
Dry hydrated potassium carbonate for effective CO2 capture

Abstract
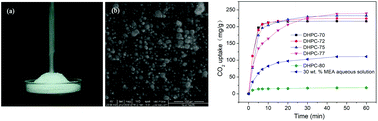
Dry hydrated potassium carbonate (DHPC) is a free-flowing powder prepared by uniformly mixing hydrated K2CO3 and hydrophobic nanosilica. We demonstrated that DHPCs can absorb CO2 rapidly because of their high surface-to-volume ratio. Their CO2 capture performance is superior to that of 30 wt% monoethanolamine (MEA), an industry standard. DHPC-75 (containing 75 wt% K2CO3) has a CO2 uptake capacity of 233 mg g−1 (90% saturation uptake within 13 min), higher than the widely used 30 wt% MEA aqueous solution (111 mg g−1, 90% saturation uptake within 25 min). DHPC-75 also exhibits an excellent cycling performance, thus becoming a promising candidate for practical application.
- This article is part of the themed collection: Inorganic Porous and Layered Material
 Please wait while we load your content...
Please wait while we load your content...

