | 521d | 9.4d | 12d | |
| L6 | 448 (2620)f | 549f | 5.4f | 8f |
| 429 (7920)d | 520d | 9.7d | 30d | |
| [Re(CO)3(L1)]BF4 | 430 (8560)d | 515d | 10.9d | 67d |
| 432 (7510)e | 503e | 9.1e | 23e | |
| [Re(CO)3(L2)]BF4 | 433 (19200)d |
519d | 9.8d | 30d |
| 430 (14300)e |
504e | 9.2e | 37e | |
| [Re(CO)3(L3)]BF4 | 430 (16300)d |
520d | 9.8d | 26d |
| 434 (10100)e |
512e | 7.7e | 29e | |
| [Re(CO)3(L4)]BF4 | 431 (13020)d |
517d | 10.2d | 40d |
| 425 (18660)e |
483e | 8.0e | 38e | |
| [Re(CO)3(L5)]BF4 | 446 (7190)f | 540f | 6.0f | 49f |
| 429 (8100)d | 526d | 10.0d | 15d | |
| [Re(CO)3(L6)]BF4 | 445 (10700)f |
539f | 5.9f | 57f |
| 448 (12240)d |
516d | 11.1d | 17d | |
Steady state emission spectra were obtained using an irradiation wavelength of 425 nm, corresponding to direct excitation of the ICT band of the ligand. All ligands and complexes showed an intense, featureless luminescence peak at 505–550 nm, giving Stokes’ shift >3500 cm−1, which is typical of this class of fluorophore (for example, Fig. 2). Again, the position of the peak was found to be moderately sensitive to the amine substituent (related to the relative electron donating power) and the solvent polarity. For example, measurements in aqueous media for L5/L6 and their complexes resulted in a bathochromic shift of the emission maxima to ca. 540 nm, typical of positive solvatochromism (Fig. 2). Time-resolved experiments revealed lifetimes <15 ns in all cases, confirming the fluorescent nature of these ICT emitting species. Quantum yields were obtained for each species in the various solvents, and ranged 8–67%. For L5/L6 and their rhenium(I) complexes these values were relatively high (around 50%) for the aqueous solutions. Taken together this gives a typical brightness value around 5000 M−1 cm−1. The naphthalimide conjugated rhenium(I) complexes are thus well suited to cell imaging via confocal fluorescence microscopy, with visible fluorescence, good Stokes’ shifts and relatively high quantum yields giving reasonably bright fluorophores.
 | ||
| Fig. 2 Luminescence spectrum for fac-[Re(CO)3(L5)]BF4 recorded in water (λex = 425 nm). | ||
Radiolabelling with technetium-99m
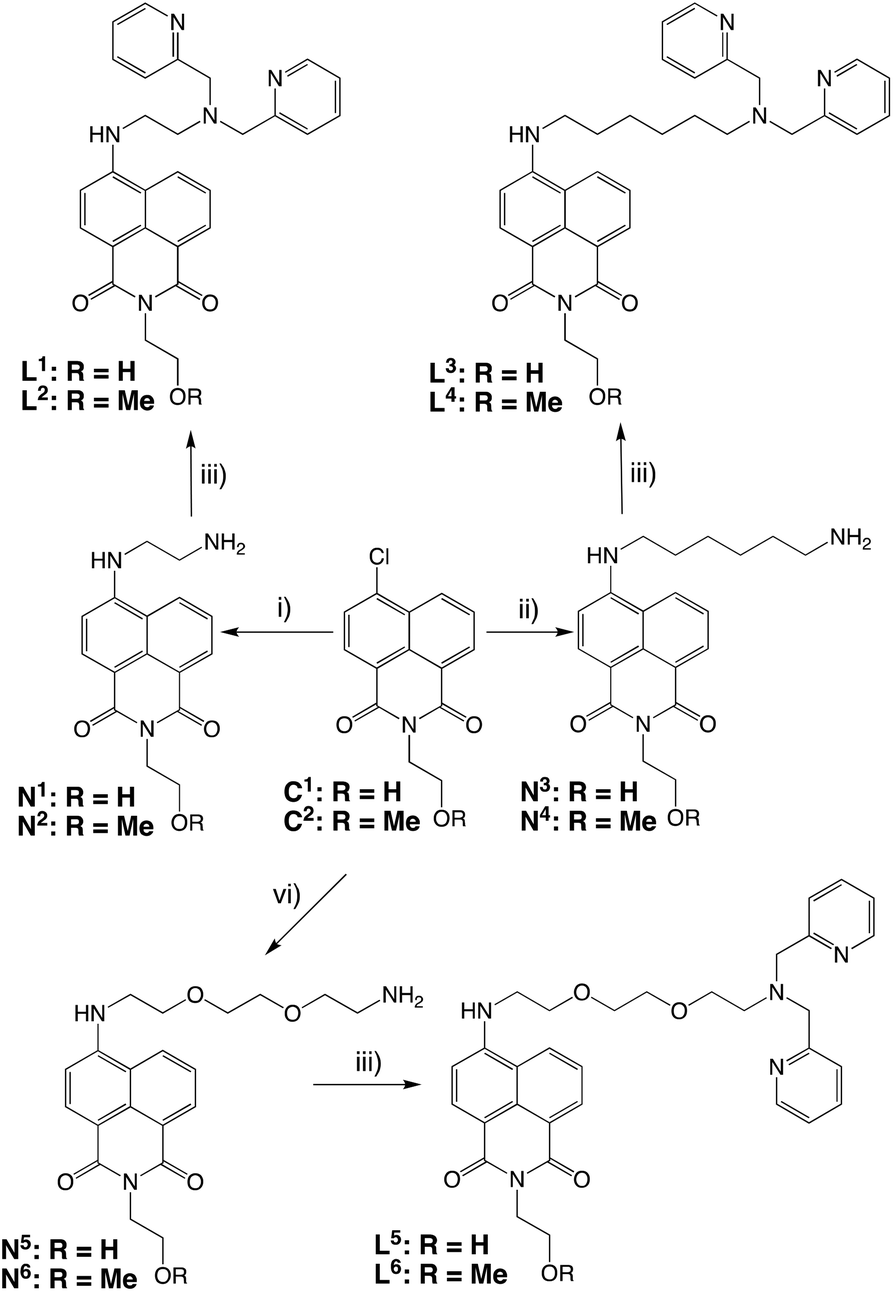
Having established the Re(I) coordination chemistry and the optical properties of the complexes, radiolabelling of selected ligand systems was attempted with the SPECT imaging (gamma emitting) isotope technetium-99m. Radiolabelling studies were conducted using fac-[99mTc(CO)3(H2O)3]+, which was obtained from the reduction of [99mTcO4]− using the method reported by Alberto et al.32,33 Initial attempts to radiolabel L1 (the ligand variant with an ethylene linker between the naphthalimide and the dipicolylamine chelate unit) led to the formation of a number of radioactive species indicative, perhaps, of the decomposition of the ligand. Certainly, the limited aqueous solubility of L1 was likely to be a hindrance to efficient radiolabelling.Therefore, the focus of further radiochemistry experiments was focussed upon the more hydrophilic naphthalimide L5 (the ligand variant incorporating the ethylenedioxodiamine linker) which demonstrated good water solubility.
Preliminary experiments with water-soluble L5 indicated much better radiolabelling yields and limited degradation (Fig. 3),34 and therefore a range of conditions were tested to optimise the radiolabelling reaction (Table 2). Varying quantities of lyophilised L5 (15 to 300 μg), were radiolabelled with freshly reduced fac-[99mTc(CO)3(H2O)3]+ (10–40 MBq, 0.13–2.6 mM final precursor concentration, pH 7–8, buffered with 5 mM PBS) at 70 °C. The radiolabelling yield was determined by integration of radio-HPLC traces and, at 30 min, varied from 23% to 57%, plateauing at 55–57% for 30–150 μg (0.26–1.32 mM) range while declining to 43% when the amount of L5 was increased to 300 μg (2.0 mM). Increasing the reaction time to 60 min only had an effect on the lowest concentration (0.13 mM), with the yield increasing from 23% to 39%. Similarly, a three-fold increase of fac-[99mTc(CO)3(H2O)3]+ (up to 124–135 MBq) did not have any observable effect on the radiolabelling yield at 0.53–1.32 mM precursor concentrations. Thus, optimised L5 radiolabelling conditions were fixed at 0.53–1.32 mM precursor concentration, pH 7–8 (PBS), 70 °C, 30 min (Fig. 3).
 | ||
| Fig. 3 Preparation (top) and stability assessments (bottom) of radiolabelled L5: comparative HPLC traces for fac-[Re(CO)3(L5)]+ and fac-[99mTc(CO)3(L5)]+. | ||
| L5 conc. (mM) | 30 min | 60 min | 90 min |
|---|---|---|---|
| 0.2 | 43 | 47 | 53 |
| 0.4 | 51 | 53 | 55 |
| 1.0 | 56 | 57 | 57 |
| 2.0 | 42 | 41 | 43 |
Stability and mitochondrial uptake of fac-[99mTc(CO)3(L5)]+
Lipophilicity is an important parameter in dictating the cellular localisation of organometallic complexes.35 Lipophilicity (logD7.4) measurements were carried out on the radiolabelled species fac-[99mTc(CO)3(L5)]+ and showed a value of 0.67 ± 0.06. This suggests a species with suitable solubility for biological studies, but passive cell membrane permeability is likely to be less favourable.36 For drug-like species, such a value can also indicate that a renal clearance pathway will be observed.37 To mimic in vivo conditions, formulated fac-[99mTc(CO)3(L5)]+ was incubated with human serum at 37 °C. HPLC analysis did not reveal any significant instability up to 3 hours, indicating that the radiolabelled compound had sufficient stability to progress to preliminary in vivo imaging experiments.
To investigate fac-[99mTc(CO)3(L5)]+ behaviour towards mitochondria, experiments were carried out using mitochondria freshly isolated from rat heart tissue. fac-[99mTc(CO)3(L5)]+ accumulated in the mitochondria to a higher extent (55.68 ± 6.5%) than complexes containing the cationic triphenylphosphonium mitochondria targeting group that has been frequently used in mitochondrial targeting.38 This uptake was highly dependent on mitochondrial membrane potential (MMP) as shown by a nearly complete abrogation (1.35 ± 0.14%) when the MMP was depolarised using carbonyl cyanide m-chlorophenylhydrazine (CCCP).
Cell imaging studies with fac-[Re(CO)3(L5)]+ and preliminary in vivo assessment of fac-[99mTc(CO)3(L5)]+
Cellular cytotoxicity was assessed using U87 cells in vitro by the MTT assay, giving a CC50 value of 72.7 μM (Fig. 4), which indicates low cytotoxicity in vitro for the compound. Whole cell mitochondrial uptake of fac-[Re(CO)3(L5)]+ was then studied using confocal fluorescence microscopy in human breast cancer cells (MCF-7) in culture. As a control, a benzyl chloride based mitochondrial stain (MitoTracker deep red, MDR) was used for dye co-localisation studies as its emission wavelength (665 nm) is sufficiently different from fac-[Re(CO)3(L5)]+ (540 nm) thus allowing independent detection. Confocal fluorescence microscopy studies into the mitochondrial localisation for fac-[Re(CO)3(L5)]+ with MCF-7 cells gave an overlap coefficient of 0.66 (Fig. 5) suggesting a good degree of co-localisation. | ||
| Fig. 4 Cell viability assay (MTT) result for fac-[Re(CO)3(L5)]+. | ||
 | ||
| Fig. 5 Confocal fluorescence microscopy images of MCF-7 cells incubated with fac-[Re(CO)3(L5)]+ (top left) and MitoTracker deep red (top right) and overlay (bottom; overlap coefficient = 0.66). | ||
Having established the mitochondrial and cellular uptake properties, a scoping in vivo experiment was carried out with fac-[99mTc(CO)3(L5)]+ in a naïve mouse using SPECT/CT imaging (Fig. 6). Firstly, there were no signs of toxicity to the mouse during the in vivo studies. The tracer showed rapid clearance, predominantly via the biliary system along with a moderate amount cleared renally. This is consistent with the logD of fac-[99mTc(CO)3(L5)]+, as some renal clearance was observed and more complex active transport mechanisms can influence hepatobiliary clearance.39 Urine analysis demonstrated that the tracer was of a relatively high stability (85.4 ± 1.4%) and suitable for further in vivo analysis. From these imaging studies, it was clear that cardiac uptake was not observed for fac-[99mTc(CO)3(L5)]+. This suggests that in its current form the tracer will not be appropriate for assessing cardiac function in vivo and so structural modification would be required to ensure uptake and retention.
 | ||
| Fig. 6 SPECT and CT overlaid images with the SPECT images acquired using the radiotracer fac-[99mTc(CO)3(L5)]+. Sagittal and coronal views show the localisation of the tracer after 22–60 min. Red arrow: gall bladder; yellow arrow: biliary clearance into the biliary tract and small intestine; green arrow: bladder; blue arrow: kidney. | ||
Conclusions
In conclusion, this study shows the design, synthesis and characterisation of a new class of radiotracer which is preferentially taken up into the mitochondria of cells. The tracer combines a naphthalimide fluorophore with a technetium(I) tricarbonyl unit to give a structure with excellent cellular and mitochondrial uptake characteristics. Cellular bioimaging shows good image quality in the MCF-7 cell line. The preliminary in vivo assessment of the aqueous soluble 99mTc(I) compound shows rapid clearance, aligning with the requirements for nuclear imaging to ensure background signal is minimised. These results now justify further investigation of the tumour uptake properties in vivo with appropriate tumour models and larger animal cohorts.In its current form, fac-[99mTc(CO)3(L5)]+ does not demonstrate cardiac uptake in vivo. Therefore, our further studies will also consider structurally modified variants that can promote and optimise this characteristic. During the course of these studies, Luyt and co-workers reported a peptide-functionalised naphthalimide-based fluorophore that can be used to target CXCR4 expressing cells for potential dual mode imaging.40 This further suggests that the inherent fluorophore–chelate design presented herein holds great promise for future biological and bioimaging applications.
Experimental
General considerations
All reagents and solvents were commercially available and were used without further purification if not stated otherwise. For the measurement of 1H and 13C NMR spectra a Bruker Fourier300 (300 MHz), Bruker AVANCE HD III equipped with a BFFO SmartProbe™ (400 MHz) or Bruker AVANCE III HD with BBO Prodigy CryoProbe (500 MHz) was used. The obtained chemical shifts δ are reported in ppm and are referenced to the residual solvent signal. Spin–spin coupling constants J are given in Hz.Low-resolution mass spectra were obtained by the staff at Cardiff University. High-resolution mass spectra were carried out at the EPSRC National Mass Spectrometry Facility at Swansea University. High resolution mass spectral (HRMS) data were obtained on a Waters MALDI-TOF mx at Cardiff University or on a Thermo Scientific LTQ Orbitrap XL by the EPSRC UK National Mass Spectrometry Facility at Swansea University. IR spectra were obtained from a Shimadzu IR-Affinity-1S FTIR. Reference to spectroscopic data are given for known compounds. UV-Vis studies were performed on a Shimadzu UV-1800 spectrophotometer as MeCN solutions (2.5 or 5 × 10−5 M). Photophysical data were obtained on a JobinYvon–Horiba Fluorolog spectrometer fitted with a JY TBX picosecond photodetection module as MeCN solutions. Quantum yield measurements were obtained on aerated MeCN solutions of the complexes using [Ru(bpy)3](PF6)2 in aerated MeCN as a standard (Φ = 0.016). Emission spectra were uncorrected and excitation spectra were instrument corrected. The pulsed source was a nano-LED configured for 295 nm or 372 nm output operating at 1 MHz. Luminescence lifetime profiles were obtained using the JobinYvon–Horiba FluoroHub single photon counting module and the data fits yielded the lifetime values using the provided DAS6 deconvolution software.
Standard radiolabelling protocol
All work carried out with ionising radiations was carried out within the legal requirements of the Ionising Radiations Regulations 2017 and the Environmental Permitting Regulations 2010. The studies followed local rules and regulations to ensure safe and compliant handling of open source radioactive materials. The labelling precursor in a glass HPLC vial was dissolved in 50 μL of MeOH and was further diluted with 100 μL of 10 mM PBS solution. The solution was degassed by sonication and purged with argon for 10 min. Up to ∼400 MBq of freshly reduced fac-[99mTc(CO)3(H2O)3]+ in a reduction medium (∼500 μL) was added, bringing the final precursor concentration to previously determined optimum 0.5–1.3 mM concentration range. The reaction mixture was incubated in a heating shaker at 70 °C mixing at 600 rpm for 30 min. After which it was cooled down to room temperature and injected onto semi-preparatory HPLC (ACE5 C18 5 Å 100 × 250). Product containing fraction (RCY = 47 ± 18%, n = 3, corrected for decay to the beginning of reaction) eluting at 12–14 min was collected, diluted twice with deionised water and loaded onto homemade SPE cartridge (Sep-Pak Light) containing 80–100 mg of Oasis C18 sorbent. The cartridge was washed with 2–3 mL of deionised water and dried by a current of inert gas. The product was eluted by 500 μL of ethanol, the solvent was evaporated under gentle heating aided by a current of inert gas and reformulated into PBS. For in vivo evaluation, the formulation was filtered using a 0.22 μm filter for sterility prior to animal administration. The decay corrected preparatory yield was 21 ± 11% (n = 3) and the full process requires less than 2 h from the start of synthesis.Lipophilicity
LogP was measured using a standard shake-flask method. Briefly, an aliquot of the tracer solution in ethanol (∼1 MBq) was dried in a 1.5 mL Eppendorf tube and dissolved in 1 mL of 1:1 mixture of octanol/PBS. Solution was vigorously mixed in vortex shaker for 5 min at room temperature and phases separated by centrifugation at 14.5 krpm for 2 min. 50 μL of each phase was diluted up to 1 mL and measured using an AGC.
Serum stability
An aliquot of the tracer in ethanol (∼3 MBq) was dried, dissolved in 0.5 mL of human blood serum in a HPLC vial and incubated while shaking at 600 rpm at 37 °C up to 3 hours. Samples were taken at time 0, 30 min, 60 min, 1 h, 2 h and 3 h; the protein was precipitated by addition of double volume of ice cold methanol and removed by centrifugation and the supernatant analysed by radio-HPLC.In vivo SPECT imaging and stability
All animal procedures were approved by the University of Hull Animal Welfare Ethical Review Body (AWERB) and carried out in accordance with the Animals in Scientific Procedures Act 1986 and the UKCCCR Guideline 201041 by approved protocols following institutional guidelines (Home Office Project License number 60/4549 held by Dr Cawthorne).Female CD1 nude mice (25–30 g, Charles River, UK) were induced with 5% and maintained under anaesthesia with 3% isoflurane (oxygen at 1 L min−1). Temperature and respiration were monitored throughout the scan. SPECT-CT acquisition was carried out on a Mediso NanoSPECT/CTPLUS, equipped with multi-pinhole pyramid collimators. Animals were injected i.v. with ≈30 MBq (200 μL) of fac-[99mTc(CO)3(L5)]+ 30 min prior to a 25 min SPECT scan (energy window: 126.4–154.6 keV). To show anatomical co-registration, a CT scan (240 projections, 1 s exposure, 55 kVp X-rays) was acquired following the SPECT scan. SPECT and CT images were reconstructed with an iterative algorithm and with exact cone beam Filtered Back Projection, respectively (HiSPECT, Scivis GmbH, Germany).
After imaging experiments, mice urine was collected, proteins were precipitated by addition of double volume of ice cold methanol and removed by centrifugation at 14.5 krpm for 5 min and supernatant was analysed by radio-HPLC indicating 85.4 ± 1.4% stability (n = 1, triplicate analyses).
In vitro analysis
:8–1:10 for MCF-7 cells. For confocal and flow cytometry experiments cells were counted and seeding densities (below) were used.
To measure the degree of overlap between subcellular localisation of fac-[Re(CO)3(L5)]+ and MDR, a colocalisation coefficient was calculated following standard methods. Single-label control samples were prepared to eliminate fluorescence overlap between two fluorescence channels; in addition to fac-[Re(CO)3(L5)]+ or MDR, untreated cells were assessed in order to eliminate background fluorescence. Single-label control samples were imaged under the same exposure settings as double-labelled samples. Exact coordinates were determined using crosshairs at X and Y coordinates from single-labelled controls and were identical for double-labelled samples. Once the exact coordinates were determined, the ZEN software gave tables containing values of co-localisation coefficient and overlap coefficient.
Mitochondrial protein concentration was shown to be 1.23 μg μl−1, as it was 40 times diluted, the original concentration was 49.46 μg μl−1. Hence, the total mitochondrial protein amount was 4946 μg in 100 μl. 150 μg protein was aliquoted into each Eppendorf. 100 μl of respiratory buffer containing 10 mM succinate added to give the control (n = 3 for each tracer). 10 μM CCCP was added into 100 μl of respiratory buffer containing 10 mM succinate and was then added in remaining Eppendorf tubes. All the Eppendorf tubes were transferred into the CO2 incubator containing 5% CO2 at 37 °C for 15 min and after that 0.5 MBq of tracer was added to the control and CCCP treated mitochondria. All of the Eppendorf tubes were transferred back into the CO2 incubator for 30 min and after that the assay was terminated by centrifugation at 8000g for 5 min at 4 °C. The supernatant was collected and the mitochondrial pellet was washed three times with ice cold respiratory buffer containing 10 mM succinate. After the final wash, the mitochondrial pellet was resuspended into 1 ml of PBS. The amount of radioactivity was measured using the gamma counter (PerkinElmer Wallac wizard 3′′). Percent accumulation was quantified by using the ratio of activity in pellet to that in the pellet plus supernatant. The experiment was carried out three times using internal triplicates. The data is presented as an average of each internal triplicate with the standard deviation among triplicates.
Ligands and precursors
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 43.
4-Chloro-1,8-naphthalic anhydride (2 g, 8.6 mmol) was dissolved in ethanol (60 mL) and ethanolamine (0.79 mL, 12.9 mmol) was added. The solution was then heated at reflux for 12 h under a nitrogen atmosphere. Upon cooling to 0 °C precipitation occurred, and filtration yielded C1 as a yellow solid (2.01 g, 85%). 1H NMR (400 MHz, CDCl3): δH 8.70 (d, J = 6.8 Hz, 1H), 8.64 (d, J = 8.4 Hz, 1H), 8.54 (d, J = 7.6 Hz, 1H), 7.88 (m, 2H), 4.48 (t, J = 4.8 Hz, 2H), 4.01 (t, J = 4.8 Hz, 2H) ppm. LRMS ES+ found m/z = 275.03 [M+].
44.
Prepared as for C1 except using 2-methoxyethylamine (0.90 g, 12.9 mmol), yielding C2 as a yellow solid (1.96 g, 79%). 1H NMR (400 MHz, CDCl3): δH 8.68 (d, J = 7.0 Hz, 1H), 8.61 (d, J = 8.1 Hz, 1H), 8.52 (d, J = 8.2 Hz, 1H), 7.91–7.80 (m, 2H), 4.44 (t, J = 5.7 Hz, 2H), 3.73 (t, J = 5.7 Hz, 2H), 3.38 (s, 3H) ppm. LRMS ES+ found m/z = 289.05 [M+].
:2 acetone hexane with 4% triethylamine as the eluent. The solvents were then removed under vacuum yielding the intermediate product as a red oil (0.40 g, 30%). 1H NMR (300 MHz, CDCl3): δH 8.37 (d, J = 6.8 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.46 (t, J = 7.5 Hz, 1H), 6.53 (d, J = 8.4 Hz, 1H), 6.03 (s, 1H), 5.00 (s, 1H), 4.36 (t, J = 4.8 Hz, 2H), 3.92 (t, J = 4.5 Hz, 2H), 3.82 (t, J = 5.1 Hz 2H), 3.64 (m, 4H), 3.57–3.45 (m, 4H), 3.28 (d, J = 4.8 Hz, 2H), 1.34 (s, 9H) ppm. LRMS found m/z = 389.18 [M + H]+. Deprotection of the intermediate product was achieved by stirring in 50% TFA in DCM (10 mL) for 24 hours under a nitrogen atmosphere. The solvents were then removed under vacuum, and the residue dissolved in methanol and the solvent removed again. This was repeated in triplicate to yield N5 as a red oil (0.29 g, 91%). 1H NMR (300 MHz, CD3OD): δH 8.31–8.19 (m, 2H), 8.09 (m, 1H), 7.43 (m, 1H), 6.61 (m, 1H), 4.23 (m, 2H), 3.80 (t, J = 5.1 Hz, 2H), 3.72–3.64 (m, 6H), 3.59–3.51 (m, 2H), 3.30–3.25 (m, 2H), 3.11–3.01 (m, 2H) ppm. 13C{1H} NMR (75 MHz, CD3OD): δC 164.78, 150.78, 149.04, 134.14, 133.04, 130.55, 128.31, 128.01, 127.62, 123.86, 108.00, 103.65, 70.03, 68.62, 66.54, 59.03, 42.69, 39.22 ppm. LRMS found m/z = 388.18 [M + H]+. HRMS expected m/z = 388.1872 for [M + H]+, found m/z = 388.1857.
600), 262 (20400) nm.
420), 280 (19800), 262 (20500) nm.
200, sh), 263 (20400) nm.
200, sh), 263 (17500) nm.
200, sh), 260 (21000) nm.
600), 263 (20600) nm.
O), 1904 (CO), 1684 (C
43.
4-Chloro-1,8-naphthalic anhydride (2 g, 8.6 mmol) was dissolved in ethanol (60 mL) and ethanolamine (0.79 mL, 12.9 mmol) was added. The solution was then heated at reflux for 12 h under a nitrogen atmosphere. Upon cooling to 0 °C precipitation occurred, and filtration yielded C1 as a yellow solid (2.01 g, 85%). 1H NMR (400 MHz, CDCl3): δH 8.70 (d, J = 6.8 Hz, 1H), 8.64 (d, J = 8.4 Hz, 1H), 8.54 (d, J = 7.6 Hz, 1H), 7.88 (m, 2H), 4.48 (t, J = 4.8 Hz, 2H), 4.01 (t, J = 4.8 Hz, 2H) ppm. LRMS ES+ found m/z = 275.03 [M+].
44.
Prepared as for C1 except using 2-methoxyethylamine (0.90 g, 12.9 mmol), yielding C2 as a yellow solid (1.96 g, 79%). 1H NMR (400 MHz, CDCl3): δH 8.68 (d, J = 7.0 Hz, 1H), 8.61 (d, J = 8.1 Hz, 1H), 8.52 (d, J = 8.2 Hz, 1H), 7.91–7.80 (m, 2H), 4.44 (t, J = 5.7 Hz, 2H), 3.73 (t, J = 5.7 Hz, 2H), 3.38 (s, 3H) ppm. LRMS ES+ found m/z = 289.05 [M+].
:2 acetone hexane with 4% triethylamine as the eluent. The solvents were then removed under vacuum yielding the intermediate product as a red oil (0.40 g, 30%). 1H NMR (300 MHz, CDCl3): δH 8.37 (d, J = 6.8 Hz, 1H), 8.28 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.46 (t, J = 7.5 Hz, 1H), 6.53 (d, J = 8.4 Hz, 1H), 6.03 (s, 1H), 5.00 (s, 1H), 4.36 (t, J = 4.8 Hz, 2H), 3.92 (t, J = 4.5 Hz, 2H), 3.82 (t, J = 5.1 Hz 2H), 3.64 (m, 4H), 3.57–3.45 (m, 4H), 3.28 (d, J = 4.8 Hz, 2H), 1.34 (s, 9H) ppm. LRMS found m/z = 389.18 [M + H]+. Deprotection of the intermediate product was achieved by stirring in 50% TFA in DCM (10 mL) for 24 hours under a nitrogen atmosphere. The solvents were then removed under vacuum, and the residue dissolved in methanol and the solvent removed again. This was repeated in triplicate to yield N5 as a red oil (0.29 g, 91%). 1H NMR (300 MHz, CD3OD): δH 8.31–8.19 (m, 2H), 8.09 (m, 1H), 7.43 (m, 1H), 6.61 (m, 1H), 4.23 (m, 2H), 3.80 (t, J = 5.1 Hz, 2H), 3.72–3.64 (m, 6H), 3.59–3.51 (m, 2H), 3.30–3.25 (m, 2H), 3.11–3.01 (m, 2H) ppm. 13C{1H} NMR (75 MHz, CD3OD): δC 164.78, 150.78, 149.04, 134.14, 133.04, 130.55, 128.31, 128.01, 127.62, 123.86, 108.00, 103.65, 70.03, 68.62, 66.54, 59.03, 42.69, 39.22 ppm. LRMS found m/z = 388.18 [M + H]+. HRMS expected m/z = 388.1872 for [M + H]+, found m/z = 388.1857.
600), 262 (20400) nm.
420), 280 (19800), 262 (20500) nm.
200, sh), 263 (20400) nm.
200, sh), 263 (17500) nm.
200, sh), 260 (21000) nm.
600), 263 (20600) nm.
O), 1904 (CO), 1684 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1638 (CO), 1057 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 430 (8560), 281 (18900, sh), (21000) nm.
O), 1910 (CO), 1684 (CO), 1638 (CO), 1055 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 433 (9800), 280 (18400, sh), 261 (22500) nm.
O), 1944 (CO), 1638 (CO), 1072 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 434 (10100), 279 (18200), 256 (22400) nm.
O), 1907 (CO), 1681 (CO), 1639 (CO), 1055 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 431 (13020), 276 (24400, sh), 260 (26100) nm.
O), 1928 (CO), 1683 (CO), 1635 (CO), 1058 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 429 (8100), 279 (13300), 262 (18000) nm.
O), 1908 (CO), 1684 (CO), 1645 (CO), 1057 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 430 (12240), 281 (21000), 261 (23100) nm.
O), 1638 (CO), 1057 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 430 (8560), 281 (18900, sh), (21000) nm.
O), 1910 (CO), 1684 (CO), 1638 (CO), 1055 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 433 (9800), 280 (18400, sh), 261 (22500) nm.
O), 1944 (CO), 1638 (CO), 1072 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 434 (10100), 279 (18200), 256 (22400) nm.
O), 1907 (CO), 1681 (CO), 1639 (CO), 1055 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 431 (13020), 276 (24400, sh), 260 (26100) nm.
O), 1928 (CO), 1683 (CO), 1635 (CO), 1058 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 429 (8100), 279 (13300), 262 (18000) nm.
O), 1908 (CO), 1684 (CO), 1645 (CO), 1057 (BF4−). UV-vis. (MeCN) λmax (ε/dm3 mol−1 cm−1): 430 (12240), 281 (21000), 261 (23100) nm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AD, IAF and SJAP thank DSTL and Ministry of Defence for financial support. The staff of the EPSRC Mass Spectrometry National Service (Swansea University) are also thanked. We also thank EPSRC for instrumentation grant EP/L027240/1. We acknowledge the Daisy Appeal Charity (grant no. DAhul2011) for funding, and thank Dr Assem Allam and his family for the generous donation to help found the PET Research Centre at the University of Hull.Notes and references
- B. P. Burke, C. Cawthorne and S. J. Archibald, Philos. Trans. R. Soc., A, 2017, 375, 0261 CrossRef PubMed.
- S. Lutje, M. Rijpkema, W. Helfrich, W. J. G. Oyen and O. C. Boerman, Mol. Imaging Biol., 2014, 16, 747–755 CrossRef CAS PubMed.
- (a) L. E. Jennings and N. J. Long, Chem. Commun., 2009, 28, 3511–3524 RSC; (b) S. Lee and X. Chen, Mol. Imaging, 2009, 8, 87–100 CrossRef CAS PubMed; (c) S. R. Cherry, Annu. Rev. Biomed. Eng., 2006, 8, 35–62 CrossRef CAS PubMed; (d) X. Li, X.-N. Zhang, X.-D. Li and J. Chang, Cancer Biol. Med., 2016, 13, 339–348 CAS; (e) F. L. Thorp-Greenwood and M. P. Coogan, Dalton Trans., 2011, 40, 6129–6143 RSC.
- A. Azhdarinia, P. Ghosh, S. Ghosh, N. Wilganowski and E. M. Sevick-Muraca, Mol. Imaging Biol., 2012, 14, 261–276 CrossRef PubMed.
- D. L. Li, J. J. Zhang, C. W. Chi, X. Xiao, J. M. Wang, L. X. Lang, I. Ali, G. Niu, L. W. Zhang, J. Tian, N. Ji, Z. H. Zhu and X. Y. Chen, Theranostics, 2018, 8, 2508–2520 CrossRef CAS PubMed.
- E. Mery, M. Golzio, S. Guillermet, D. Lanore, A. Le Naour, B. Thibault, A. F. Tilkin-Mariame, E. Bellard, J. P. Delord, D. Querleu, G. Ferron and B. Couderc, Oncotarget, 2017, 8, 109559–109574 CrossRef PubMed.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed.
- (a) A. Mahmood and A. G. Jones, Technetium Radiopharmaceuticals, in Handbook of Radiopharmaceuticals, ed. M. J. Welch and C. S. Redvanly, Wiley, 2005, ch. 10 Search PubMed; (b) P. J. Blower, Dalton Trans., 2015, 44, 4819–4844 RSC.
- (a) M. H. Kim, C. G. Kim, S.-G. Kim and D.-W. Kim, J. Labelled Compd. Radiopharm., 2017, 60, 649–658 CrossRef CAS PubMed; (b) M. H. Kim, S.-G. Kim and D.-W. Kim, J. Labelled Compd. Radiopharm., 2018, 61, 557–566 CrossRef CAS PubMed; (c) J. Y. Lee, H. J. Kim, Y.-S. Lee and J. M. Jeong, Sci. Rep., 2018, 8, 13636 CrossRef PubMed; (d) M. Stalnionis and R. Rotomskis, Phys. Med., 2014, 30, e120 CrossRef.
- M. P. Coogan, R. P. Doyle, J. F. Valliant, J. W. Babich and J. Zubieta, J. Labelled Compd. Radiopharm., 2014, 57, 255–261 CrossRef CAS PubMed.
- (a) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- M. Sagnou, B. Mavroidi, A. Shegani, M. Paravatou-Petsotas, C. Raptopoulou, V. Psycharis, I. Permettis, M. S. Papadopoulos and M. Pelecanou, J. Med. Chem., 2019, 62, 2638–2650 CrossRef CAS PubMed.
- T. Storr, K. H. Thompson and C. Orvig, Chem. Soc. Rev., 2006, 35, 534–544 RSC.
- For example: (a) D. J. Hayne, A. J. North, M. Fodero-Tavoletti, J. M. White, L. W. Hung, A. Rigopoulos, C. A. McLean, P. A. Adlard, U. Ackermann, H. Tochon-Danguy, V. L. Villemagne, K. J. Barnham and P. S. Donnelly, Dalton Trans., 2015, 44, 4933–4944 RSC; (b) C. Xiong, W. Lu, R. Zhang, M. Tian, W. Tong, J. Gelovani and C. Li, Chem. – Eur. J., 2009, 15, 8979–8984 CrossRef CAS PubMed.
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kuhn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS.
- (a) S. Jurgens, W. A. Herrmann and F. E. Kuhn, J. Organomet. Chem., 2014, 751, 83–89 CrossRef; (b) P. S. Donnelly, Dalton Trans., 2011, 40, 999–1010 RSC; (c) B. Lambert, K. Bacher and L. Defreyne, J. Nucl. Med. Mol. Imaging, 2009, 53, 305–310 CAS.
- F.-X. Wang, J.-H. Liang, H. Zhang, Z.-H. Wang, Q. Wan, C.-P. Tan, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2019, 11, 13123–13133 CrossRef CAS PubMed.
- A. François, C. Auzanneau, V. Le Morvan, C. Galaup, H. S. Godfrey, L. Marty, A. Boulay, M. Artigau, B. Mestre-Voegtlé, N. Leygue, C. Picard, Y. Coulais, J. Robert and E. Benoist, Dalton Trans., 2014, 43, 439 RSC.
- For example: (a) L. Wei, J. W. Babich, W. Oullette and J. Zubieta, Inorg. Chem., 2006, 45, 3057–3066 CrossRef CAS PubMed; (b) N. Agorastos, L. Borsig, A. Renard, P. Antoni, G. Viola, B. Springler, P. Kurz and R. Alberto, Chem. – Eur. J., 2007, 13, 3842–3853 CrossRef CAS PubMed; (c) P. Haefliger, N. Agorastos, A. Renard, G. Giambonini-Brugnoli, C. Marty and R. Alberto, Bioconjugate Chem., 2005, 16, 582–587 CrossRef CAS PubMed; (d) E. E. Langdon-Jones, A. B. Jones, C. F. Williams, A. J. Hayes, D. Lloyd, H. J. Mottram and S. J. A. Pope, Eur. J. Inorg. Chem., 2016, 759–766 Search PubMed.
- F. J. Dai, H. Y. He, X. J. Xu, S. Chen, C. J. Wang, C. Y. Feng, Z. Y. Tian, H. Y. Dong and S. Q. Xie, Bioorg. Chem., 2018, 77, 16–24 CrossRef CAS PubMed.
- E. Calatrava-Perez, S. A. Bright, S. Achermann, C. Moylan, M. O. Senge, E. B. Veale, D. C. Williams, T. Gunnlaugsson and E. M. Scanlan, Chem. Commun., 2016, 52, 13086–13089 RSC.
- E. E. Langdon-Jones, C. F. Williams, A. J. Hayes, D. Lloyd, S. J. Coles, P. N. Horton, L. M. Groves and S. J. A. Pope, Eur. J. Inorg. Chem., 2017, 5279–5287 CrossRef CAS.
- C. Satriano, G. T. Sfrazzetto, M. E. Amato, F. P. Ballistreri, A. Copani, M. L. Giuffrida, G. Grasso, A. Pappalardo, E. Rizzarelli, G. A. Tomaselli and R. M. Toscano, Chem. Commun., 2013, 49, 5565–5567 RSC.
- E. E. Langdon-Jones, D. Lloyd, A. J. Hayes, S. D. Wainwright, H. J. Mottram, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2015, 54, 6606–6615 CrossRef CAS PubMed.
- S. Nigam, B. P. Burke, L. H. Davies, J. Domarkas, J. F. Wallis, P. G. Waddell, J. S. Waby, D. M. Benoit, A. M. Seymour, C. Cawthorne, L. J. Higham and S. J. Archibald, Chem. Commun., 2016, 52, 7114–7117 RSC.
- W. L. Turnbull, E. Murrell, M. Bulcan-Gnirss, M. Majeed, M. Milne and L. G. Luyt, Dalton Trans., 2019, 48, 14077–14084 RSC.
- T. Storr, Y. Sugai, C. A. Barta, M. J. Adam, S. Yano and C. Orvig, Inorg. Chem., 2005, 44, 2698–2705 CrossRef CAS PubMed.
- J. A. Czaplewska, F. Theil, E. Altuntas, T. Niksch, M. Freesmeyer, B. Happ, D. Pretzel, H. Schafer, M. Obata, S. Yano, U. S. Schubert and M. Gottschaldt, Eur. J. Inorg. Chem., 2014, 6290–6297 CrossRef CAS.
- L. A. Mullice, R. H. Laye, L. P. Harding, N. J. Buurma and S. J. A. Pope, New J. Chem., 2008, 32, 2140–2149 RSC.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- E. E. Langdon-Jones, N. O. Symonds, S. E. Yates, A. J. Hayes, D. Lloyd, R. Williams, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2014, 53, 3788–3797 CrossRef CAS PubMed.
- R. Alberto, J. Organomet. Chem., 2007, 692, 1179–1186 CrossRef CAS.
- R. Alberto, R. Schibli, A. Egli, A. P. Schubiger, U. Abram and T. A. Kaden, J. Am. Chem. Soc., 1998, 120, 7987–7988 CrossRef CAS.
- B. B. Kasten, X. Ma, K. Cheng, L. Bu, W. S. Slocumb, T. R. Hayes, S. Trabue, Z. Cheng and P. D. Benny, Bioconjugate Chem., 2016, 27, 130–142 CrossRef CAS PubMed.
- A. J. Hallett, E. Placet, R. Prieux, D. McCafferty, J. A. Platts, D. Lloyd, M. Isaacs, A. J. Hayes, S. J. Coles, M. B. Pitak, S. Marchant, S. N. Marriott, R. K. Allemann, A. Dervisi and I. A. Fallis, Dalton Trans., 2018, 47, 14241–14253 RSC.
- L. Di and E. H. Kerns, Curr. Opin. Chem. Biol., 2003, 7, 402–408 CrossRef CAS PubMed.
- J. E. A. Comer, High throughput measurement of logD and pKa, in Methods and Principlesin Medicinal Chemistry, ed. P. Artursson, H. Lennernas and H. van de Waterbeemd, Wiley-VCH, Weinheim, 2003, vol. 18, p. 21 Search PubMed.
- C. Moura, F. Mendes, L. Gano, I. Santos and A. Paulo, J. Inorg. Biochem., 2013, 123, 34–45 CrossRef CAS PubMed.
- R. A. Fearn and B. H. Hirst, Environ. Toxicol. Pharmacol., 2006, 21, 168 CrossRef CAS PubMed.
- W. L. Turnbull, L. Yu, E. Murrell, M. Milne, C. L. Charron and L. G. Luyt, Org. Biomol. Chem., 2019, 17, 598 RSC.
- P. Workman, E. O. Aboagye, F. Balkwill, A. Balmain, G. Bruder, D. J. Chaplin J. A. Double, J. Everitt, D. A. H. Farningham, M. J. Glennie L. R. Kelland, V. Robinson, I. J. Stratford G. M. Tozer, S. Watson, S. R. Wedge S. A. Eccles, V. Navaratnam, S. Ryder and Committe of the National Cancer Research Institute, Br. J. Cancer, 2010, 102, 1555 CrossRef CAS PubMed.
- E. A. Boehm, B. E. Jones, G. K. Radda, R. L. Veech and K. Clarke, Am. J. Physiol.: Heart Circ. Physiol., 2001, 280, 977 CrossRef PubMed.
- K. Sakayori, Y. Shibasaki and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5571 CrossRef CAS.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt04024b |
| This journal is © The Royal Society of Chemistry 2020 |