
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing 31P NMR relaxation rates with a kinetically inert gadolinium complex†
Louise R.
Tear
ab,
Mahon L.
Maguire
bc,
Manuel
Tropiano
a,
Kezi
Yao
a,
Nicola J.
Farrer
 a,
Stephen
Faulkner
*a and
Jurgen E.
Schneider
bd
a,
Stephen
Faulkner
*a and
Jurgen E.
Schneider
bd
aChemistry Research Laboratory, University of Oxford, Mansfield Road, OX1 3TA, UK. E-mail: stephen.faulkner@chem.ox.ac.uk; Tel: +44 (0)1865 272640
bBritish Heart Foundation Experimental MR Unit (BMRU), University of Oxford, Roosevelt Drive, Oxford, OX3 7BN, UK
cCentre for Preclinical Imaging, University of Liverpool, Nuffield Wing, Sherrington Building, Crown Street, Liverpool, L69 3BX, UK
dLeeds Institute of Cardiovascular and Metabolic Medicine, Biomedical Imaging Science Department, University of Leeds, Clarendon Way, Leeds, LS2 9JT, UK
First published on 20th February 2020
Abstract
The kinetically stable heptadentate gadolinium complex Gd.pDO3A (1.Gd) demonstrates significant 31P nuclear magnetic resonance (NMR) relaxation enhancement of biologically relevant phosphate species; adenosine triphosphate (ATP), phosphocreatine (PCr) and inorganic phosphate. Gd.pDO3A (1.Gd) binds these species in fast exchange, enabling the relaxation of the bulk phosphate species in solution. This gives rise to 31P relaxation enhancements up to 250-fold higher than those observed for 31P relaxation enhancements with the commercial MRI contrast agent Gd.DOTA (DOTAREM), 2. Gd.pDO3A-like complexes may have potential applications as 31P magnetic resonance contrast agents, since shortening the T1 relaxation time of phosphate species would reduce the time needed to acquire 31P-MR spectra.
Introduction
Lanthanide complexes are powerful tools for imaging and assay, and have played key roles in a variety of applications from contrast enhanced MRI1–4 to time-gated optical imaging and bioassay.5–7While the physical properties of open-shell lanthanide ions lend themselves to such applications – since the 4f electrons have little role in bonding – their chemical properties present a challenge when using lanthanides for biological applications. Lanthanide ions are too toxic to administer as free ions, form labile complexes with simple ligands, and tend to form insoluble salts with a variety of common ions. This challenge can be addressed through coordination chemistry.8–10 Multidentate ligands derived from macrocycles can offer high kinetic and thermodynamic stability, removing free lanthanide ions from solution, and eliminating lanthanide toxicity. Kinetic control is particularly important in this context, since precipitation of lanthanide species can act as a kinetic trap. Kinetically labile complexes can give rise to hazards in biology: for instance nephrogenic systemic fibrosis (NSF) has been correlated with the use of labile gadolinium complexes in clinical imaging.11–14
More than thirty years after the clinical approval of the first generation of MRI contrast agents, the clinical use of MRI contrast media remains broadly focused on the use of blood pool contrast agents.15 These operate as T1-shortening magnetic resonance contrast agents by using the paramagnetism of gadolinium to cause rapid relaxation of water protons in bulk solution, through fast exchange between lanthanide-bound solvent and bulk solvent.1,16
A variety of reports detail the preparation and use of gadolinium complexes as responsive contrast agents, where the relaxation enhancement varies depending on the interaction of the complex with an analyte of interest.6 However, these have found little traction in clinical applications due to difficulties in quantifying the observed response: since the contrast agent changes the behaviour of bulk water, it is very difficult to distinguish between a small quantity of complex in its “on” state (where relaxation is enhanced greatly) and a large quantity of complex in the “off” state. This dichotomy has been successfully addressed in the case of luminescent complexes, where ratiometric imaging methods can be used to quantify behaviour on the basis of taking the ratio between two different emission wavelengths.7,17 However, such an approach is very challenging in the context of MRI, though Caravan and co-workers have had some success in quantitative imaging using the DREMR protocol, which measures relaxation at two different fields and relies on differences in field dependence of relaxivity between the “off” and “on” forms of a complex.18
In this manuscript, we describe how 31P NMR methods can be used to probe the interaction of a lanthanide complex with some ionic phosphate species.6,19–21 Relatively weak and rapidly reversible phosphate binding gives rise to 31P relaxation enhancement for bulk phosphate species, while different species respond to the complex to different degrees. Such results potentially open up a new strategy for imaging.
Here we report our investigation into using the kinetically inert lanthanide complex (Gd.pDO3A (1.Gd) Fig. 1)22–26 as a 31P contrast agent for enhancing phosphorus relaxation. 1.Gd comes from a family of pDO3A complexes which we have previously used as building blocks for more complex architectures: it combines high kinetic stability with charge neutrality: we reasoned that this combination would reduce the affinity for phosphate, permitting us to anticipate relatively rapid exchange of bound phosphate with bulk. As such we chose this system to test the hypothesis of using 1.Gd as a contrast medium for 31P relaxation enhancement.
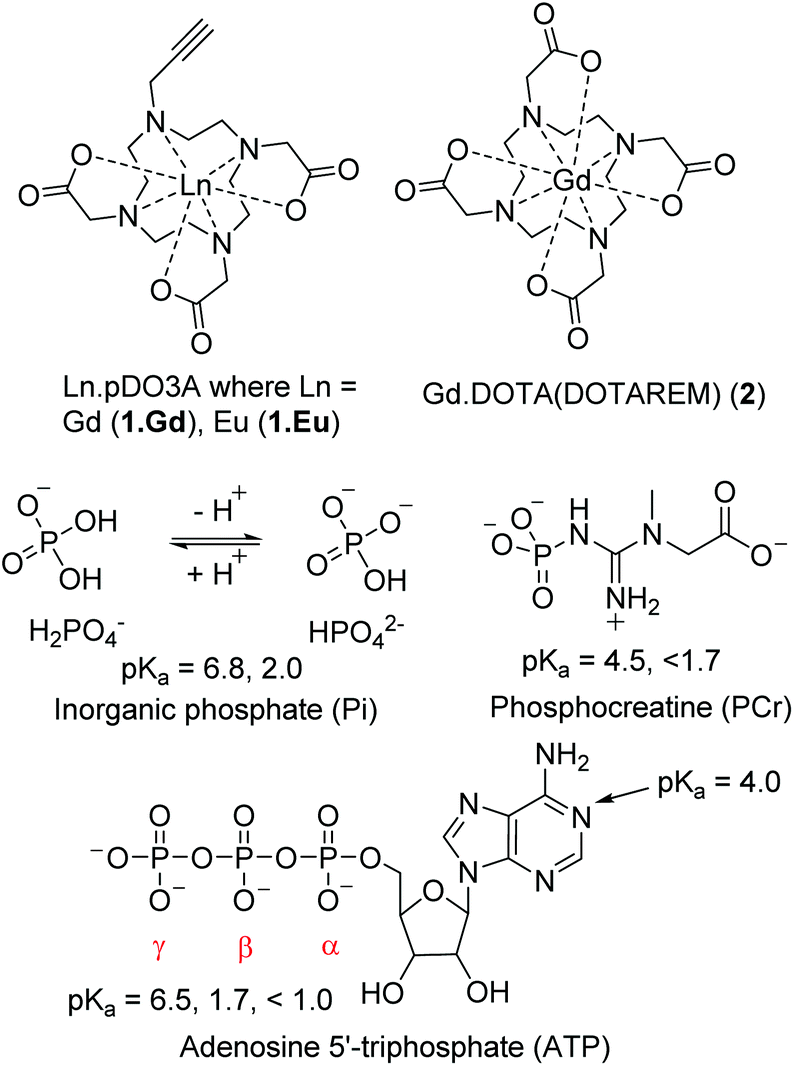 | ||
| Fig. 1 Structures of complexes discussed in this study; commercial contrast agent Gd.DOTA; complexes of p.DO3A complexes of Gd (1.Gd) and Eu (1.Eu) which are investigated in this study; relevant phosphate species with reported pKa values (H2O) of the phosphate groups.27–29 | ||
Results and discussion
Both gadolinium (1.Gd) and europium (1.Eu) pDO3A complexes were synthesized and characterised using established procedures.22 Given the importance of maintaining the integrity of these complexes and the issues associated with the formation of phosphate colloids with free lanthanide ions,11–14 we resolved to explore their stability before embarking on a more detailed study. In such a system, precipitation constitutes a kinetic trap, and absolute measurements of thermodynamic stability can be misleading as they do not guarantee safety of complexes where the system is under thermodynamic control.11 Under such circumstances, kinetic stability is essential, and our previous studies on heterometallic systems26 led us to believe that these systems would be kinetically inert. The method of Tóth30 was used to explore the stability of the complexes by challenging the europium complex with an excess of free gadolinium ions in aqueous solution. No change in the form of the observed spectrum was observed over a period of five days, indicating that the complex is kinetically inert (since the luminescence spectra of free and complexed europium are very different, as can be seen in the ESI† to this paper).The interaction of 1.Eu with three phosphate metabolites – inorganic phosphate (Pi), PCr and ATP – was assessed by luminescence spectroscopy. Titration of each metabolite with 1.Eu in HEPES buffer at pH 7.4 resulted in modulation of the Eu emission intensity. Changes to the emission spectra clearly reveal a change to the local coordination environment at the metal centre (Fig. 2, and Fig. S1 and S2†), from which it is possible to infer phosphate displacing water at the metal centre. In the case of inorganic phosphate (Fig. 2), the consequences of binding are clear, with significant changes to the local ligand field being evident from changes to the fine structure of the 5D0–7F1 transition (around 595 nm) and to the relative intensity of the 5D0–7F2 transition, which is hypersensitive to local symmetry.
 | ||
| Fig. 2 Main figure: changes to the total emission spectrum (λex = 394 nm) of a solution of 1.Eu in HEPES buffer with increasing concentration of inorganic phosphate. Inset: the binding isotherm obtained from changes in the intensity of the 5D0–7F2 emission band centred on 617 nm and fitted using Dynafit®.31 | ||
The integrated intensity of the 5D0–7F2 transition (λem = 617 nm) was plotted against the phosphate species concentration to determine the strength of the binding interaction (the inset to Fig. 2 shows a binding isotherm for inorganic phosphate: the others are recorded in Fig. S1 and S2†).
These were fitted using Dynafit®31 and modelled to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding equilibrium (ESI, eqn (S1)†) to determine the association constants Ka listed in Table 1. Since the values calculated were measured in the presence of HEPES, which has limited ionic strength and can potentially interact with lanthanide complexes, these values represent effective binding constants, but clearly reveal consistent binding of a variety of phosphate species, with slightly weaker binding of PCr as a result of its zwitterionic nature.
:1 binding equilibrium (ESI, eqn (S1)†) to determine the association constants Ka listed in Table 1. Since the values calculated were measured in the presence of HEPES, which has limited ionic strength and can potentially interact with lanthanide complexes, these values represent effective binding constants, but clearly reveal consistent binding of a variety of phosphate species, with slightly weaker binding of PCr as a result of its zwitterionic nature.
The observed values of Ka are fully consistent with fast exchange between bulk phosphate and phosphate bound to 1.Eu. The three phosphate species all exhibit very similar affinity constants. Binding of phosphate was further demonstrated by measuring the luminescence lifetimes in H2O and D2O, and determining the number of inner-sphere water molecules using the modified Horrocks equation.32 This revealed that q = 2 for 1.Eu in the absence of phosphate, but q = 0.9 in the presence of phosphate – consistent with monodentate coordination of phosphate in line with literature precedent.6
To further explore exchange, we used 1.Gd to explore the possibility of observing changes to the bulk relaxation rates of phosphate species in the presence of increasing concentrations of 1.Gd, reasoning that fast exchange would lead to clear concentration dependent enhancement of the 31P longitudinal relaxation rates, in the same way that conventional MRI contrast agents enhance the relaxation of bulk water. 31P relaxation rates (R1 = 1/T1) were measured for ATP, PCr and Pi with increasing concentrations of 1.Gd (Fig. 3).
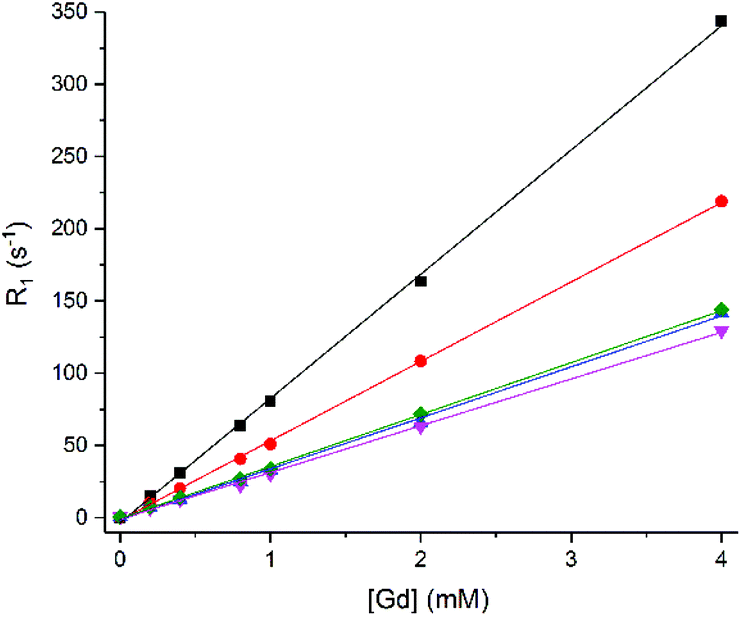 | ||
Fig. 3
31P Relaxation rate (R1) of Pi (■), PCr ( ) and α-( ) and α-( ), β-( ), β-( ), γ-( ), γ-( ) ATP (8.7, 5.5 and 7.9 mM) versus concentration of 1.Gd, pH 7.2, 162 MHz, 298 K. ) ATP (8.7, 5.5 and 7.9 mM) versus concentration of 1.Gd, pH 7.2, 162 MHz, 298 K. | ||
1.Gd showed a significant linear relaxation enhancement of all phosphate species (Fig. 3), suggesting that fast exchange is indeed occurring. This linear enhancement also confirms that the gadolinium complex is kinetically stable under the conditions of the experiment, since exchange of gadolinium and formation of a new gadolinium bound species results in significant curvature to the plot, as previously observed by Muller and co-workers (who observed significant competition between ATP and gadolinium complexes of DTPA bis amide ligands).33
The relaxivities obtained (r1 = 1/([Gd]T1)) are displayed in Table 2 for 1.Gd. The table also shows the 31P relaxivity properties for 2 (Gd.DOTA-, Dotarem®) for comparison. These values for 1.Gd are all greater than 30 mM−1 s−1, with the most significant effect observed for Pi followed by PCr and then ATP. This is most likely to be a result of the size of the phosphate molecule and the consequently easy access to the Gd binding site.
| Phosphate speciesa |
r
1b (mM Gd−1 s−1) |
|
|---|---|---|
| 1.Gd | 2 | |
| a Solution contains [PBS] = 8.7 mM, [PCr] = 5.5 mM, [ATP] = 7.9 mM. b ± Standard error of linear fit, R2 values >0.99 for all fits. | ||
| Pi | 85.96 ± 1.00 | 0.35 ± 0.01 |
| PCr | 54.97 ± 0.48 | 0.24 ± 0.01 |
| α-ATP | 32.35 ± 0.46 | 0.39 ± 0.03 |
| β-ATP | 35.97 ± 0.35 | 0.27 ± 0.01 |
| γ-ATP | 35.30 ± 0.64 | 0.25 ± 0.01 |
Increasing concentrations of contrast agent 2 resulted in a much smaller, though still linear, relaxation enhancement across all five phosphate resonances (Pi, PCr, α-ATP, β-ATP, γ-ATP) (Table 2 and Fig S4†). The 31P relaxivity values (r1 = 1/([Gd]T1) of all phosphate species were below 0.5 mM−1 s−1. These relaxivity values for 2 are more than 60 times smaller than the relaxivity values measured for 1.Gd.
Taking the observations on 1.Gd together with those on 2, it is clear that phosphate affinity is playing an important role in relaxation rate enhancement. For 2, which displays negligible affinity for phosphate, the small enhancements observed are likely to be the consequence of an outer sphere interaction.
Outer-sphere relaxation is defined by diffusion, the distance of closest approach and the electronic relaxation time of the metal ion.34 For complexes of a similar size and molecular weight, the outer-sphere contribution to longitudinal relaxation rate is considered to be comparable and independent of differences in functional groups on the ligand. On this basis, we can use the relaxivity measurements obtained using 2 to estimate the inner sphere contribution to 31P relaxivity with 1.Gd, using eqn (1).
| r1 = rIS1 + rOS1 | (1) |
From this equation the inner-sphere contribution to the overall observed 31P relaxivity for 1.Gd is estimated to be ≥99% and is clearly the dominant effect. This is much greater than is commonly observed for 1H relaxivity, where the outer sphere contribution for a q = 1 complex can be around 40% (at imaging field strengths).35,36 The much lower outer-sphere contribution for phosphates undoubtedly reflects that there are invariably many water molecules in the outer coordination sphere of a complex. Since the total concentration of phosphate nuclei will be around three orders of magnitude lower than that of water protons (25 mM total phosphate species versus ∼55 M water) the contribution of second-sphere phosphate molecules is always likely to be relatively small.
Since phosphate binds to 1.Gd, it was anticipated that phosphate binding would exclude water from the inner coordination sphere. To confirm this, the 1H water relaxation rate was measured with increasing concentrations of 1.Gd in distilled water and in phosphate solution (Fig. 4, values in Table 3). The relaxivity in water is very similar to that for DO3A37 and greater than that for Dotarem, as would be expected for a complex with q = 2.1
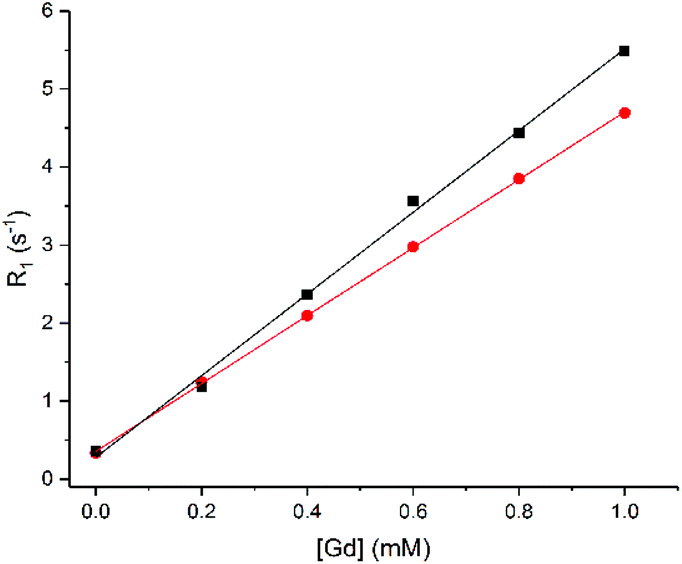 | ||
Fig. 4
1H water relaxation rate (R1) in DI water (■) or phosphate solution ( ) (8.7 mM PBS, 5.5 mM PCr and 7.9 mM ATP) versus concentration of 1.Gd, pH 7.2, 400 MHz, 298 K. Error bars are within the size of the data points. ) (8.7 mM PBS, 5.5 mM PCr and 7.9 mM ATP) versus concentration of 1.Gd, pH 7.2, 400 MHz, 298 K. Error bars are within the size of the data points. | ||
The 1H relaxivity was found to be about 16% lower in phosphate solution than in distilled water. This is due to the competition between water and phosphate species for the gadolinium centre, which reduces the concentration of bound water molecules relative to bulk. This suggests that a combination of the decrease in 1H signal and increase in 31P signal may allow the possibility for ratiometric determination of phosphate from multinuclear relaxometric measurements – though it is clear that current instrumentation would make such simultaneous dual measurements extremely challenging.
Experimental
For materials, methods and procedures see ESI.†Conclusions
In this study, we have shown how binding and fast exchange between a gadolinium complex and anionic phosphate species can be exploited to enhance relaxation of bulk phosphate anions in solution. We have demonstrated significant 31P relaxation enhancement using a kinetically inert heptadentate lanthanide complex 1.Gd. We observe dramatic enhancements in relaxation for a variety of phosphorus containing species (particularly phosphate itself, ATP and phosphocreatine). Preliminary studies suggest that this 1.Gd is not internalised by cells to any significant extent,38 limiting the biological application of this complex to extracellular investigation without further derivation. The use of phosphate contrast media may prove a profitable avenue for investigation if these issues of cellular uptake can be addressed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the Engineering and Physical Sciences Research Council (EPSRC), the Medical Research Council (MRC) [grant number EP/L016052/1] and the British Heart Foundation (FS/11/50/29038). NF thanks the Wellcome Trust (201406/Z/16/Z), Cancer Research UK (C5255/A18085) through the Cancer Research UK Oxford Centre, L‘Oréal (Women in Science Fellowship) and Singapore Chemical Group (Innovation Fund Young Researcher Award) for funding.Notes and references
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS PubMed.
- J. Lohrke, T. Frenzel, J. Endrikat, F. C. Alves, T. M. Grist, M. Law, J. M. Lee, T. Leiner, K. C. Li, K. Nikolaou, M. R. Prince, H. H. Schild, J. C. Weinreb, K. Yoshikawa and H. Pietsch, Adv. Ther., 2016, 33, 1 CrossRef PubMed.
- L. M. De León-Rodríguez, A. F. Martins, M. C. Pinho, N. M. Rofsky and A. D. Sherry, J. Magn. Reson. Imaging, 2015, 42, 545 CrossRef PubMed.
- G.-P. Yan, L. Robinson and P. Hogg, Radiography, 2007, 13, e5 CrossRef.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770 CrossRef CAS PubMed.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652 RSC.
- S. H. Hewitt and S. J. Butler, Chem. Commun., 2018, 54, 6635 RSC.
- T. J. Sørensen and S. Faulkner, Acc. Chem. Res., 2018, 51, 2493 CrossRef PubMed.
- D. Messeri, M. P. Lowe, D. Parker and M. Botta, Chem. Commun., 2001, 1, 2742 RSC.
- M. Tropiano, O. A. Blackburn, J. A. Tilney, L. R. Hill, M. P. Placidi, R. J. Aarons, D. Sykes, M. W. Jones, A. M. Kenwright, J. S. Snaith, T. J. Sørensen and S. Faulkner, Chem. – Eur. J., 2013, 19, 16566 CrossRef CAS PubMed.
- M. Le Fur and P. Caravan, Metallomics, 2019, 11, 240 RSC.
- W. A. High, R. A. Ayers, J. Chandler, G. Zito and S. E. Cowper, J. Am. Acad. Dermatol., 2007, 56, 21 CrossRef PubMed.
- P. Marckmann, L. Skov, K. Rossen, A. Dupont, M. B. Damholt, J. G. Heaf and H. S. Thomsen, J. Am. Soc. Nephrol., 2006, 17, 2359 CrossRef PubMed.
- T. Grobner, Nephrol., Dial., Transplant., 2006, 21, 1104 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957 CrossRef CAS PubMed.
- V. C. Pierre, M. J. Allen and P. Caravan, J. Biol. Inorg. Chem., 2014, 19, 127–131 CrossRef CAS PubMed.
- T. J. Sørensen, A. M. Kenwright and S. Faulkner, Chem. Sci., 2015, 6, 2054 RSC.
- J. K. Alford, A. G. Sorensen, T. Benner, B. A. Chronik, W. B. Handler, T. J. Scholl, G. Madan and P. Caravan, Proc. Int. Soc. Magn. Reson. Med., 2011, 19, 452 Search PubMed.
- P. Atkinson, Y. Bretonnière, D. Parker and G. Muller, Helv. Chim. Acta, 2005, 88, 391 CrossRef CAS.
- J. I. Bruce, R. S. Dickins, L. J. Govenlock, T. Gunnlaugsson, S. Lopinski, M. P. Lowe, D. Parker, R. D. Peacock, J. J. B. Perry, S. Aime and M. Botta, J. Am. Chem. Soc., 2000, 122, 9674 CrossRef CAS.
- R. N. Muller, B. Radüchel, S. Laurent, J. Platzek, C. Piérart, P. Mareski and L. Vander Elst, Eur. J. Inorg. Chem., 1999, 1949 CrossRef CAS.
- M. Jauregui, W. S. Perry, C. Allain, L. R. Vidler, M. C. Willis, A. M. Kenwright, J. S. Snaith, G. J. Stasiuk, M. P. Lowe and S. Faulkner, Dalton Trans., 2009, 6283 RSC.
- A. K. R. Junker, M. Tropiano, S. Faulkner, T. J. Sørensen, A. Kathrine, R. Junker, M. Tropiano, S. Faulkner and T. Just Sørensen, Inorg. Chem., 2016, 55, 12299 CrossRef CAS PubMed.
- M. Tropiano, N. L. Kilah, M. Morten, H. Rahman, J. J. Davis, P. D. Beer and S. Faulkner, J. Am. Chem. Soc., 2011, 133, 11847 CrossRef CAS PubMed.
- C. Allain, P. D. Beer, S. Faulkner, M. W. Jones, A. M. Kenwright, N. L. Kilah, R. C. Knighton, T. J. Sørensen and M. Tropiano, Chem. Sci., 2013, 4, 489 RSC.
- M. Tropiano, A. M. Kenwright and S. Faulkner, Chem. – Eur. J., 2015, 21, 5697 CrossRef CAS PubMed.
- P. Oesper, in Phosphorus Metabolism, ed. W. D. McElroy and B. Glass, John Hopkins University Press, Baltimore, 1951, vol. I, pp. 523–536 Search PubMed.
- L. Chen, Biomed. Res., 2017, 28, 8195 CAS.
- P. Kaczmarek, W. Szczepanik and M. Jezowska-Bojczuk, Dalton Trans., 2005, 3653 RSC.
- É. Tóth, R. Király, J. Platzek, B. Radüchel and E. Brücher, Inorg. Chim. Acta, 1996, 249, 191 CrossRef.
- P. Kuzmič, Methods Enzymol., 2009, 467, 247 Search PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- L. Vander Elst, Y. Van Haverbeke, J. F. Goudemant and R. N. Muller, Magn. Reson. Med., 1994, 31, 437 CrossRef CAS PubMed.
- S. Aime, M. Botta, M. Fasano and E. Terreno, Chem. Soc. Rev., 1998, 27, 19 RSC.
- S. Aime, A. S. Batsanov, M. Botta, J. A. K. Howard, D. Parker, K. Senanayake and G. Williams, Inorg. Chem., 1994, 33, 4696 CrossRef CAS.
- M. Botta, Eur. J. Inorg. Chem., 2000, 399 CrossRef CAS.
- P. Placidi, L. S. Natrajan, D. Sykes, A. M. Kenwright and S. Faulkner, Helv. Chim. Acta, 2009, 92, 2427 CrossRef.
- L. R. Tear, Molecular Imaging Probes for 31P Contrast, DPhil Thesis, University of Oxford, Oxford, UK, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods including spectroscopy, determination of binding constants and isotherms. See DOI: 10.1039/c9dt03761f |
| This journal is © The Royal Society of Chemistry 2020 |