
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrogenation of fluorinated arenes using rhodium nanoparticles on molecularly modified silica†
Souha
Kacem
ab,
Meike
Emondts
bc,
Alexis
Bordet
 *a and
Walter
Leitner
*ab
*a and
Walter
Leitner
*ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: alexis.bordet@cec.mpg.de; walter.leitner@cec.mpg.de
bInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
cDWI-Leibniz Institute for Interactive Materials, Forckenbeckstrasse 50, 52056 Aachen, Germany
First published on 26th October 2020
Abstract
The production of fluorinated cyclohexane derivatives is accomplished through the selective hydrogenation of readily available fluorinated arenes using Rh nanoparticles on molecularly modified silica supports (Rh@Si–R) as highly effective and recyclable catalysts. The catalyst preparation comprises grafting non-polar molecular entities on the SiO2 surface generating a hydrophobic environment for controlled deposition of well-defined rhodium particles from a simple organometallic precursor. A broad range of fluorinated cyclohexane derivatives was shown to be accessible with excellent efficacy (0.05–0.5 mol% Rh, 10–55 bar H2, 80–100 °C, 1–2 h), including industrially relevant building blocks. Addition of CaO as scavenger for trace amounts of HF greatly improves the recyclability of the catalytic system and prevents the risks associated to the presence of HF, without compromising the activity and selectivity of the reaction.
Introduction
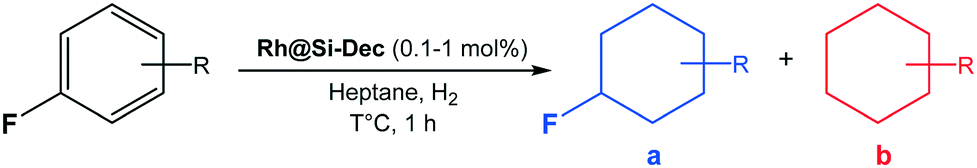
While fluorinated cycloalkanes are of great interest as building blocks for the production of materials,1 agrochemicals2 and pharmaceuticals,3 their synthesis remains particularly challenging. The most prominent methods for alkane fluorination4 typically require several steps and the use of stoichiometric quantities of toxic and difficult-to-handle reagents such as elemental fluorine,5 cesium fluoroxysulfate,6 anhydrous hydrofluoric acid,1b or cobalt trifluoride for polyfluorination.7 Recent progress in catalytic fluorination was achieved using homogeneous catalysts (e.g. Cu,8 Mg,9 Ag10) in the presence of complex fluoride sources (e.g. AgF/TBAF·3H2O,9 Selectfluor®8,10). In contrast, fluorinated arenes are widely accessible through a variety of very efficient routes,11 especially thanks to recent advances in late stage fluorination techniques.12 Consequently, the hydrogenation of fluorinated arenes appears as an attractive and benign approach to access fluorocycloalkane derivatives.13 However, the competing hydrodefluorinating pathway significantly reduces the efficiency and selectivity of this transformation, largely hampering its application and exploration so far (Scheme 1).14a In the past decades, several studies investigated the use of heterogeneous catalysts for the hydrogenation of fluorinated arenes to fluorinated cyclohexanes.13a,14a–c In many cases, the C–F bond cleavage was predominant and the catalysts were used mostly for hydrodefluorination.14 As a notable exception, a study by Stanger and Angelici evidenced the possibility of using non-polar solvents to limit hydrodefluorination.14a | ||
| Scheme 1 (a) Scheme illustrating the catalytic hydrogenation of fluorinated arenes and the competing hydrodefluorination pathway. Examples of applications for fluorinated cyclohexane derivatives: (b) 5-HT2 (c) receptor modulator,3b (c) potassium channel modulator.3d | ||
In heptane as solvent, the authors were able to hydrogenate fluorobenzene to a mixture of 73% fluorocyclohexane and 27% cyclohexane using large (10–15 nm) and polydisperse Rh NPs immobilized on SiO2. They showed that the release of HF can promote the conversion of fluorocyclohexane to cyclohexane with negative impact on selectivity. More recently, Glorius et al. reported the use of a Rh-cyclic(alkyl)(amino)carbene complex for the selective hydrogenation of various fluoroaromatics to fluorocycloalkanes using hexane as solvent and molecular sieves or SiO2 as additives.15 While the present study was in progress, the Glorius group reported that the Rh–carbene complex acts as pre-catalyst (in agreement with studies by Bullock et al.),16 decomposing under reaction conditions to form polydisperse Rh NPs (2–10 nm) on SiO2.17 It was demonstrated that the presence of the carbene ligand in the precursor was required to obtain materials exhibiting high chemoselectivity. High yields of fluorinated cyclohexanes were obtained using the resulting Rh@SiO2 material at Rh loading of 1–5 mol% (relative to substrate) and H2 pressure of 50 bar within 24 h. The stability and recyclability of the catalytic system was not yet addressed in this study.17
Despite these promising advances, the development of catalytic systems possessing high activity, selectivity, stability, and recyclability for the hydrogenation of fluoroarenes to fluorocyclohexanes remains a major challenge. In this context, working with metal NPs immobilized on molecularly modified supports seems particularly attractive since these materials were demonstrated to be versatile and suitable for the production of catalytic systems with tailor-made reactivity for challenging hydrogenation and hydrodeoxygenation reactions.18 We report here the rational design of Rh nanoparticles on hydrophobic silica supports (Rh@Si–R), exploiting the molecular control over the NPs environment at the support as determining factor to tune the selectivity of the reaction. In addition, we address the formation of traces of HF over the course of the reaction which is a serious issue with regards to safety as well as catalyst and product stability. The potential of this catalytic system is demonstrated for the synthesis of a broad range of fluorinated cycloalkanes from readily available substrates including building blocks used for the production of fine chemicals and pharmaceuticals.
Results and discussion
The design and synthetic methodology for the catalyst materials is depicted in Fig. 1a. The preparation of molecularly functionalized silica supports (Si–R) was accomplished through the condensation of commercial alkyl-triethoxysilanes on dehydroxylated SiO2 (see ESI† for full synthetic details).19 The hydrophobicity of the surface was modulated by variation of the length of the alkyl chains between C3 to C10 including also a perfluorinated C10-chain. The generation of Rh NPs on the Si–R supports to produce Rh@Si–R catalysts was accomplished by adapting a method previously reported for NPs@SILP materials.20 This involved the wet impregnation of the Si–R supports (0.50 g) with a solution of [Rh(allyl)3] (11.3 mg, 0.05 mmol) in dichloromethane (DCM) (2 mL). After evaporation of the solvent under vacuum, the impregnated powder was subjected to an atmosphere of H2 (50 bar) at 100 °C for 16 h. Under these conditions, the bright yellow powder turned black indicating the reduction of the organometallic precursor and the formation of Rh NPs. The same method was used to synthesize Rh NPs on the unmodified silica, Rh@SiO2, for direct comparison.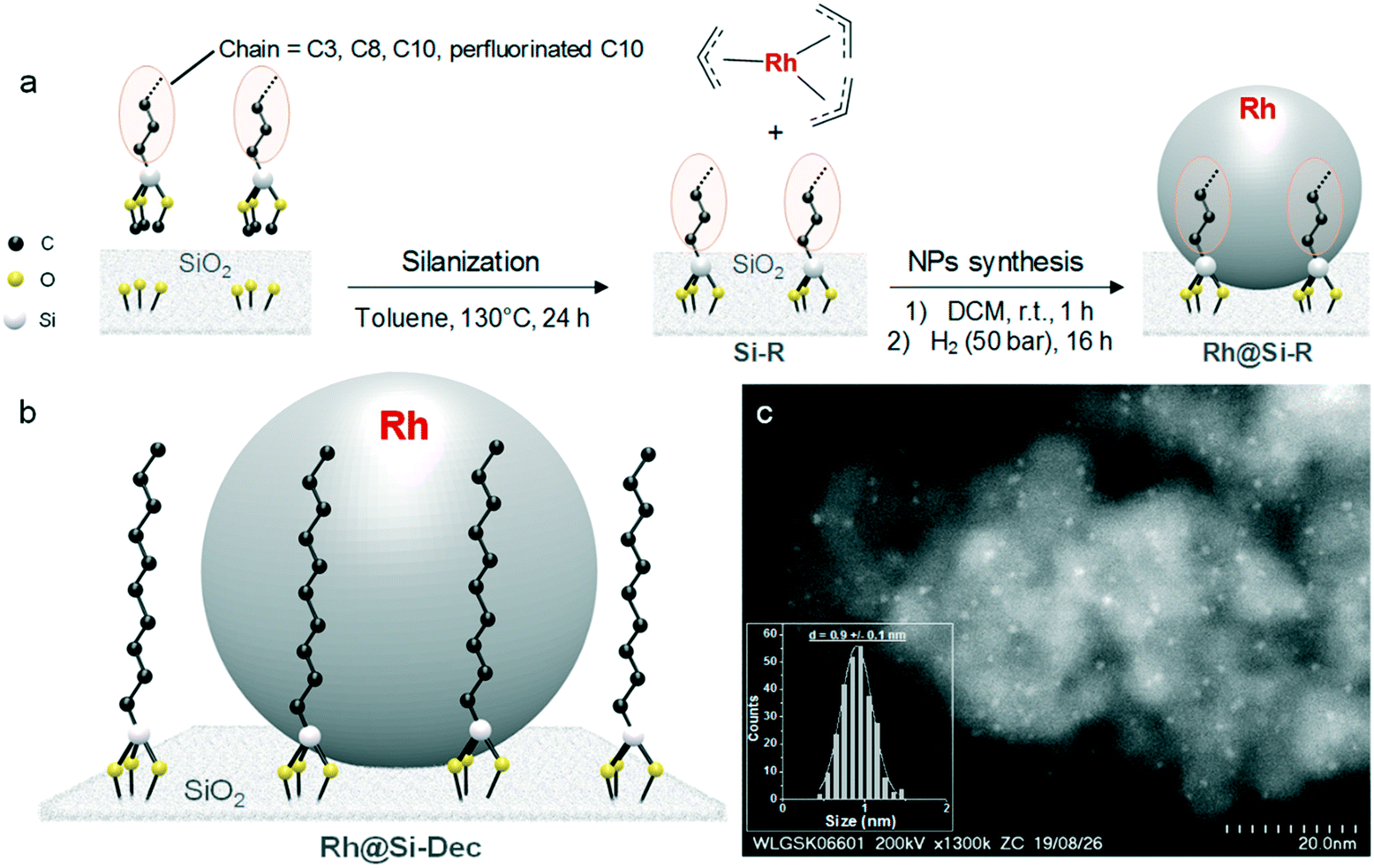 | ||
| Fig. 1 (a) Design and synthetic methodology for the preparation of Rh@Si–R materials with R = Prop (n-C3H7); Oct (n-C8H19), Dec (n-C10H21) and F-Dec (n-(C2H4)C8F19); (b) schematic representation of Rh@Si–Dec, and (c) scanning transmission electron microscopy with high angle annular dark field (STEM-HAADF) image of Rh@Si–Dec and the corresponding particle size distribution histogram. | ||
Characterization of Rh@Si–Dec by STEM-HAADF revealed the formation of small, uniform, and well-dispersed NPs with a mean diameter of 0.9 (±0.1) nm (Fig. 1). The Rh loading on Rh@Si–Dec was determined to be 0.92 wt% by ICP-AAS, well in agreement with the theoretical value (1 wt%). Surface area as determined by BET analysis was with 311 m2 g−1 somewhat lower than for the starting SiO2 (342 m2 g−1), as expected due to the chemisorption of the alkylsilanes. Characterization of the other catalysts gave similar results with comparable nanoparticles sizes, showing no significant variation of the NPs size depending on the alkyl chain length. (Fig. S1–S4 and Table S1†). Solid-state 29Si NMR of Si–Dec and Rh@Si–Dec (Fig. S5†) showed the presence of two types of Si species: (1) tetra-functionalized (Q) signals at −109 ppm (Q4 = Si(OSi)4) and −101 ppm (Q3 = Si(OSi)3OH); and (2) tri-functionalized signals at −56 ppm (T2 = R-Si(OSi)2OR′) and – 49 ppm (T1 = R-Si(OSi)(OR′)2). The T2 and T1 signals correspond to the Si atoms of the alkyl-triethoxysilanes bound to the SiO2 surface and thus provide evidence for the covalent attachment of the alkyl chains on the silica support.
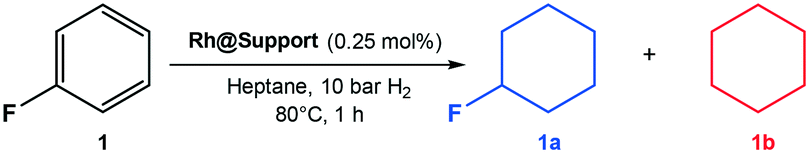
The catalytic activity of Rh@SiO2 and Rh@Si–R catalysts was tested using fluorobenzene (1) as a model substrate (Table 1). A preliminary screening of the reaction parameters using Rh@Si–Dec as catalyst (Tables S2–S5†) resulted in the definition of a standard set of conditions: 10 bar H2, 80 °C, 1 h, 500 rpm in heptane. In agreement with previous literature reports,14a,15 heptane was found to be the most suitable among the solvents tested. Using Rh@SiO2, a fairly good conversion of 1 (75%) and selectivity (84%) to the fluorinated cycloalkane 1a were observed corresponding to a yield of 63% for the desired product (entry 1) for a 1 h reaction. Full conversion was reached after extending the reaction time to 2 h, with 79% selectivity for the desired product (entry 2). The molecularly modified Rh@Si–R catalysts proved to be more active, reaching full conversion in 1 h in all cases at significantly higher selectivity. The selectivity toward the formation of 1a increased significantly when increasing length size of the alkyl chain from n-propyl to n-octyl and n-decyl (entry 3–5). Using SiO2 modified with 1H,1H,2H,2H-perfluorodecyltriethoxysilane as support did not lead to improvement of the selectivity (entry 6). An excellent yield of 1a of 92% was obtained with Rh@Si–Dec, exceeding the values obtained previously with any other catalyst.14,15 The superior performance of the molecularly modified support was confirmed also using ethylfluorobenzene (2), for which Rh@SiO2 gave much lower selectivity than the Rh@Si–R catalysts (Table S6†). Again, Rh@Si–Dec showed with 85% the highest selectivity at full conversion, while the use of Rh@SiO2 resulted in the lowest selectivity (65%, 61% yield). These results demonstrate that the use of molecularly modified supports allows control of the NPs environment to promote the selective hydrogenation over the hydrodefluorination. Therefore, Rh@Si–Dec was selected as catalyst for further studies.
|
|
|||||
|---|---|---|---|---|---|
| # | Catalyst | X (%) | S 1a (%) | Y 1a (%) | Y 1b (%) |
| Reaction conditions: catalyst (5 mg, 0.0005 mmol Rh), fluorobenzene (0.2 mmol, 19.0 mg, 400 eq.), n-heptane (750 mg, ≈1 mL), 10 bar H2, 80 °C, 1 h, 500 rpm.a 2 h. Conversion and selectivity determined by GC-FID using tetradecane as an internal standard (X: conversion/S: selectivity/Y: yield). | |||||
| 1 | Rh@SiO2 | 75 | 84 | 63 | 12 |
| 2a | Rh@SiO2 | >99 | 79 | 79 | 21 |
| 3 | Rh@Si–Prop | 99 | 69 | 68 | 31 |
| 4 | Rh@Si–Oct | >99 | 81 | 81 | 19 |
| 5 | Rh@Si–Dec | >99 | 92 | 92 | 8 |
| 6 | Rh@Si–Fdec | >99 | 82 | 82 | 18 |
Despite the high selectivity achieved with Rh@Si–Dec, small amounts of HF will still be released due to the competing hydrodefluorination.14a For practical applications, this raises concerns regarding safety aspects and material compatibility of the reactors (stainless steel autoclaves). Furthermore, it has been noted that this can lead to acid catalyzed side-reactions.14a Importantly, HF can also react with the support and/or the Rh-particles18d altering and ultimately depleting the catalyst performance. Indeed, it was found that catalyst stability was limited using the Rh@Si–Dec under standard conditions. Upon recycling the catalyst material, 77% of selectivity for the fluorinated product (1a) were obtained during the first two runs, after which a significant drop to 74% (third run) and 61% (fourth run) were noted (Fig. 2a). Analysis by STEM-HAADF of the spent catalyst after the second run revealed a significant increase in the particle size (2.1 ± 0.3 nm) with the formation of some aggregates (Fig. S6†). After the fourth run, the Rh NPs were found completely aggregated (Fig. 2b), and characterization by solid-state 29Si NMR showed major changes with the almost complete disappearance of the tri-functionalized T1 and T2 signals observed on the starting Rh@Si–Dec material, indicating a loss of the molecular modifiers (Fig. S7a†).
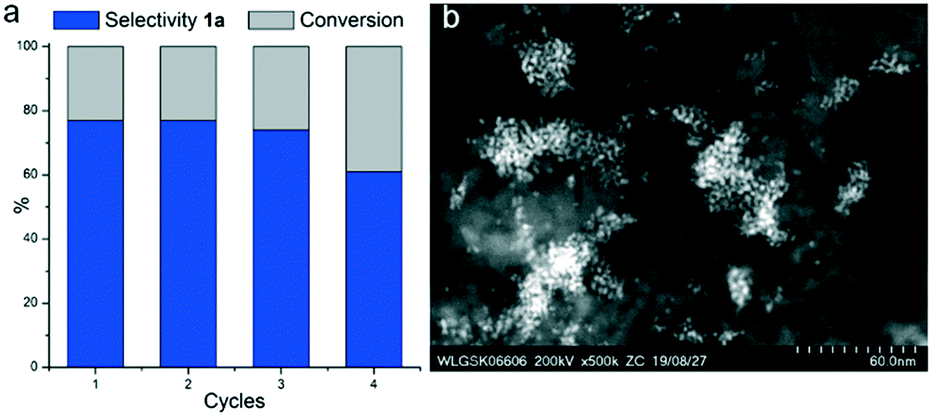 | ||
| Fig. 2 Study of the stability of Rh@Si–Dec through recycling experiments. a) Catalytic results, b) STEM-HAADF of Rh@Si–Dec after 4 cycles. Reaction conditions: Rh@Si–Dec (20 mg, 0.002 mmol Rh), fluorobenzene (2 mmol, 192 mg, 1000 eq.), n-heptane (1300 mg, ≈2 mL), 10 bar H2, 80 °C, 1 h, 500 rpm. Catalyst decanted, washed with heptane (1 mL) and dried under argon flow for 1 h between each run. The first cycle was performed using a fresh catalyst. Product yield determined by GC-FID using tetradecane as an internal standard. | ||
In order to address these limitations in a straightforward manner, we attempted to use catalytic amounts of CaO as additive to the catalytic system. While CaO is well established as sorbent for SO2 capture in many industrial processes,21 a few reports also highlighted its efficiency as scavenger for HF in waste streams22 leading to formation of CaF2 and water.
The addition of CaO to the reaction mixture did not affect the product distribution in the hydrogenation of 1. This shows that the performance of Rh@Si–Dec is not influenced negatively by the presence of this fluoride scavenger (Table S7†). This is in contrast to other bases such as HCOOK and K2CO3 which have been shown to promote hydrodefluorination by facilitating the formation of HF in homogeneous and heterogeneous catalyzed processes.23 Characterization by XRD of the mixture after catalysis evidenced diffraction peaks characteristic of CaF2, confirming the ability of CaO to trap HF under these conditions (Fig. S8†). Thus, CaO was systematically evaluated as additional stabilization component in the catalytic system.
To get further insight into the reaction pathway, a time profile was recorded for the conversion of 1 in the presence of CaO. For that, the conditions were slightly modified to slow down the reaction and allow the collection of sufficient data points (Fig. 3). The results show a mixture of 1a (44%) and 1b (5%) after 30 min, and full conversion with 91% yield of 1a after 120 min, with high TOF. Thus, the hydrogenation and hydrodefluorination occur in parallel whereby the hydrodefluorination takes place either before or during the hydrogenation of the aromatic ring. In addition, no consecutive hydrodefluorination of the fluorocyclohexane 1a to 1b was observed.
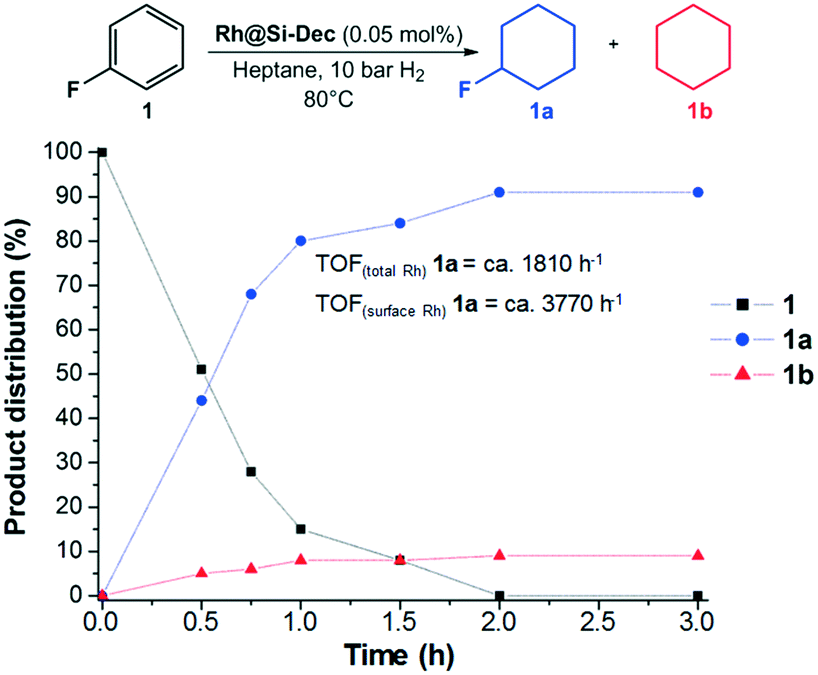 | ||
| Fig. 3 Time profile for the conversion of fluorobenzene (1) using Rh@Si–Dec in the presence of CaO. Reaction conditions: Rh@Si–Dec (6 mg, 0.0006 mmol Rh, 0.05 mol% relative to substrate), fluorobenzene (1.2 mmol, 115 mg, 2000 eq.), n-heptane (750 mg, ≈1 mL), CaO (4.5 mol% relative to substrate), 10 bar H2, 80 °C, 500 rpm. Conversion and selectivity determined by GC-FID using tetradecane as an internal standard. TOFs calculated based on the total amount of Rh as well as on the estimated percentage of Rh atoms at the surface of the NPs, see ESI† for details. | ||
Most importantly, the addition of CaO greatly increased the stability of the Rh@Si–Dec catalyst allowing for effective recycling. As shown on Fig. 4, the Rh@Si–Dec/CaO catalyst system was able to produce fluorocyclohexane (1a) in high yield and constant selectivity for at least 5 cycles. Characterization of Rh@Si–Dec by TEM after 5 cycles showed that the Rh NPs remained small and well dispersed on the support, despite a small increase in size (1.8 ± 0.6 nm). Elemental analysis by ICP-AAS did not evidence any leaching of the metal during the reaction (Table S1†). Characterization by solid-state 29Si NMR showed similar tri-functionalized T1 and T2 signals as observed on the starting Rh@Si–Dec material, indicating that the molecular modification of the surface was conserved under these conditions (Fig. S7b†). To the best of our knowledge, this is the first catalytic system possessing high activity (0.1 mol% catalyst) and selectivity for the hydrogenation of fluoroarenes that can be effectively reused over several cycles.
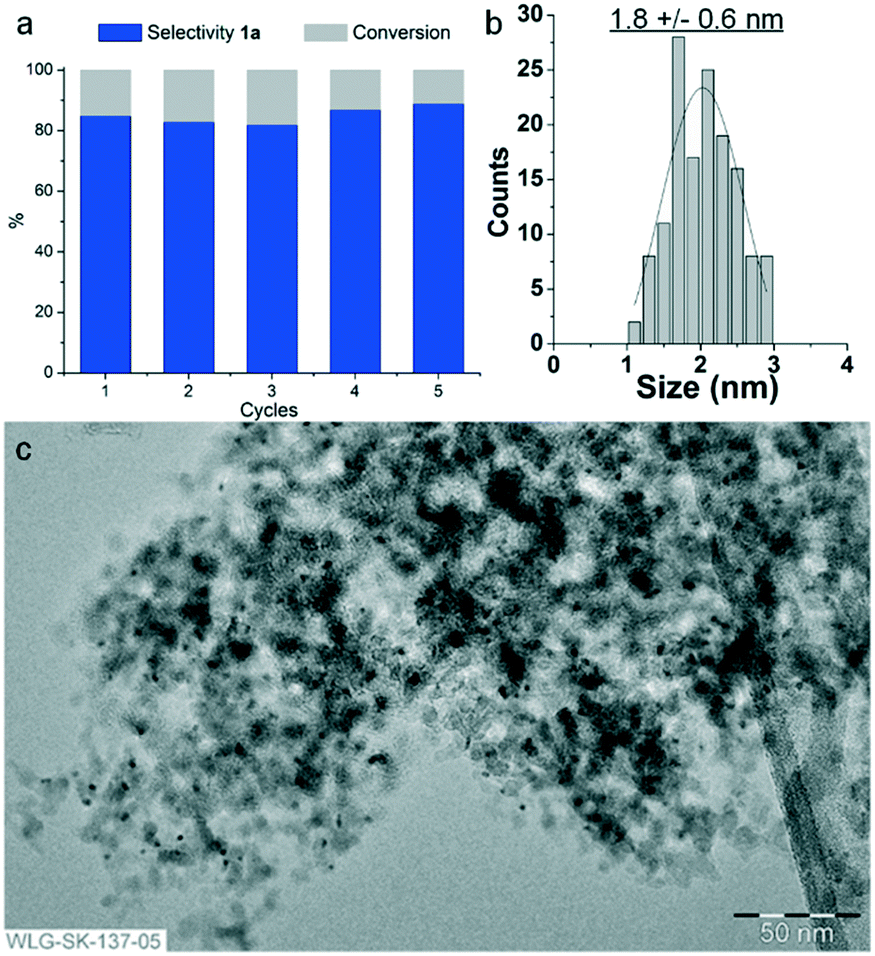 | ||
| Fig. 4 Study of the stability of Rh@Si–Dec through recycling experiments in the presence of CaO. (a) Catalytic results, (b) particle size distribution histogram corresponding to (c) TEM of Rh@Si–Dec after 5 cycles. Reaction conditions: Rh@Si–Dec (20 mg, 0.002 mmol Rh), fluorobenzene (2 mmol, 192 mg, 1000 eq.), CaO (36 mg, 0.64 mmol), n-heptane (1300 mg, ≈2 mL), 10 bar H2, 80 °C, 1 h, 500 rpm. Catalyst decanted, washed with heptane (1 mL) and dried under argon flow for 1 h between each run. The first cycle was performed using a fresh catalyst. Product yield determined by GC-FID using tetradecane as an internal standard. | ||
The scope of potential applications of Rh@Si–Dec was studied for a selection of fluorinated aromatic substrates (Table 2). All the substrates considered were effectively hydrogenated under optimized reaction conditions, giving fluorinated cyclohexane derivatives in good to excellent yields with stereoselectivities in the range of what is typically observed for arene hydrogenation.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Eq. | T °C | P (bar) | X (%) | Product a | Y a (%) |
cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans :trans |
Y b (%) |
| Reaction conditions: Rh@Si–Dec (5 mg, 0.0005 mmol Rh), n-heptane (750 mg, ≈1 mL), substrate (0.05–0.5 mmol), CaO (4.5 mol% relative to substrate), 80–100 °C, 10–55 bar H2, 1 h, 500 rpm.a 2 h. Eq: refers to the equivalence number. Conversion and yield determined by GC-FID using tetradecane as an internal standard. X: conversion/Y: yield. | |||||||||
| 1 |

|
1000 | 80 | 10 | >99 |

|
92 | — | 8 |
| 2 |

|
400 | 80 | 10 | >99 |

|
85 | 77:23 |
15 |
| 3 |
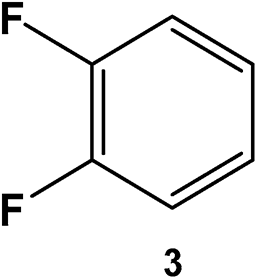
|
500 | 100 | 10 | >99 |

|
77 | 88:12 |
23 |
| 4 |
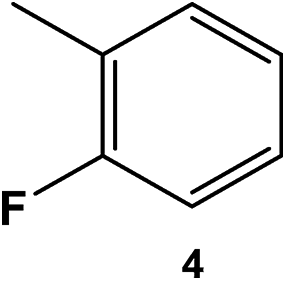
|
400 | 80 | 10 | >99 |
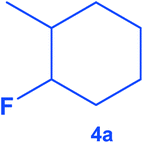
|
84 | 91:9 |
16 |
| 5 |

|
400 | 80 | 10 | >99 |

|
75 | 75:25 |
25 |
| 6 |
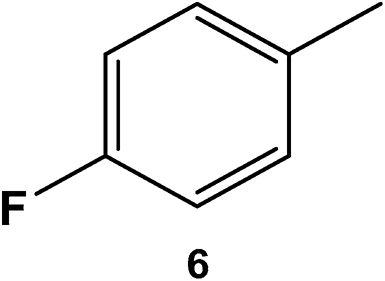
|
400 | 100 | 10 | >99 |
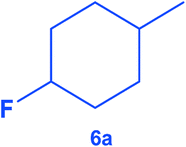
|
85 | 73:27 |
15 |
| 7 |

|
800 | 80 | 10 | >99 |

|
92 | 89:11 |
8 |
| 8 |

|
100 | 80 | 55 | >99 |

|
88 | 79:21 |
12 |
| 9a |

|
200 | 80 | 55 | >99 |

|
75 | 71:29 |
25 |
| 10a |

|
200 | 100 | 55 | >99 |

|
70 | 80:20 |
30 |
| 11a |
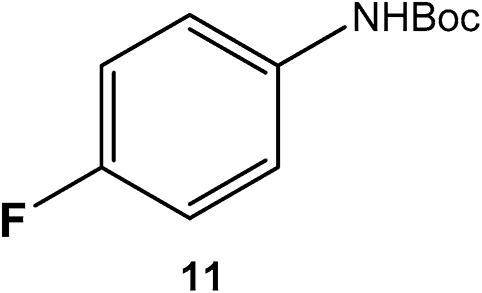
|
250 | 80 | 55 | >99 |
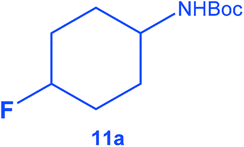
|
70 | 78:22 |
30 |
The catalyst was found tolerant to various functional groups. In some cases, an increase of the temperature to 100 °C was necessary to reach higher selectivity for the fluorinated product (substrates 3, 6 and 10). No significant change in stereoselectivity was observed when tuning the temperature in the 80–100 °C range. A low H2 pressure of 10 bar was sufficient for the selective conversion of substrates 1 to 7. Substrates 8–11 could only be effectively converted when raising the pressure to 55 bar.
To the best of our knowledge, this is the first report of the selective hydrogenation of substrates 2, 4–7 and 9 (see ESI† for characterization). In particular, this new single-step pathway for the production of 4-fluorocyclohexane carboxylic acid (9a) is noteworthy since 9a is a key intermediate for the preparation of various bio-active molecules.24 Interestingly, 9a, the (TMS)-protected alcohol (10a) and the (Boc)-protected amine (11a) fluorinated cyclohexane could be also isolated and characterized by NMR (see ESI†). These compounds are important building blocks used in the synthesis of pharmaceuticals3e including those highlighted in Scheme 1 that serve as 5-HT2(c) receptor modulator drug3b and potassium channel modulators,3d respectively. Moreover, 11a is used in the synthesis of analogs of the anticancer drug lomustine and of the mucolytic agent bromhexine.15
Conclusions
In conclusion, Rh nanoparticles (NPs) on molecularly modified silica supports (Rh@Si–R) were developed as catalysts for selective hydrogenation of fluorinated arenes to fluorocyclohexanes. Our results demonstrate that controlling the hydrophobicity of the intimate environment of the NPs is of critical importance to favor the hydrogenation over the hydrodefluorination pathway. The molecular approach to material preparation offers rational control over the properties of the support and the generation of well-defined metal particles. Adding CaO as HF scavenger to the catalytic system was found highly effective to prevent side-reactions and catalyst degradation, thus improving the recyclability of the catalytic system while avoiding the risks associated to the presence of HF. Overall, the Rh@Si–Dec (Dec = n-C10H21) catalyst combines high activity, selectivity and stability/recyclability for the hydrogenation of a large scope of fluorinated arenes, providing access to a broad range of fluorinated cyclohexane derivatives. We believe that this approach combining NPs on molecularly modified supports and HF scavenger will facilitate the development of next-generation catalysts for this challenging transformation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support by the Max Planck Society and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832. Furthermore, the authors thank Yasmin Eisenmann for her support with the experiments, Alina Jakubowski, Annika Gurowski, Justus Werkmeister, Norbert Pfänder (MPI-CEC, Mülheim/Ruhr), Adrian Schlüter, Claudia Weidenthaler, Jan Ternieden (MPI-Kofo, Mülheim/Ruhr) for their support with the analytics. Open Access funding provided by the Max Planck Society.Notes and references
-
(a) M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC
; (b) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications., Wiley-VCH, 2nd edn, 2013 CrossRef
- T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS
-
(a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS
- M. Rueda-Becerril, C. Chatalova Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS
-
(a) W. T. Miller and A. L. Dittman, J. Am. Chem. Soc., 1956, 78, 2793–2797 CrossRef CAS
- S. Stavber and M. Zupan, Tetrahedron, 1989, 45, 2737–2742 CrossRef CAS
-
R. D. Chambers, Fluorine in Organic Chemistry, Blackwell Publishing Ltd., Oxford, 2004 Search PubMed
- S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580–10583 CrossRef CAS
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS
- F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS
-
(a) D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398–8427 RSC
-
(a) J. Li, J. Chen, R. Sang, W.-S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56–62 CrossRef CAS
-
(a) M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS
-
(a) K. J. Stanger and R. J. Angelici, J. Mol. Catal. A: Chem., 2004, 207, 59–68 CrossRef CAS
- M. P. Wiesenfeldt, Z. Nairoukh, W. Li and F. Glorius, Science, 2017, 357, 908–912 CrossRef CAS
-
(a) B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2019, 8, 8441–8449 CrossRef
- D. Moock, M. P. Wiesenfeldt, M. Freitag, S. Muratsugu, S. Ikemoto, R. Knitsch, J. Schneidewind, W. Baumann, A. H. Schäfer, A. Timmer, M. Tada, M. R. Hansen and F. Glorius, ACS Catal., 2020, 10, 6309–6317 CrossRef CAS
-
(a) K. L. Luska, A. Bordet, S. Tricard, I. Sinev, W. Grünert, B. Chaudret and W. Leitner, ACS Catal., 2016, 6, 3719–3726 CrossRef CAS
-
(a) A. Katz and M. E. Davis, Nature, 2000, 403, 286–289 CrossRef CAS
- K. L. Luska, J. Julis, E. Stavitski, D. N. Zakharov, A. Adams and W. Leitner, Chem. Sci., 2014, 5, 4895–4905 RSC
-
(a) V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2007, 41, 4435–4440 CrossRef CAS
-
(a) A. F. Shaaban, Thermochim. Acta, 1991, 180, 9–21 CrossRef CAS
-
(a) Y. Sawama, Y. Yabe, M. Shigetsura, T. Yamada, S. Nagata, Y. Fujiwara, T. Maegawa, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2012, 354, 777–782 CrossRef CAS
-
(a) P. Peng, H. Chen, Y. Zhu, Z. Wang, J. Li, R.-H. Luo, J. Wang, L. Chen, L.-M. Yang, H. Jiang, X. Xie, B. Wu, Y.-T. Zheng and H. Liu, J. Med. Chem., 2018, 61, 9621–9636 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01716g |
| This journal is © The Royal Society of Chemistry 2020 |