
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDicerium letterbox-shaped tetraphenolates: f-block complexes designed for two-electron chemistry†
Polly L.
Arnold
 *a,
Kai
Wang
a,
Steven J.
Gray
a,
Liane M.
Moreau
b,
Corwin H.
Booth
b,
Massimiliano
Curcio
a,
Jordann A. L.
Wells
a and
Alexandra M. Z.
Slawin
c
*a,
Kai
Wang
a,
Steven J.
Gray
a,
Liane M.
Moreau
b,
Corwin H.
Booth
b,
Massimiliano
Curcio
a,
Jordann A. L.
Wells
a and
Alexandra M. Z.
Slawin
c
aSchool of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: polly.arnold@ed.ac.uk
bLawrence Berkeley National Laboratory, University of California, Berkeley, CA, USA
cSchool of Chemistry and EaStCHEM, University of St Andrews, St Andrews, Fife KY16 9S, UK
First published on 20th December 2019
Abstract
Rare examples of molecular, dinuclear CeIII and PrIII complexes with robust Ln-coordination are accessible by use of the tetraphenolate pTP as a supporting, chelating O-donor ligand platform, pTP = [{2-(OC6H2R2-2,4)2CH}-C6H4-1,4]4− that favours the higher formal oxidation states accessible to rare earths. Two classes of complexes have been made from the platforms; one metallacyclic 2 + 2 [Ln2(pTP)2] framework with a rigid, letterbox-shaped geometry and [Ln(aryloxide)4] core, and one more flexible [(LnX)2(pTP)] with one rare earth ion at either end of the platform. The LnIII letterbox complexes have two K+ counter-cations, one of which sits inside the letterbox, binding the two central arenes of the platform sufficiently strongly that it cannot be displaced by solvent molecules (THF and pyridine) or crown ethers. Oxidation of the CeIII lettterboxes is facile and forms the unusual neutral molecular (CeIV)2 letterbox in which the CeIV reduction potential is −1.83 V vs. Fc/Fc+. The electronic structure of the Ce(III/IV) complexes was investigated using HERFD-XAS (high energy resolution fluorescence detection X-ray absorption spectroscopy).
Introduction
Cerium is a cheap, non-toxic, redox-active, early lanthanide. It is earth-abundant, being more common than copper or nickel, its salts are six times less toxic than those of iron, and it is the only rare earth with a readily accessible +III/+IV redox couple.1–4 It has been used widely as a stoichiometric oxidant in organic chemistry, as a redox active heterogeneous catalyst support, and increasingly in the development of homogeneous catalysts for a range of small molecule transformations.1–4 The CeIII/IV redox potential can be easily tuned across an extremely large window by appropriate ligand choice, for example from E° = +1.30 V vs. Fc/Fc+ for [Ce(ClO4)4] in 8 M HClO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5–8 to Epc = −2.39 V vs. Fc/Fc+ in [CeL(OtBu)2(THF)2], [L = 1, 10-di(2-tert-butyl-6-diphenylphosphiniminophenolate)ferrocene].9 In coordination and organometallic chemistry, various results have shown that the redox potential of the couple is tuneable by introducing different anionic ligands to the cerium ion10–13 or by forming ionic ‘ate’ complexes.7,14 For example, the alkali metal CeIII ate complexes, [M3(THF)n][Ce(BINOLate)3] (M = Li, Na, K, and Cs, BINOLate = 1,1′-binaphtholate) are readily oxidised to form two types of stable CeIV complexes.15,16 There is a significant research effort to find complexes that can replace expensive platinum group metal homogeneous catalysts that have traditionally been used in so much of organic chemistry due to their useful and ready two-electron reaction processes, namely oxidative addition and reductive elimination. However, the 3d-metal analogues that are proposed as their obvious cheap, less-toxic replacements undergo one-electron redox processes, hampering progress in this area. Cerium, and its earth-abundant f-block neighbours, have significant under-studied potential to act as new catalyst alternatives if their reactivity can be controlled by strongly binding ancillary ligand sets.
5–8 to Epc = −2.39 V vs. Fc/Fc+ in [CeL(OtBu)2(THF)2], [L = 1, 10-di(2-tert-butyl-6-diphenylphosphiniminophenolate)ferrocene].9 In coordination and organometallic chemistry, various results have shown that the redox potential of the couple is tuneable by introducing different anionic ligands to the cerium ion10–13 or by forming ionic ‘ate’ complexes.7,14 For example, the alkali metal CeIII ate complexes, [M3(THF)n][Ce(BINOLate)3] (M = Li, Na, K, and Cs, BINOLate = 1,1′-binaphtholate) are readily oxidised to form two types of stable CeIV complexes.15,16 There is a significant research effort to find complexes that can replace expensive platinum group metal homogeneous catalysts that have traditionally been used in so much of organic chemistry due to their useful and ready two-electron reaction processes, namely oxidative addition and reductive elimination. However, the 3d-metal analogues that are proposed as their obvious cheap, less-toxic replacements undergo one-electron redox processes, hampering progress in this area. Cerium, and its earth-abundant f-block neighbours, have significant under-studied potential to act as new catalyst alternatives if their reactivity can be controlled by strongly binding ancillary ligand sets.
Anionic oxygen-donor alkoxide and aryloxide ligands have shown most use in stabilising the higher oxidation state in the CeIII/IV redox couple,7,12,17–21 and applications of cerium reagents in molecular chemistry have largely been focused on mononuclear cerium complexes.6,20,22 We have used functionalised aryloxide ligands to support cerium(III) catalysts [Ce(LR)3] (LR = ortho-NHC-aryloxide = O(o-C6H2-tBu2-2,6-CN(C2H2)NMes)) for the formation of cyclic carbonates from CO2 and epoxides,23 and cerium(IV) catalysts [Me3SiOCe(OArP)3], (OArP = ortho-phosphino-aryloxide = OC6H2-6-tBu-4-Me-2-PPh2) for the ring-opening polymerisation (ROP) of the bio-renewable ester L-lactide.3
The development of systems that can combine two cerium cations in a molecule has received considerably less attention. However, it has been shown by EXAFS spectroscopy that the active form of the classical cerium oxidant, aqueous Ce(IV), is dinuclear,24 so the development of robust and well-defined molecular [Ce]2 complexes that can combine two readily accessible CeIV states is a potentially important target for developing catalytic cerium oxidation chemistry.25–29
Here we report the use of a tetrakis(aryloxide) ligand platform that makes the first robust molecular [Ce]2 complexes ligated by aryloxides and shows how the letterbox structures strongly favour the CeIV oxidation state. We are only able to observe two-electron separated redox states in the system, a feature not usually achievable in molecular f-block chemistry.
Results and discussion
The tetraphenol ligands H4(pTPR), [α,α,α′,α′-tetra(3-tert-butyl-5-R-2-hydroxyphenyl)-p-xylene, R = Me, tBu], (Scheme 1) are synthesised via a straightforward condensation reaction. We have previously reported the synthesis of their UIII/III and UIV/IV complexes, and others have reported the use of pTP ligands to support catalysis by both partially deprotonated potassium salts [K2H2(pTPR)],30 or VV and MoVI-imido complexes which have shown catalytic reactivity for the ring-opening polymerisation of ε-caprolactone.26,31 In the case of the potassium complexes, the authors attributed the remarkable stability of the doubly deprotonated salt [K2H2(pTPR)] to the formation of potassium–arene interactions with the central arene of the platform, and capacity for the remaining protons to bridge the two aryloxide O atoms on each side.30 In our hands, the tetra-potassium salt 1R, ([K4(pTPR)]2(THF)11, R = Me), is readily isolated from the reaction between H4(pTPMe) with four equivalents of KN′′ (N′′ = N(SiMe3)2) in THF at room temperature, Scheme 1, although we note that it is extremely sensitive to hydrolysis. It has been fully characterised, including by a single crystal X-ray diffraction study, but the syntheses of the cerium complexes below are most straightforward when samples of 1R are made in situ (R = Me, tBu).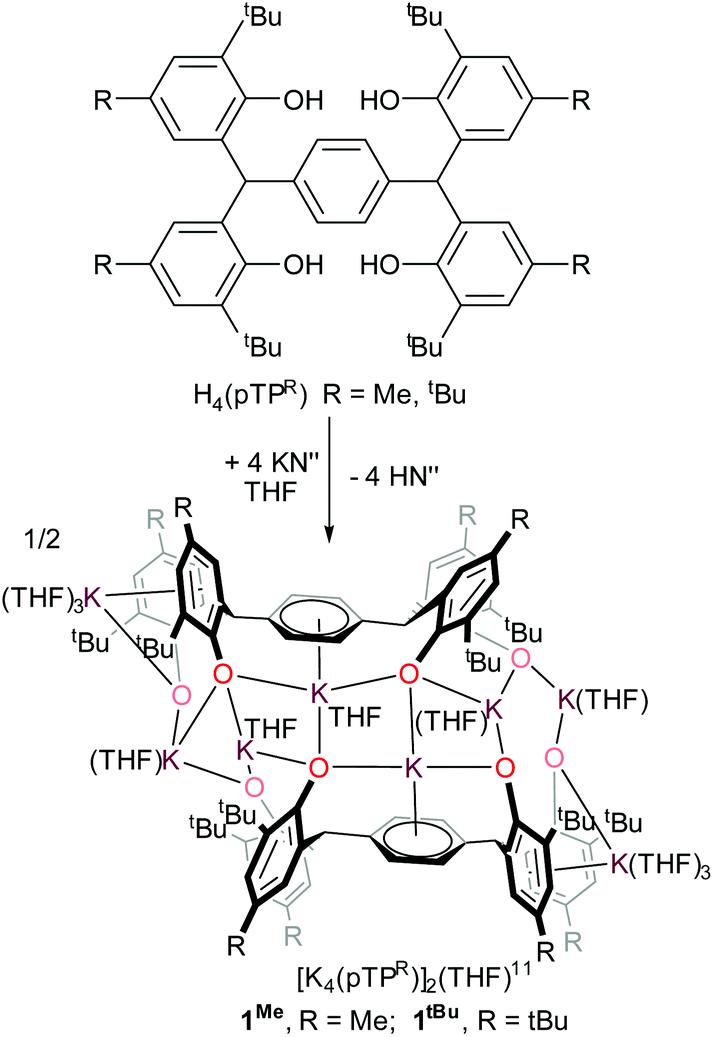 | ||
| Scheme 1 Synthesis of tetrapotassium salt 1R. | ||
The tetrapotassium salt 1Me crystallises as a THF-solvated dimer [K8(pTPMe)2(THF)11] in the monoclinic space group P2(1)/c, with four molecules in the unit cell. In the crystal structure (Fig. 1), four K+ ions (K3–K6) and four oxygen atoms (O3–O6) form a near-planar ladder-like [K4O4] skeleton. This type of coordination has previously been reported in the family of K(OAr)(sol) salts, for various aryl groups such as 2,6-dimethyl, and potassium p-halide-substituted aryloxides, [(4-X-C6H4OK)6·(dioxane)6], (X = F, Cl, Br). Each of these K+ coordinates to three phenolate oxygen atoms while oxygen atoms bridge two K+ ions. The K–O bond distances range from 2.552(4) Å (K4–O5) to 3.132(4) Å (K6–O5), falling in the reported range of K–O bonds of 2.432(6) Å to 3.194 (Å). The two ions in the middle of the skeleton of the structure, K4 and K5, coordinate to the phenyl linker via π interactions with an average distance of 2.868 Å and 3.046 Å, respectively.
 | ||
| Fig. 1 Solid-state structure of complex 1Me. Thermal ellipsoids of non-carbon atoms are shown at 30% probability. All hydrogen atoms and lattice solvent molecules are omitted for clarity. | ||
Syntheses of rare earth complexes of pTP
Reactions of complex 1R and [LnCl3(THF)2] (Ln = Ce, Pr) in a 1:1 Ln:pTP ratio in THF affords the targeted binuclear rare earth metal letterbox complexes as their ate salts [K(THF)n][KLn2(pTPR)2(THF)4] 2R-Ln, (Ln = Ce, Pr; R = Me, tBu) in good yields (∼80%). Analogous reactions of 1tBu with [LnCl3(THF)2] (Ln = Ce, Pr) in a 2:1 Ln:pTP ratio in THF affords the binuclear (LnIII)2 complexes [{LnCl(thf)n}2(pTPtBu)]. The products are purified by evaporation of the filtered solution and recrystallisation from pyridine to afford microcrystalline [{LnX(py)4}2(pTPtBu)] 3-Ln (LnX = CeCl, Ce(BH4), PrCl) in similar yields (∼80%), shown in Scheme 2.
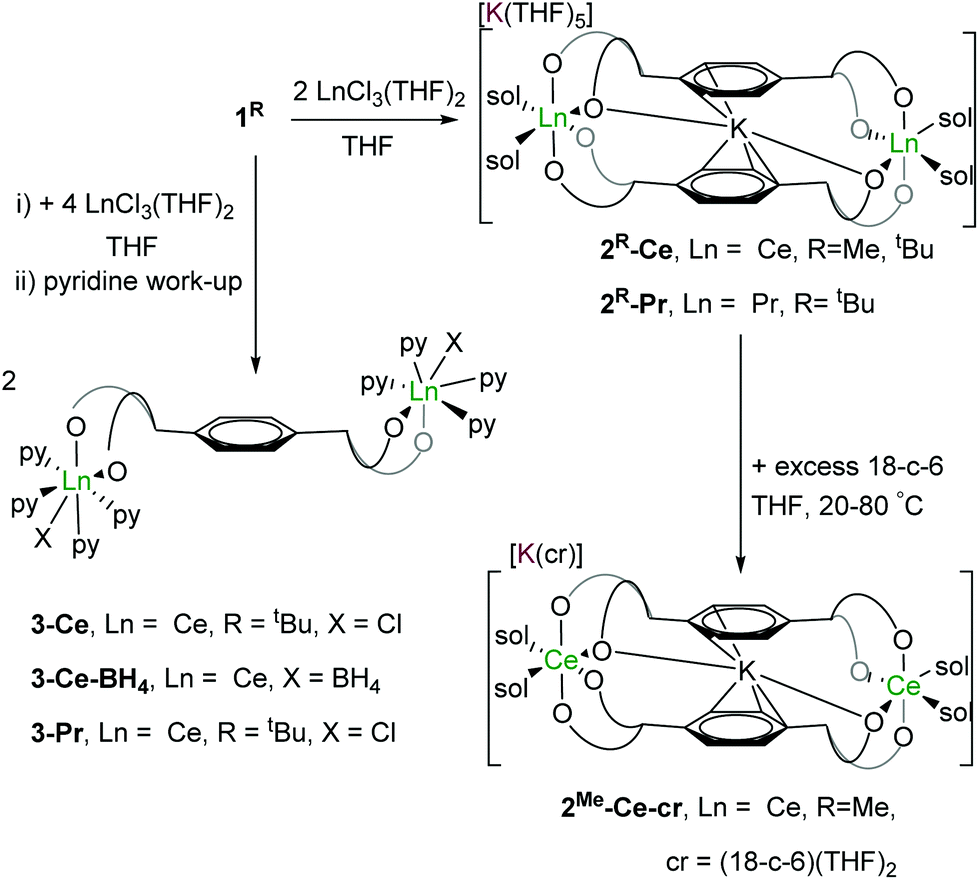 | ||
| Scheme 2 Synthesis of dinuclear CeIII and PrIII complexes. | ||
Crystals of complexes 2Me-Ce and 2tBu-Ce can be grown from concentrated THF solutions stored at −30 °C. The solid-state structure of 2Me-Ce is shown in Fig. 2. Each CeIII cation is coordinated by four oxygen atoms from the phenolate ligands and two THF molecules, displaying a distorted octahedral geometry. The Ce–OAr bonds range from 2.235(5) to 2.394(5) Å; comparable to previously reported Ce(III) aryloxide complexes such as [{Li(THF)2}Ce(BMP)2(THF)2] (BMP = 2,2′-methylenebis(6-tert-butyl-4-methylphenolate)) which have an average Ce–OAr bond length of 2.3570 Å.9,15,16 One K+ counter-cation sits in the lattice, coordinated by seven THF molecules, while the other occupies the centre of the ‘letterbox’ shaped rectangular void formed by the two Ce ions and the two pTP ligand platforms. The K+ inside the letterbox has close contacts to one aryloxide oxygen atom from each Ce-coordinated pTP with a distance of 3.021(6) Å and an approximately η6-coordination to both phenyl groups of the platform giving a bent bis(arene) sandwich geometry, (right, Fig. 2). The average distance between the K+ and two ring centroids is 2.969 Å while the dihedral angle between the planes of the two phenyl rings, denoted θ in Fig. 2, is 62.94° (2Me-Ce) and 72.08° (2tBu-Ce). The inter-centroid distance between the phenyl rings is calculated to be 5.500(6) Å and 5.382(4) Å, respectively.
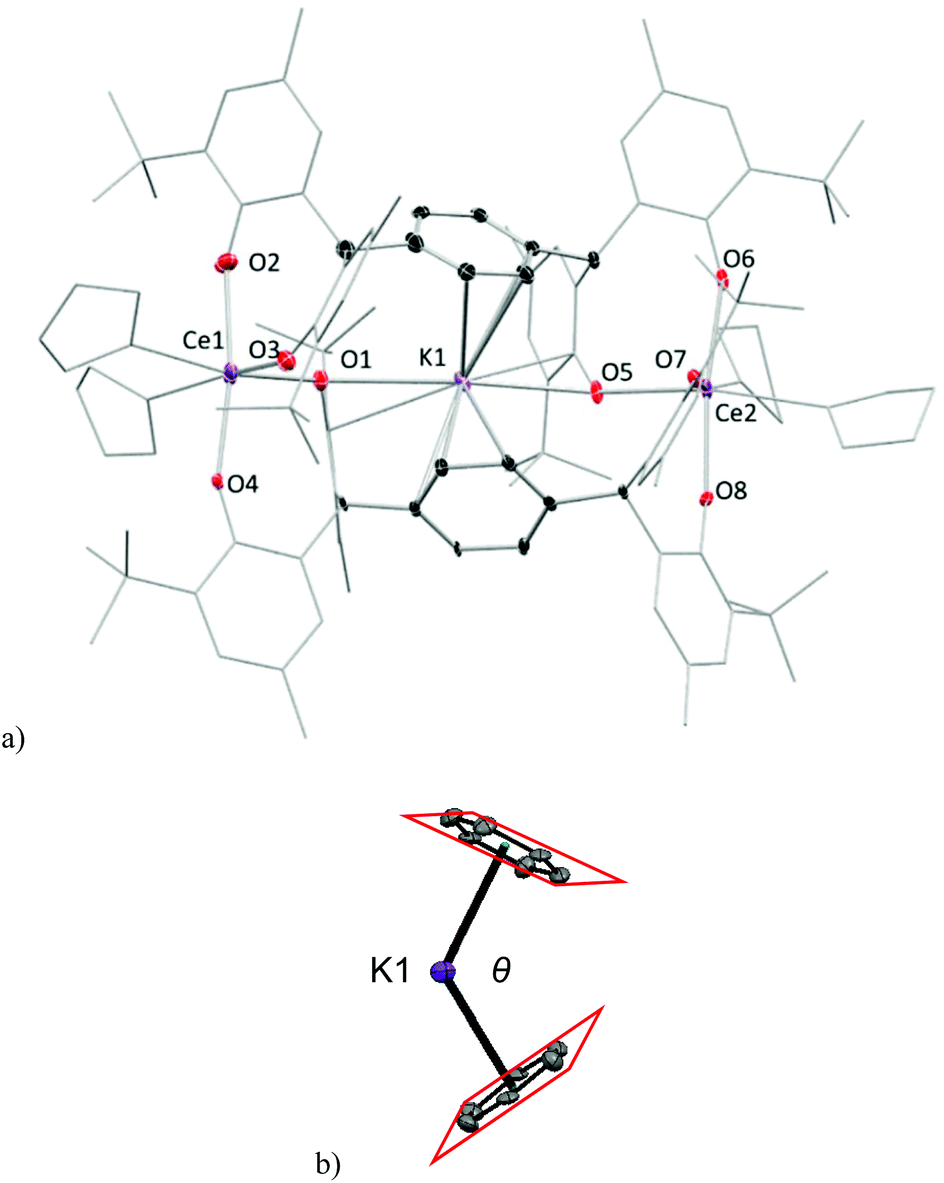 | ||
| Fig. 2 Solid state structure of complex 2Me. (a) core of anion with K(THF)7 counter-cation and all H and two lattice THF solvent molecules omitted for clarity. Coordinated solvent shown as wireframe, and ligands shown as capped stick for clarity; thermal ellipsoids of other atoms are shown at 30% probability. (b) Bis(η6-arene) coordination of the endo K+ cation with the angle between the two arene planes labelled as θ. | ||
The complexes of 2tBu-Pr and 2tBu-Ce are essentially isostructural (see ESI†) in accordance with the similar ionic radii of Ce3+ and Pr3+ cations. The Pr–OAr bonds (2.348(10) Å and 2.396(7) Å) are longer than the reported values (average 2.16 Å) in complex [Pr(O-2,6-iPr2C6H3)3(THF)2],32 but shorter than those in the complexes of [(EtZn)3(THF)2(BINOLate)3-Pr(THF)] at 2.412(32) Å.32,33,33 The dihedral angle between the two phenyl rings is θ = 71.7°, which is comparable to that in complex 2tBu-Ce.
The complexes 2-Ce are paramagnetic so 1H NMR spectra of the complexes contain broadened and shifted, but still assignable, resonances for the ligands. The CeIII complexes should have one unpaired electron on each f-block cation. Accordingly, they were analysed by EPR spectroscopy. As anticipated, no EPR signal was visible in solutions of 2tBu-Ce at room temperature or 100 K. However, an EPR resonance was observed at 9 K for a solid-state sample; a weak resonance is observed at 200 mT which is attributed to the disallowed ΔS = 2 half-field signal that corresponds to the S = −1 to S = 1 state of the [CeIII2] system, see ESI.†
The letterbox-encapsulated K+ is remarkably difficult to remove: addition of pyridine or excess 18-crown-6 in THF solution to 2R-Ce yields the pyridine-solvated 2R-Ce-py, [K(py)5][KCe2(pTPR)2(py)4] (R = Me, tBu) (see ESI†), or external-K-18-crown-6 solvate 2Me-Ce-cr, [K(18-c-6)(THF)2][KCe2(pTPR)2(THF)4] (R = Me), respectively. Almost no change in the 1H NMR chemical shifts other than those of the solvating donor ligands is observed even when the THF solution of 2Me-Ce-cr is heated at 60 °C for 24 hours. The structures of 2R-Ce-py and 2Me-Ce-cr (see ESI†) have been confirmed by single crystal XRD.
Single crystals of complex 3tBu-Ln, [{LnX(py)4}2(pTPtBu)] (LnX = CeCl, PrCl), suitable for X-ray diffraction were grown by vapour diffusion of hexane into a saturated pyridine solution at room temperature. The solid-state structure of 3tBu-Ce is shown in Fig. 3a. That of the pyridine solvate of 3tBu-Ce, grown from a saturated pyridine solution at −30 °C, is shown in the ESI.†
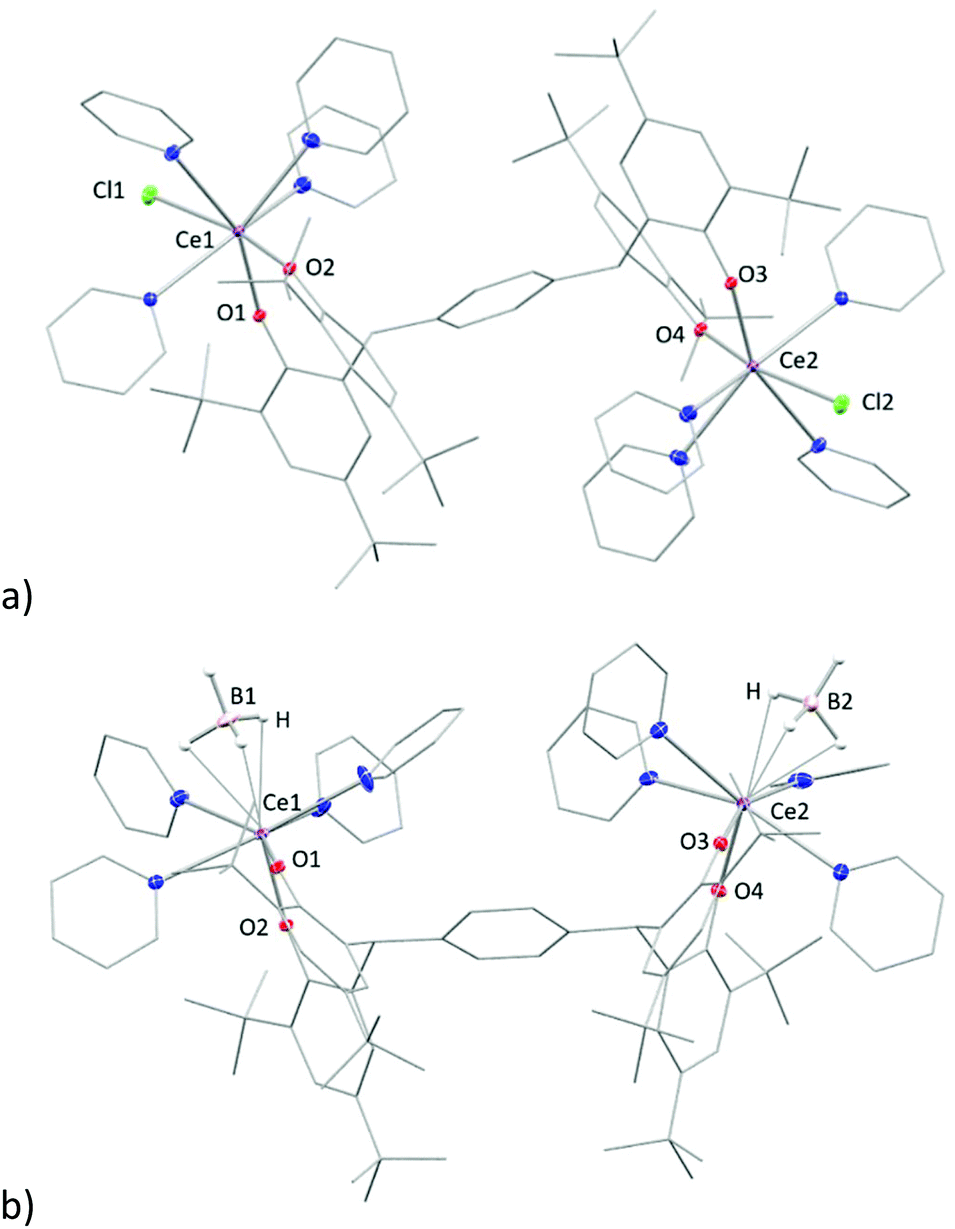 | ||
| Fig. 3 Solid state structure of complex (a) 3-Ce (b) 3-Ce-BH4. Thermal ellipsoids are shown at 30% probability. All hydrogen atoms (except for BH4) and solvents are omitted for clarity. Selected bond lengths in (a): Ln1–Cl1: 2.7789(6) Å (Ce), 2.7601(14) Å (Pr). | ||
The Ce and Pr analogues are isostructural (see ESI†), with a trans-disposition of the two metal bis(aryloxide) fragments, on either side of the phenyl-linker, displaying a trans-configuration. The Ln1–Cl1 bond in 3-Ce is 2.7789(6) Å, 0.02 Å longer than in 3-Pr in line with the similarity between their ionic radii. However, a cis-configuration is observed in the borohydride analogue, [{Ce(BH4)(py)4}2(pTPtBu)] (Fig. 3b), where two borohydride groups reside on the same side of the phenyl ring. The average Ce–B distance of 2. 832 Å is slightly longer than the reported value of 2.678(6) Å and 2.704(7) Å in the complex [Ce(BH4)2(THF)5][BPh4].34
The CeIII complexes 2-Ce are extremely sensitive to oxidation by even trace amounts of O2. Accordingly, stoichiometric reactions with a variety of oxidants, such as I2 or CuX2 (X = Cl or OTf), leads to the instant formation of intensely blue-coloured products characterised as the CeIV complexes 4R, [Ce2(pTPR)2(THF)4] (R = Me, tBu) (Scheme 3). The blue colour observed in these complexes is attributed to a ligand–π to vacant Ce-4f charge-transfer band (LMCT) that is observed in many CeIV complexes.12,17,35,36 In the UV-Vis spectrum of a THF solution of 4tBu the broad absorption band is centred at 576 nm (see ESI†), a relatively low energy compared to other CeIV aryloxide complexes (e.g. 487 nm in the complex [Li3(thf)3CeIV(BINOLate)Cl]).20,37
 | ||
| Scheme 3 Concerted two-electron oxidation of complex 2 with oxidants. | ||
Redox chemistry
Reactions of 2R-Ce with a range of oxidants have been studied. The cleanest oxidations of 2R-Ce are with CuX2 (X = Cl or OTf), affording Cu0 metal and KX by-products. Reactions with other oxidants (I2, XeF2 and HgX2 (X = Cl, I, OAc)) are described in the ESI.† This reaction can be monitored by 1H NMR spectroscopy as the paramagnetically shifted resonances of the starting material 2 disappear immediately and are replaced by a set of diamagnetic ligand resonances attributable to a CeIV/CeIV complex.The reaction with just a single equivalent of I2 or CuX2 (X = Cl or OTf) generates the new (CeIV)2 product 4R and unreacted (CeIII)2 starting material 2R-Ce in equal amounts. This represents a rare, concerted, two-electron redox process for a single molecular lanthanide complex.
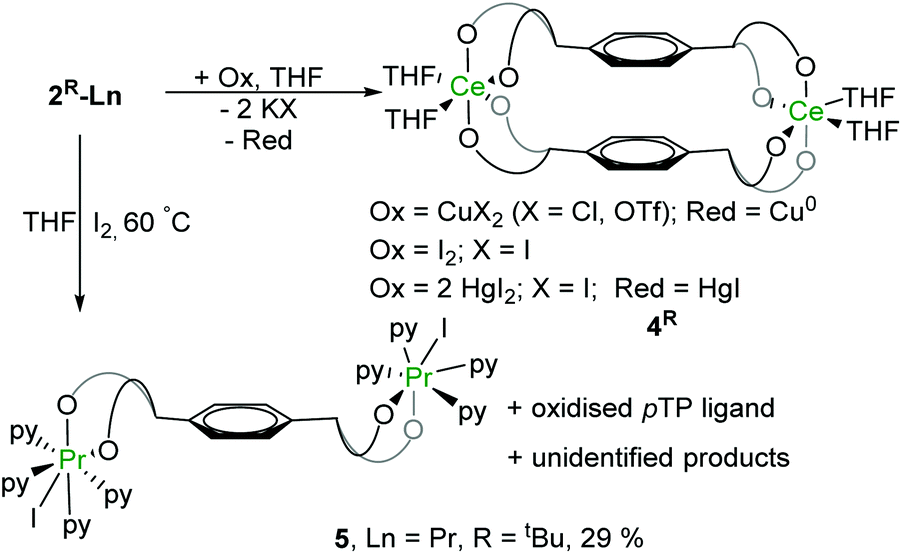
Single crystals of 4tBu were grown by slow evaporation of hexane into a concentrated THF solution but the diffraction data are of poor quality, so only the connectivity can be deduced (Fig. 4). Complex 4Me was analysed only by NMR spectroscopy, see ESI.† In the molecular structure of 4tBu each Ce atom is coordinated by four phenolate oxygen atoms and two THF molecules. With the loss of K+ from the letterbox, the dihedral angle between the two central arene rings decreases to 25° while their inter-centroid distance decreases to 4.538 Å. The X-ray data are poor, and the precision of the metrics is not reliable, but the average Ce–OAr bond length is around 2.15 Å so appreciably shorter than that in Ce(III) complexes (2.382 Å for 2Me-Ce, 2.350 Å for 2tBu-Ce), consistent with the decrease in Ce radius upon oxidation to f0 (from 1.01 Å to 0.87 Å).38
 | ||
| Fig. 4 Solid-state structure of complex 4tBu. All hydrogen atoms and lattice solvent molecules are omitted for clarity. The inter-arene distance (4.538 Å) is represented by the blue double-headed arrow. | ||
The cyclic voltammograms of CeIII complex 2tBu-Ce shows a small current increase corresponding to an irreversible oxidation at +0.76 V which is tentatively assigned to the CeIII–IV process, but is increasingly difficult to observe with additional scans; this may be the result of decomposition of the complex in supporting electrolyte solution (see ESI† for details). However, the CeIV complex is considerably more robust, and cyclic voltammetry of a THF solution of 4tBu using 0.1 M [nBu4N][PF6] as supporting electrolyte shows a quasi-reversible reduction at Epc = −1.31 V with a small return oxidation wave at Epa = −1.02 V. This may correspond to the one-electron reduction of one CeIV ion. A larger reduction wave is observed at Epc = −1.83 V vs. Fc/Fc+ (Fig. 5). It is not clear yet whether in the electrochemical experiment, the return oxidation wave is due to an impurity or a process occurring at the ligand first. It has been observed on multiple occasions in repeated measurements and using different batches of material, and in two different laboratories. The O-donor ligands, and the observed higher stability of the CeIV complex 4 compared with the CeIII complex 2 all support the expected stabilisation of the +4 oxidation state in the complex, although more in-depth characterisation is warranted. Our previous electrochemical analyses of the free ligand, and the potassium salt, were not helpful.39 The couples can be compared to a range of related O-ligated CeIV complexes such as [Ce(OtBu)4(py)2] (Epc = −1.99 V vs. Fc/Fc+ in DCM),40 Ce(2-(tBuNO)py)4 (Epc = −1.95 V vs. Fc/Fc+ in DCM),12,40 CeL(OtBu)4 (Epc = −2.39 V vs. Fc/Fc+ in THF)9 and imidophosphorane supported complexes Ce(NPPip3)4 (reduction range of −2.30 < Epc < −2.47 V vs. Fc/Fc+ in THF).10
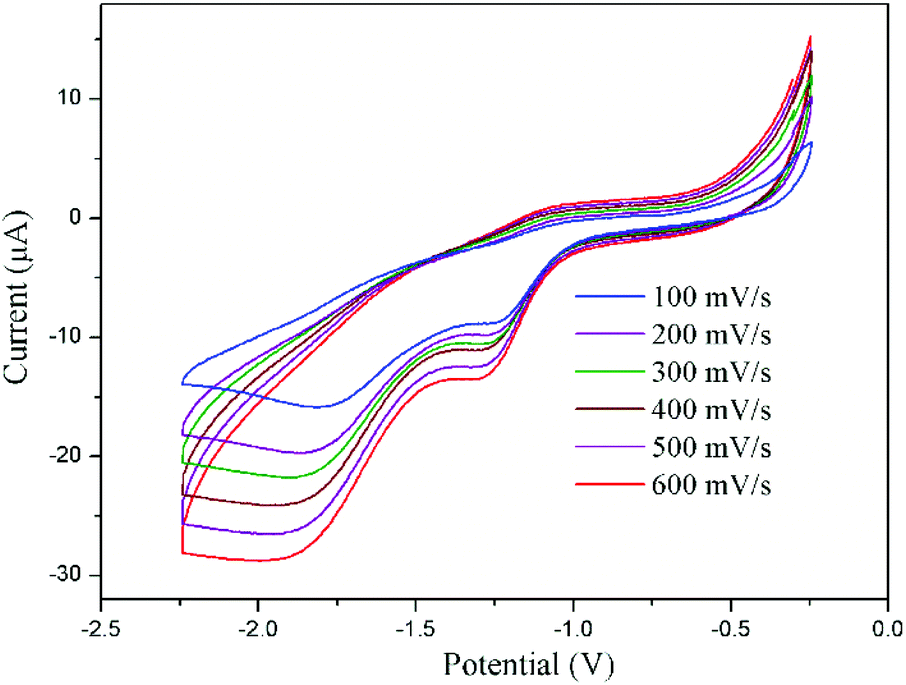 | ||
| Fig. 5 Cyclic voltammogram of complex 4tBu at different scan rates versus Fc/Fc+ measured in THF with 0.1 M [nBu4N][PF6]. | ||
In order to obtain a more chemically accurate view on the Ce complex oxidation state, HERFD-XAS (high energy resolution fluorescence detection X-ray absorption spectroscopy) was employed. HERFD-XAS provides a method to probe the 5d density of states in detail. Specifically, this enables a fingerprinting determination of whether a complex can be formally considered Ce(III) vs. Ce(IV). HERFD-XAS spectra (Fig. 6) of sample 2tBu-Ce shows a single peak, indicative of formal Ce(III). Sample 4tBu, however, shows two main peak features approximately 10 eV apart. This doublet peak is indicative of formal Ce(IV), and is also observed in CeO2, which serves as a fingerprinting standard (Fig. 6). Thus, electronically, 2tBu can be referred to as Ce(III) and 4tBu as Ce(IV), as it contains considerable f0 character.41,42
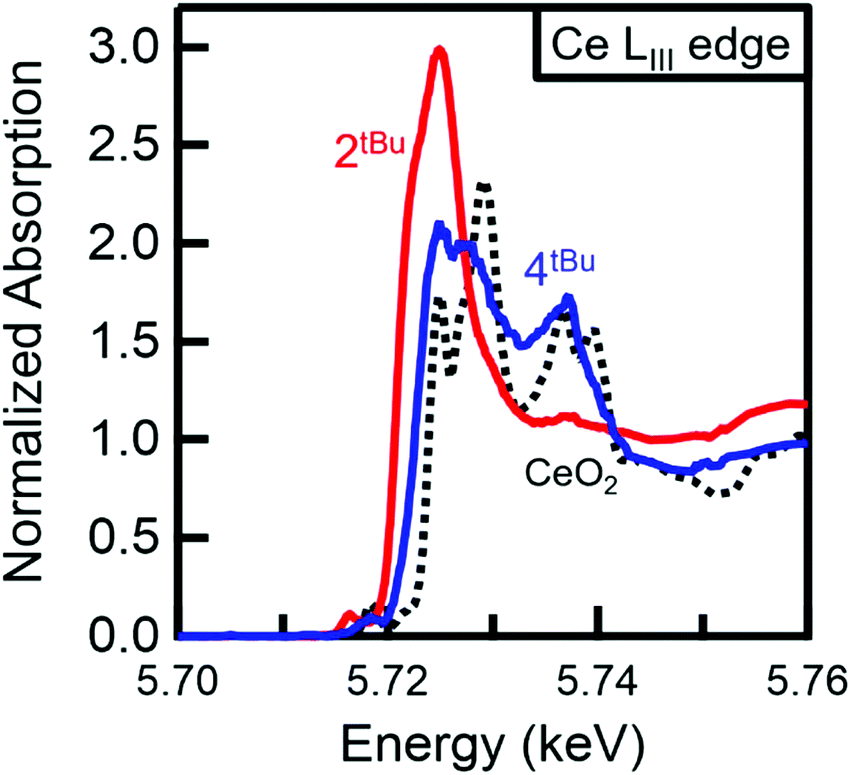 | ||
| Fig. 6 Normalised HERFD-XAS spectra at the Ce LIII absorption edge. The increased edge energy and doublet peak for 4tBu (blue) compared with 2tBu-Ce (red) confirm the Ce(IV) oxidation state of 4tBu. The doublet peaks of 4tBu also match those observed for CeO2 (black, dotted), which serves as a Ce(IV) fingerprinting standard. Peak splitting in the CeO2 HERFD spectrum has been attributed to 5d state splitting.43 | ||
Under certain conditions, praseodymium can exist in the formal PrIV oxidation state in some solid-state compounds such as NaPrF5,44 PrF4,45 and Pr oxides or even in the +V oxidation state in the gas-phase.46–50 However, molecular PrIV complexes remain an elusive and interesting target. Recent reports on the synthesis and isolation of the TbIV complexes51,52 have shown great potential for the stabilisation of rare earth metals in the +IV oxidation state with bespoke ligand systems. Here, unlike the cerium counterpart, the (PrIII)2 complex 2tBu-Pr is inert to most of the oxidants under the same reaction conditions. No reactivity with oxidants such as O2, CuX2 (X = Cl, OTf), Ph3CCl or benzoquinone was observed in solutions monitored by 1H NMR spectroscopy. Addition of I2 to solutions of complex 2tBu-Pr showed no reaction at room temperature, but the mixture changes colour from brown to green when heated at 60 °C for 8 hours. A reaction monitored by solution NMR spectroscopy shows the full transformation of starting material into several different products (see ESI†), from which, work-up yields a white powder was that is characterised as the dinuclear PrIII complex [{PrI(thf)3}2(pTPR)] (R = tBu), 5, with a yield of 29% (Fig. 7). It is evident from this that one of the chelating ligand platforms has been de-coordinated, and the other material that is isolated from the reaction is the product of ligand oxidation, a bicyclic ether that we have also characterised by X-ray crystallography (see ESI†).53
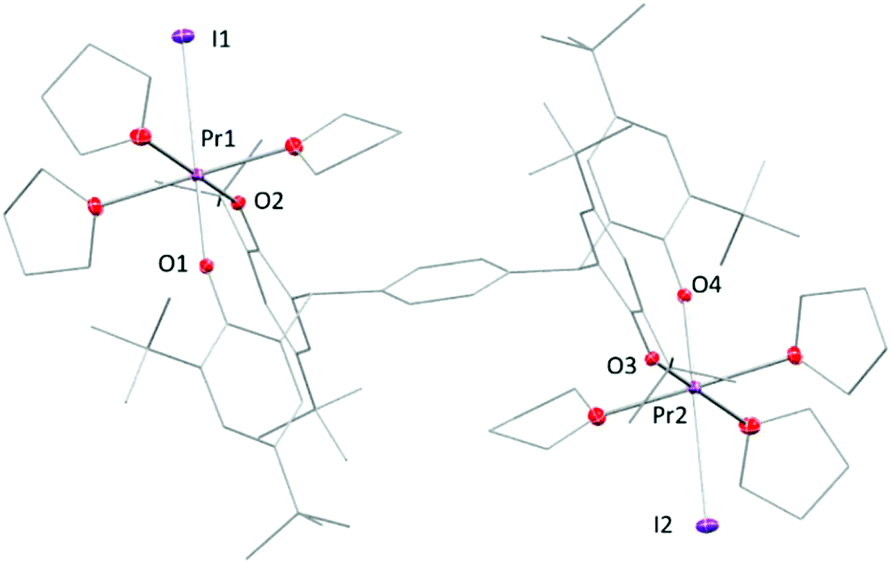 | ||
| Fig. 7 Solid-state structure of complex 5. All hydrogen atoms and lattice solvent molecules are omitted for clarity. | ||
In the solid-state structure of 5, each Pr atom displays a pseudo-octahedral configuration with three THF molecules, two phenolate oxygen atoms and one iodine atom, similarly to 3-Pr. The two metal centres are bonded to the opposite ends of the tetraphenolate ligand, in a trans-geometry. The Pr–I bond is 3.1697(5) Å while two Pr–O bonds are labelled in the figure as 2.176(4) Å and 2.202(4) Å. These are slightly longer than those observed in the homoleptic aryloxide [Pr(O-2,6-iPr2C6H3)3(THF)232] but are ∼0.1 Å shorter than the average length (2.30 Å) measured in 2tBu-Pr.
Conclusions
In summary, we have successfully synthesised a series of tetraphenolate supported bi-metallic CeIII and PrIII complexes with robust Ln-coordination and which favour the higher formal oxidation states accessible for rare earths. Two types of geometry are accessible; complexes with the new, rigid, letterbox-shaped geometries and [Ln(aryloxide)4] cores in a 2 + 2 [Ln2(pTP)2] framework, and flexible complexes with one rare earth ion at either end of the single ligand platform in the form [(LnX)2(pTP)] are readily accessible. The binding of one K+ cation inside the letterbox shape of the [Ln2(pTP)2] complexes in a bis(arene) motif is sufficiently strong that it cannot be extracted by crown ethers, although it can be removed through salt elimination by oxidation of the complex to the neutral CeIV letterbox complex. Solution electrochemical experiments showed that the CeIV cation is particularly well stabilised by the ligand with a measured CeIV reduction potential of −1.83 V vs. Fc/Fc+. HERFD-XAS data on the CeIV complex confirms the formal +4 oxidation state for complex 4tBu based on a doublet peak that indicates considerable f0 character. Chemical oxidation reactions show that only two-electron redox processes occur at the bimetallic letterbox-shaped complexes. Oxidation of the PrIII complexes to target molecular PrIV yields products of ligand oxidation although there may be opportunities for judicious oxidant choice to enable the stabilisation of reaction intermediates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 740311, PLA), and the support of the Technische Universität München – Institute for Advanced Study, funded by the German Excellence Initiative. K. W. thanks the China Scholarship Council (CSC) for a postgraduate fellowship. We thank the EPSRC CRITICAT Centre for Doctoral Training (Ph.D. studentships to S. G. and M. C.; Grant No. EP/L016419/1). We also thank the University of Edinburgh and the EPSRC-UK for funding under grants EP/N022122/1 and EP/M010554/1. T. L. was supported by the NSF under the Center for Enabling New Technologies through Catalysis CCI (CENTC) (CHE-1205189). We thank Dr M. Seymour, University of Edinburgh, and Prof K. Meyer, University of Erlangen- Nuremberg, for the EPR data. Work at Lawrence Berkeley National Laboratory was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences Heavy Element Chemistry Program of the U.S. Department of Energy (DOE) at LBNL under Contract No. DE-AC02-05CH11231. The Stanford Synchrotron Radiation Lightsource is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. We thank Dimosthenis Sokaras and Julian Rees for their assistance with HERFD-XAS data collection and Pieter Glatzel and the European Synchrotron Radiation Facility for the use of their Ge(331) analyzer crystals.Notes and references
- V. Sridharan and J. C. Menendez, Chem. Rev., 2010, 110, 3805–3849 CrossRef CAS PubMed
.
- V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1862–1891 CrossRef CAS
- F. Sinclair, J. A. Hlina, J. A. L. Wells, M. P. Shaver and P. L. Arnold, Dalton Trans., 2017, 46, 10786–10790 RSC
- Y.-M. So, Y. Li, K.-C. Au-Yeung, G.-C. Wang, K.-L. Wong, H. H. Y. Sung, P. L. Arnold, I. D. Williams, Z. Lin and W.-H. Leung, Inorg. Chem., 2016, 55, 10003–10012 CrossRef CAS PubMed
- M. S. Sherrill, C. B. King and R. C. Spooner, J. Am. Chem. Soc., 1943, 65, 170–179 CrossRef CAS
- L. A. Solola, P. J. Carroll and E. J. Schelter, J. Organomet. Chem., 2018, 857, 5–9 CrossRef CAS
- N. A. Piro, J. R. Robinson, P. J. Walsh and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed
- E. M. Broderick, P. S. Thuy-Boun, N. Guo, C. S. Vogel, J. Sutter, J. T. Miller, K. Meyer and P. L. Diaconescu, Inorg. Chem., 2011, 50, 2870–2877 CrossRef CAS PubMed
- N. T. Rice, J. Su, T. P. Gompa, D. R. Russo, J. Telser, L. Palatinus, J. Bacsa, P. Yang, E. R. Batista and H. S. La Pierre, Inorg. Chem., 2019, 58, 5289–5304 CrossRef CAS PubMed
- C. Morton, N. W. Alcock, M. R. Lees, I. J. Munslow, C. J. Sanders and P. Scott, J. Am. Chem. Soc., 1999, 121, 11255–11256 CrossRef CAS
- J. A. Bogart, A. J. Lewis, S. A. Medling, N. A. Piro, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2013, 52, 11600–11607 CrossRef CAS PubMed
- Y. M. So, Y. Li, K. C. Au-Yeung, G. C. Wang, K. L. Wong, H. H. Y. Sung, P. L. Arnold, I. D. Williams, Z. Y. Lin and W. H. Leung, Inorg. Chem., 2016, 55, 10003–10012 CrossRef CAS PubMed
- L. Clark, M. G. Cushion, H. E. Dyer, A. D. Schwarz, R. Duchateau and P. Mountford, Chem. Commun., 2010, 46, 273–275 RSC
- J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS PubMed
- J. R. Levin, W. L. Dorfner, P. J. Carroll and E. J. Schelter, Chem. Sci., 2015, 6, 6925–6934 RSC
- B. D. Mahoney, N. A. Piro, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2013, 52, 5970–5977 CrossRef CAS PubMed
- J. A. Bogart, C. A. Lippincott, P. J. Carroll, C. H. Booth and E. J. Schelter, Chem. – Eur. J., 2015, 21, 17850–17859 CrossRef CAS PubMed
- D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Dalton Trans., 2017, 46, 6265–6277 RSC
- J. R. Robinson, P. J. Carroll, P. J. Walsh and E. J. Schelter, Angew. Chem., Int. Ed., 2012, 51, 10159–10163 CrossRef CAS PubMed
- P. Drose, A. R. Crozier, S. Lashkari, J. Gottfriedsen, S. Blaurock, C. G. Hrib, C. Maichle-Mossmer, C. Schadle, R. Anwander and F. T. Edelmann, J. Am. Chem. Soc., 2010, 132, 14046–14047 CrossRef CAS PubMed
- Y. M. So and W. H. Leung, Coord. Chem. Rev., 2017, 340, 172–197 CrossRef CAS
- P. L. Arnold, R. W. F. Kerr, C. Weetman, S. R. Docherty, J. Rieb, F. L. Cruickshank, K. Wang, C. Jandl, M. W. McMullon, A. Pöthig, F. E. Kühn and A. D. Smith, Chem. Sci., 2018, 9, 8035–8045 RSC
- A. Ikeda-Ohno, S. Tsushima, C. Hennig, T. Yaita and G. Bernhard, Dalton Trans., 2012, 41, 7190–7192 RSC
- S. Shirase, K. Shinohara, H. Tsurugi and K. Mashima, ACS Catal., 2018, 8, 6939–6947 CrossRef CAS
- M. Paul, S. Shirase, Y. Morimoto, L. Mathey, B. Murugesapandian, S. Tanaka, S. Itoh, H. Tsurugi and K. Mashima, Chem. – Eur. J., 2016, 22, 4008–4014 CrossRef CAS PubMed
- M. P. Coles, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert, Z. Li and A. V. Protchenko, Dalton Trans., 2010, 39, 6780–6788 RSC
- G.-C. Wang, H. H. Y. Sung, I. D. Williams and W.-H. Leung, Inorg. Chem., 2012, 51, 3640–3647 CrossRef CAS PubMed
- A. Mustapha, J. Reglinski and A. R. Kennedy, Inorg. Chim. Acta, 2009, 362, 1267–1274 CrossRef CAS
- J. J. Zhang, C. L. Jian, Y. Gao, L. Wang, N. Tang and J. C. Wu, Inorg. Chem., 2012, 51, 13380–13389 CrossRef CAS PubMed
- Y. Al-Khafaji, T. J. Prior, M. R. J. Elsegood and C. Redshaw, Catalysts, 2015, 5, 1928–1947 CrossRef CAS
- D. M. Barnhart, D. L. Clark, J. C. Gordon, J. C. Huffman, R. L. Vincent, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 3487–3497 CrossRef CAS
- A. J. Wooten, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2008, 130, 7407–7419 CrossRef CAS PubMed
- T. Arliguie, L. Belkhiri, S. E. Bouaoud, P. Thuery, C. Villiers, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2009, 48, 221–230 CrossRef CAS PubMed
- J. A. Bogart, A. J. Lewis, M. A. Boreen, H. B. Lee, S. A. Medling, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2015, 54, 2830–2837 CrossRef CAS PubMed
- A. Vogler and H. Kunkely, Inorg. Chim. Acta, 2006, 359, 4130–4138 CrossRef CAS
- G. Mandel, R. P. Bauman and E. Banks, J. Chem. Phys., 1960, 33, 192–193 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- J. A. Wells, M. L. Seymour, M. Suvova and P. L. Arnold, Dalton Trans., 2016, 45, 16026–16032 RSC
- U. J. Williams, D. Schneider, W. L. Dorfner, C. Maichle-Mossmer, P. J. Carroll, R. Anwander and E. J. Schelter, Dalton Trans., 2014, 43, 16197–16206 RSC
- C. Paun, O. V. Safonova, J. Szlachetko, P. M. Abdala, M. Nachtegaal, J. Sa, E. Kleymenov, A. Cervellino, F. Krumeich and J. A. van Bokhoven, J. Phys. Chem. C, 2012, 116, 7312–7317 CrossRef CAS
- A. Bianconi, A. Marcelli, H. Dexpert, R. Karnatak, A. Kotani, T. Jo and J. Petiau, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 806–812 CrossRef CAS PubMed
- A. Kotani, K. O. Kvashnina, S. M. Butorin and P. Glatzel, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 210–215 CrossRef CAS
- L. B. Asprey and T. K. Keenan, J. Inorg. Nucl. Chem., 1961, 16, 260–262 CrossRef CAS
- T. Vent-Schmidt and S. Riedel, Inorg. Chem., 2015, 54, 11114–11120 CrossRef CAS PubMed
- B. G. Hyde, E. E. Garver, U. E. Kuntz and L. Eyring, J. Phys. Chem., 1965, 69, 1667–1675 CrossRef CAS
- C. L. Sieglaff and L. Eyring, J. Am. Chem. Soc., 1957, 79, 3024–3026 CrossRef CAS
- Q. Zhang, S.-X. Hu, H. Qu, J. Su, G. Wang, J.-B. Lu, M. Chen, M. Zhou and J. Li, Angew. Chem., Int. Ed., 2016, 55, 6896–6900 CrossRef CAS PubMed
- S.-X. Hu, J. Jian, J. Su, X. Wu, J. Li and M. Zhou, Chem. Sci., 2017, 8, 4035–4043 RSC
- J. Jian, Q. Zhang, X. Wu and M. Zhou, J. Phys. Chem. A, 2017, 121, 7861–7868 CrossRef CAS PubMed
- C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS PubMed
- N. T. Rice, I. A. Popov, D. R. Russo, J. Bacsa, E. R. Batista, P. Yang, J. Telser and H. S. La Pierre, J. Am. Chem. Soc., 2019, 141, 13222–13233 CrossRef CAS PubMed
- C. Redshaw, M. J. Walton, M. R. J. Elsegood, T. J. Prior and K. Michiue, RSC Adv., 2015, 5, 89783–89796 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization of new compounds, crystallographic and computational details and NMR and HRMS spectra. CCDC 1919080 (1Me), 1919083 (2Me), 1919084 (2Me-py), 1919085 (2Me-18-c-6), 1919086 (2tBu-Pr), 1919081 (3tBu-Ce), 1920898 (3tBu-Ce-BH4), 1919082 (3tBu-Pr), 1919087 (4tBu), 1919088 (5tBu-Pr) and 1919089 (5tBu-byproduct). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03291f |
| This journal is © The Royal Society of Chemistry 2020 |