
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in the control of selectivity in hydrogenation reactions by confined metal functionalities†
Moussa
Zaarour‡
 ,
Jurjen
Cazemier‡
and
Javier
Ruiz-Martínez
*
,
Jurjen
Cazemier‡
and
Javier
Ruiz-Martínez
*
King Abdullah University of Science and Technology, KAUST Catalysis Center (KCC), Catalysis Nanomaterials and Spectroscopy (CNS), Thuwal 23955, Saudi Arabia. E-mail: Moussa.zaarour@kaust.edu.sa; jurjen.cazemier@kaust.edu.sa; javier.ruizmartinez@kaust.edu.sa; Tel: +966128084530
First published on 27th October 2020
Abstract
Confining metal active species in the voids of porous solid matrices such as zeolites, metal–organic frameworks (MOFs), and carbon nanotubes (CNTs) can bring fascinating key advantages in the field of selective hydrogenation reactions. Confined metal species act as intermolecular selective catalysts capable of discriminating reagents based on their molecular size and shape. They also exhibit intramolecular selectivity by converting one or more functional groups selectively in the presence of others. In this review, we present a comprehensive overview of the different synthetic methods for confining active metal species in the voids of zeolites, MOFs, CNTs, and other porous structures. We then emphasize the strong influence of metal confinement on steering catalytic selectivity in a wide range of selective hydrogenation reactions. Finally, we share our opinion on the different synthesis methods for potential practical applications and on future research directions.
 Moussa Zaarour | Moussa Zaarour received his International Master's degree in Catalysis, Molecules, and Green Chemistry (2009) and his PhD in Organometallic Chemistry (2012) from the University of Rennes 1 (France). He worked as a post-doctoral fellow (2012–2015), and as a researcher (2016–2019) at the Laboratory of Catalysis and Spectroscopy (LCS-ENSICAEN, France), his research focused on the preparation and in-depth spectroscopic characterization of zeolites and zeolite-like materials. Currently, he is a post-doctoral fellow in the group of Prof. Javier Ruiz-Martínez at King Abdullah University of Science and Technology (KAUST). His ongoing research focuses on designing, characterizing, and testing confined metal catalysts for the selective hydrogenation reactions. |
 Jurjen Cazemier | Jurjen Cazemier received his BSc in Chemistry from the University of Groningen, Groningen (2015, The Netherlands), and his MSc in chemistry from the University of Groningen, Groningen (The Netherlands, 2018). Currently, he is a PhD student in the group of Prof. Javier Ruiz-Martínez at King Abdullah University of Science and Technology (KAUST). His current research focuses on the development and testing of heterogeneous catalysts. |
 Javier Ruiz-Martínez | Javier Ruiz-Martínez received his Master degree from the University of Granada and Norwegian University of Science and Technology (Spain, 2003) and his PhD from the University of Alicante (Spain, 2009). He held the position of Assistant Professor of Inorganic Chemistry and Catalysis at Utrecht University (The Netherlands, 2013–2015) then became a Heterogeneous Catalysis Specialist at AkzoNobel Specialty Chemicals (The Netherlands, 2015–2019). Currently, he is the head of the group Catalysis, Nanomaterials, and Spectroscopy (CNS), at the KAUST Catalysis Center (KCC). His research focuses on the study of solid catalysts and chemical reactions using advanced spectroscopic techniques. The topics of his studies include C1 chemistry, petrochemistry, selective hydrogenation, and environmental catalysis. |
1. Introduction
Catalytic hydrogenation is undeniably one of most important conversion steps in the chemical industry. In numbers, around a quarter of all chemical processes involve a catalytic hydrogenation step1,2 and many key industrial chemical sectors have relevant examples of selective hydrogenation reactions. For instance, the selective hydrogenation of alkynes (also described as semihydrogenation or half-hydrogenation) and dienes are important reactions in ethylene, propylene and butylene streams for the polymerization of olefins3 and in fine chemicals for the synthesis of biological active compounds such as insect sex pheromones and vitamins.2 Other relevant examples are found in the synthesis of high added-value fragrances, flavors, agrochemicals, and pharmaceuticals where there is often a step where a functional group such as –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –NO2, –C
O, –NO2, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, –COOH, and –CONH2 is selectively hydrogenated in a molecule with multifunctional groups.4
N, –COOH, and –CONH2 is selectively hydrogenated in a molecule with multifunctional groups.4
Several parameters have a significant effect in steering selectivity during catalytic hydrogenation reactions. Solvents are able to strongly influence selectivity due to their polar and acid/base properties.5,6 Optimization of other parameters, such as temperature, pressure, composition and type of reactor can promote selectivity by tuning kinetics and thermodynamics of the reaction. Notwithstanding, the design and development of advanced catalytic materials is of paramount importance for the ultimate control in hydrogenation selectivity. In this respect, homogeneous catalysts have shown extraordinary selectivity owing to their molecular design optimized by the steric and electronic effects of ligands. However, the ease of separation, regeneration, and stability of heterogeneous catalysts have urged the developments of solid materials for selective hydrogenation reactions.
In solid catalysts, hydrogenation reactions are typically performed by two general families of metals: (a) noble metals such as Pt, Pd, Ru, Ir, Rh, and Ru and (b) group VIII transition metals, for example Ni, Cu, Cr, and Co. The metal active component is often in the form of ultrafine particles and is deposited on a high surface area support. Although high hydrogenation activities can be achieved by bare metal nanoparticles, poor selectivities are observed due to the uncontrolled hydrogenation of untargeted functional groups. Selectivity is traditionally modified by the addition of a second metal or the use of a support that interacts strongly with the hydrogenation functionality.7–11 The promotional effect of the second metal or support is explained by changes in the electronic density of the hydrogenation metal12 or by a geometric effect where the active sites have a conformation more favorable for the hydrogenation of the targeted functionality. A relevant commercial example is the Lindlar catalyst for selective alkyne hydrogenations where a Pd/CaCO3 catalysts is modified by a dilution effect of the active sites by lead.13 More recent investigations have brought a new class of hybrid organic–inorganic catalysts where the metal active sites are modified by organic ligands.14,15 These new advances have even been translated into a new family of ligand-modified supported catalysts, Nanoselect™, commercialized by BASF16,17 and applied in reactions such as the selective alkyne hydrogenation. Despite the remarkable promoting effects of a second metal and ligands, it normally goes hand in hand with a drop in catalytic activity due to the loss of surface active sites. Another disadvantage, specially from a practical perspective, is the often unsatisfactory catalyst stability due to a dynamic structural modification of the active sites during working conditions. For instance, metal aggregation/segregation and ligand leaching may occur during reaction, leading to a strong negative impact on catalytic performance. These non-optimal performances encourage the search for catalysts with high selectivity and stability without jeopardizing catalytic activity. In this line, another emerging field of research is focused on heterogeneous single-atom catalysts. In these materials, the support stabilizes mononuclear metal species and modifies the metal properties in a similar fashion as ligands in homogeneous catalysts do, leading to a theoretically maximum catalytic efficiency. As a drawback, single site instability due to metal segregation is a general trend hampering their practical application. For more information about the fundamentals and examples of these materials, the reader is referred to excellent and recent review papers.18–20
An elegant approach to promote selectivity is the confinement or encapsulation of the hydrogenation functionality in a porous structure that typically exposes well defined pores. Confined nanoparticles can bring fascinating key advantages to the field of selective hydrogenation. (a) When pores have identical dimensions, the synthesis of nanoparticles with a well-defined size is facilitated with high precision. (b) They can efficiently disperse and stabilize metal nanoparticles and commonly prevent metal restructuring and sintering. (c) A uniform distribution of pores can also be used to induce selectivity by the molecular sieving effect, which occludes molecules larger than the pore apertures. (d) A chemoselective hydrogenation can also be promoted when the encapsulant structure contains well-defined pore cavities imposing steric constrains that solely allow adsorption modes and transitions states leading to desired products. (e) Confinement creates an additional protection layer against poisoning when catalyst pores are sufficiently small to hamper or even completely block the diffusion of poisons. (f) Enhanced stability of the active phase by confinement also opens up the possibility for efficient catalyst reusability and regeneration, which positively impacts catalyst costs. (g) Finally, confining different catalytic functionalities with a well-defined spatial distribution unlocks the possibility of performing sequential catalytic reactions in a tandem fashion.
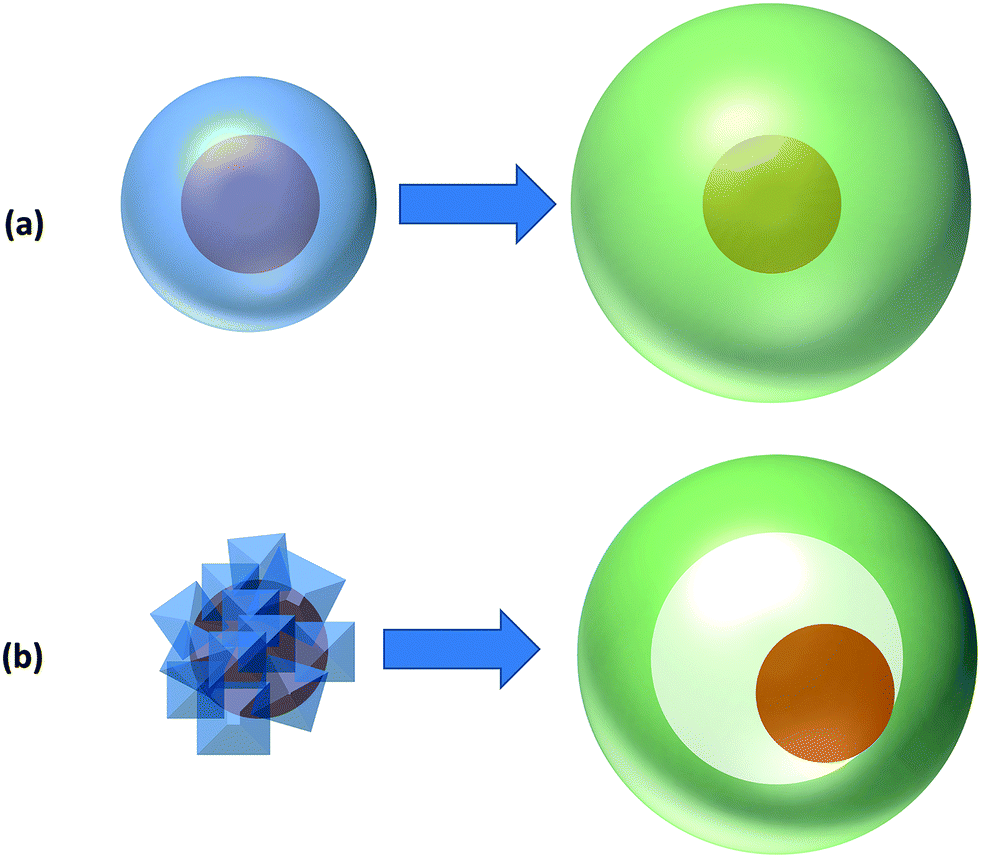
The encapsulation of single atoms (SAs), atomic clusters (ACs), and nanoparticle (NPs) as metal functionalities results in two modes of selective hydrogenation reactions, named as intermolecular and intramolecular selectivity. In the former, the encapsulant porosity controls the access of reagents present in the reaction medium based on their size and shape. In this way, the reagents possessing dimensions smaller than the encapsulant apertures are exclusively allowed to diffuse and react with the confined metal functionality, as exemplified in Fig. 1a. Intramolecular selectivity (Fig. 1b), involves multifunctional molecules where only targeted functional groups are hydrogenated based on pore steric constrains, favored transition states, and/or electronic effects.
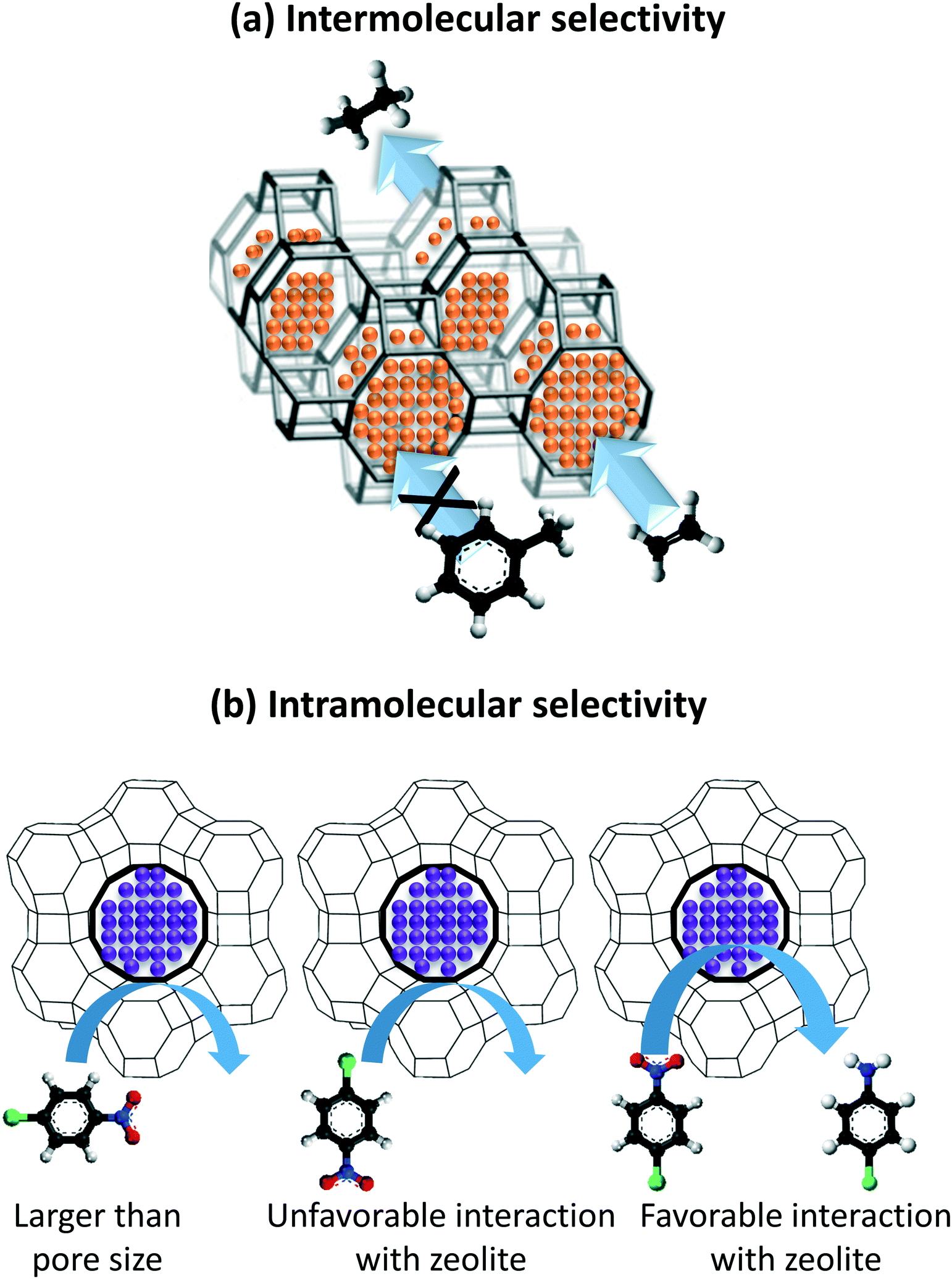 | ||
| Fig. 1 (a) Intermolecular selective hydrogenation exemplified by the hydrogenation of ethylene versus toluene on Pt/GIS, taken from ref. 30 and (b) an example of intramolecular selective hydrogenation of the nitro functional group of 4-nitrochlorobenzene on Pd@Y.31 | ||
Recent review papers have been published on metal active species confined21 within zeolites,22–24 MOFs,25,26 and CNTs27 for a myriad of catalytic applications. Considering the variety of catalytic reactions and related scientific contributions, it would be challenging to comprehensively describe the catalytic behavior of this new family of materials. Consequently, this review has focused on the advances over the last decade on the synthesis of confined metal active species and their application in hydrogenation reactions where inter- and/or intramolecular selectivity are targeted. The contribution mainly focusses on solid catalysts where the encapsulant is a structure with well-defined pores, i.e., a zeolite, a metal–organic framework, and carbon nanotubes. A few other examples are described where encapsulants with a less defined porous structure, such as silica and porous organic polymers, are used. Although we acknowledge the importance of transfer hydrogenation reactions8,28 and CO2 hydrogenation,29 this contribution only targets hydrogenation reactions of organic substrates involving H2. With more than one hundred contributions reviewed, we compare the activity and selectivity of confined catalysts prepared by different workgroups for specific hydrogenation reactions. In addition, a thorough overview of the preparation methods for confined catalysts and their variations is described. Finally, we will share our opinion on the different synthesis methods for potential practical applications and on future research directions for proper catalyst characterization, new applications, and for a more rational design of encapsulated catalysts.
2. Confining metal catalysts in zeolites
Owing to their well-defined porous structure and thermal stability, zeolites are ideal encapsulants of gases, organic molecules, organometallic complexes, metal cations, and metal nanoparticles. On the basis of the synthesis of metal-encapsulated zeolites, four main methods are considered and sketched in Fig. 2: in situ preparation, core–shell method, host–guest assembly, and reassembling and transformation. Below, we introduce the most recent examples of each confinement mode and illustrate the influence on the activity toward selective hydrogenation reactions.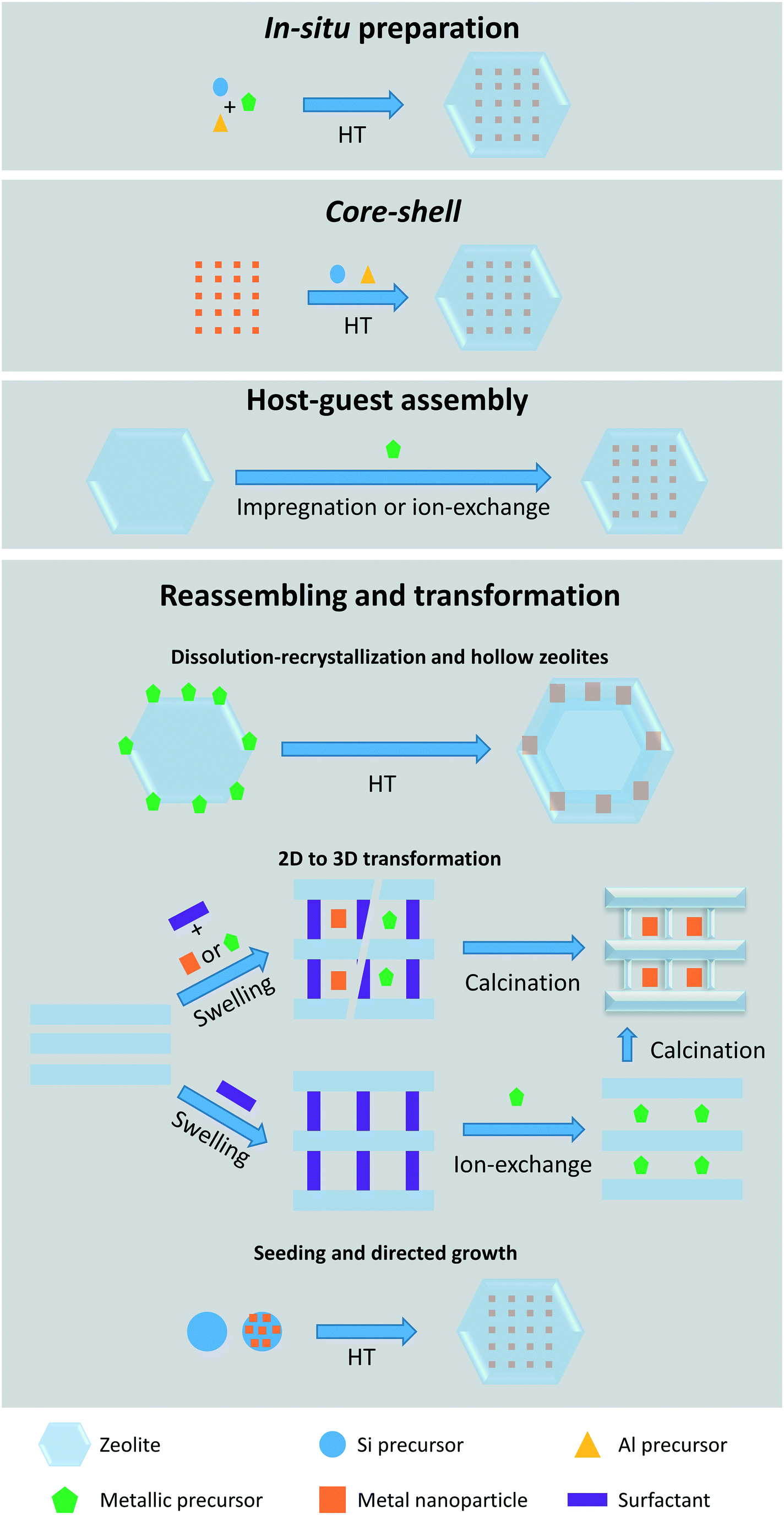 | ||
| Fig. 2 Summary of the four main methods used for the confining metal species in zeolites. | ||
2.1. Synthesis methods for confining metal species within zeolites
Using the core–shell method, pre-synthesized metal NPs40–45 or supported NPs46,47 are coated with single or multiple zeolite layers of controllable thickness following a secondary growth synthesis step. The use of pre-synthesized nanoparticles adds an additional synthetic strategy to control metal particle size and distribution. In addition, the use of zeolite seeds48 or hierarchical zeolites49 as core directs the synthesis of the desired zeolite shell.
2.2. Intermolecular selectivity
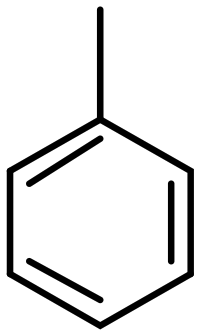
Taking the advantage of the strong sieving properties of zeolites, several research groups reported the confinement of metal active species in the voids of zeolites to induce intermolecular selective hydrogenation. For instance, size exclusion is used to selectively hydrogenate light olefins by using metal catalyst encapsulated in 8-membered ring (small pore) zeolites. One of the first examples was done by the Iglesia's group on the hydrogenation of ethylene (kinetic diameter = 0.39 nm) and toluene (kinetic diameter = 0.58 nm) over Pt clusters encapsulated in gismondine (GIS) and analcime (ANA) zeolites using the in situ approach.30 The small ethylene molecule could diffuse and interact with the metal clusters confined in both zeolites, whereas the big toluene molecule was blocked on the surface (Fig. 1a). Consequently, high ethylene/toluene turnover frequency (TOF) ratios of 242 and 182 were obtained during the catalytic hydrogenation on Pt/GIS and Pt/ANA, respectively. Examples of intermolecular selectivity in the hydrogenation of ethylene with other molecules are summarized in Table 1. In addition to promoting selectivity, the zeolite also provided a protective environment against poisoning. The hydrogenation ceased under competitive thiophene flow due to blocking of the pores by the physisorbed poison. Directly after the thiophene feed was stopped, 70% (Pt/ANA) to 85% (Pt/GIS) of the initial hydrogenation rates were recovered owing to the high dispersion of the clusters in the regions not accessible to thiophene. On the other hand, the catalytic hydrogenation of ethylene and toluene over Pt NPs supported on SiO2 did not exhibit intermolecular selectivity, and permanent deactivation took place following thiophene flow.| Catalyst | Ethylene | Control molecule | Reaction conditions | Ref. | |||
|---|---|---|---|---|---|---|---|
| TOF (s−1) | Molecule | TOF (s−1) | P alkene | P H2 | T (K) | ||
| TOF determined at conversion:a n/a.b <10%.c 80%.d 2%. Reaction conditions fore Ethylene hydrogenation.f Toluene hydrogenation. Catalyst nomenclature: metal/zeolite. GIS stands for gismondine, ANA for analcime, NaA and LTA for Linde type A zeolite. | |||||||
| Pt/GIS | 2.90a |
 Toluene Toluene |
0.012a | 0.95 kPae | 5 kPae | 308e | 30 |
| Pt/ANA | 2.00a | 0.011a | 0.35 kPaf | 99.65 kPaf | 473f | 30 | |
| Pt/NaA | 0.57b |
 Isobutene Isobutene |
0.0021b | 1.5 kPa | 5 kPa | 294 | 35 |
| Pd/NaA | 0.59b | 0.013b | 35 | ||||
| Rh/NaA | 0.29b | 0.007b | 35 | ||||
| Pt/LTA | 0.81a | 0.051a | 36 | ||||
| Pd/LTA | 0.56a | 0.014a | 36 | ||||
| Rh/LTA | 0.39a | 0.0084a | 36 | ||||
| Ir/NaA | 0.27b | 0.003b | 1.5 kPa | 5 kPa | 373 | 35 | |
| Pt-CHA-2 | 0.0843c |
 Propylene Propylene |
0.0021d | 3.2 kPa | 16.1 kPa | 353 | 38 |
Selective hydrogenation effects were further explored with ethylene and isobutene, whose kinetic diameter (0.50 nm)35 is smaller than toluene. The hydrogenation was carried out over Pt ACs encapsulated in Linde type A (LTA) zeolite (Pt/NaA). The catalyst was synthesized by in situ preparation using a sulfur-containing ligand, (3-mercaptopropyl)trimethoxysilane, as a metal stabilizer. The turnover frequency observed with the confined clusters was 271 times higher for the ethylene hydrogenation than for the isobutene hydrogenation. This behavior was explained by the faster diffusion of ethylene relative to isobutene through the zeolite micropores (pore opening 0.41 × 0.41 nm). Similar size discrimination was demonstrated by using other noble metal clusters (Pd, Ir, and Rh) confined in LTA: their relative hydrogenation rates follow the trends: Pt (271) > Ir (90) > Pd (45) > Rh (41).35 Iglesia's group also investigated the ethylene/isobutene hydrogenation over Pt, Pd, and Rh clusters (1.1–1.9 nm) confined in LTA and prepared in the presence of N-ligands (NH3 and NH2CH2CH2NH2) rather than S-based ones. The resulting catalysts were highly selective towards ethylene hydrogenation owing to the restricted diffusion of isobutene through the LTA micropore network.36 Moreover, the confined clusters were resistant to poisoning, with 70% of activity maintained under flow of thiophene.
A more challenging intermolecular selective hydrogenation was investigated by Corma's group testing ethylene and propylene, an olefin with a similar kinetic diameter (0.45 nm). Pt NPs (1.3 nm) encapsulated in the pores of chabazite (CHA) converted more than 80% of ethylene versus only 2% of propylene under identical experimental conditions. These results are explained by differences in effective diffusivities, as both molecules can fit in the CHA pores. In clear contrast, both ethylene and propylene were almost equally converted over SiO2-supported NPs in the absence of diffusion limitations.38
The zeolite sieving effect was also exploited to promote the selective hydrogenation of molecules with larger dimensions by using 10- (medium pore) and 12-membered ring zeolites. In this regard, several catalysts were tested for the hydrogenation of nitrobenzene versus other larger molecules and are summarized in Table 2. In one example, Pd NPs encapsulated in silicalite-1 selectively hydrogenated nitrobenzene but not nitronaphtalene.34 This selectivity is attributed to the successful diffusion of nitrobenzene (molecular size: 0.46 × 0.66 nm) through the MFI pore structure (size: 0.53 × 0.56 nm), whereas the larger nitronaphthalene (molecular size: 0.73 × 0.66 nm) failed to enter the micropore system. The selective hydrogenation of nitrobenzene was also achieved over Pt NPs confined in faujasite Y crystals (Pt@Y) with a 155 times higher reaction turnover frequency than in the hydrogenation of 2,4,6-tri-tert-butylnitrobenzene.31 Pt NPs impregnated on hierarchical ZSM-5 and coated with a shell of silicalite-1 (Pt@MFI(0.8)) also demonstrated a high selectivity towards the hydrogenation of nitrobenzene versus 2,3-dimethylnitrobenzene.46 While the smaller nitrobenzene molecule crossed the zeolite-shell and was fully converted into aniline, 2,3-dimethylnitrobenzene (with a kinetic minimal cross-sectional diameter of 0.73 nm) could only achieve a maximum of 50% conversion.46 The selectivity was further enhanced by narrowing the pore openings via silanization (Si-Mod Pt@MFI(0.8)); this modification did not alter the catalytic activity towards nitrobenzene, yet the conversion of 2,3-dimethylnitrobenzene was decreased to less than 20%. Replacing the silicalite-1 shell with a ZSM-5 one provided additional acidic groups in the proximity of the Pt NPs; this modification changed the pathway of nitrobenzene hydrogenation and gave rise to a distinct product, para-aminophenol.47 This product was produced in a 50–60% yield (Pt@HZSM-5(0.5)), with an additional increase up to 74% after passivating the surface NPs by diphenylthiobenzene (poisoned Pt@HZSM-5(0.5)). Meanwhile, only 11% yield was obtained over the surface supported Pd/ZSM-5 catalyst. The intermolecular selectivity was also tested over a Pt–CeO2@SG-ZSM-5 catalyst prepared by the core–shell method. The catalyst was prepared by introducing Pt–CeO2 binary metallic species into the interdomain mesopores of hierarchical ZSM-5 followed by a secondary growth using a synthetic gel of equal volume to the hierarchical pore volume. This catalyst fully converted nitrobenzene with a limited conversion of the bulkier 1,3-dimethyl-2-nitrobenzene.49 In another example, Pt NPs incorporated in ZSM-5 zeolite, denoted as Pt@MFI-b, was tested for the hydrogenation of nitrobenzene versus 3-nitrotoluene with comparable molecular dimensions (0.58 × 0.87 nm). Conversions of 86% were obtained for nitrobenzene versus 58% for 3-nitrotoluene. This intermolecular selectivity was completely lost, and both substrates were hydrogenated when the reactions were performed over the non-encapsulated impregnated homologue catalysts.48 The hydrogenation of both molecules was also tested over a bimetallic 0.8Pd0.2Ni(OH)2@S-1 catalyst. This catalyst was prepared via the in situ method by introducing palladium and nickel precursors into the zeolite synthetic gel. In this case, a better intermolecular selectivity was obtained by using the same silicalite-1 encapsulant and a negligible conversion of 3-nitrotoluene was afforded.57
| Catalyst | Nitrobenzene (NB) | Control molecule for intermolecular selectivity | Reaction conditions | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| Conversion (%) or TOF (s−1) | Molecule | Conversion (%) or TOF (s−1) | Substrate | Catalyst | H2 | T | Time | ||
| a NaBH4. Reaction conditions for the hydrogenation of:b Nitrobenzene.c 2,4,6-Tri-tert-butylnitrobenzene.d Value estimated from the plot of conversion versus time.e 60% selectivity towards the formations p-aminophenol.f 74% selectivity towards the formations p-aminophenol.g Based on nitro group. Catalyst nomenclature: metal@zeolite. “S-1”, “MFI”, and “ZSM-5”: stand for zeolites with MFI structure, “Y” stands for faujasite-Y zeolite. | |||||||||
| Pd@mnc-S1 | 94% |
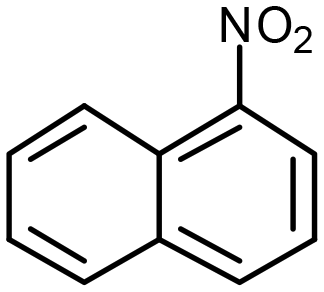 Nitronaphthalene (NNp) Nitronaphthalene (NNp) |
3.5% | 0.1 mmol | 20 mg | 0.2 mmola | RT | 5 min | 34 |
| Pt@Y | 0.1580 s−1 |
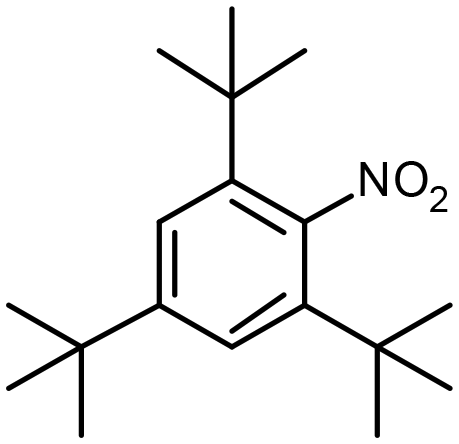 2,4,6-Tri-tert-butylnitrobenzene (tBNB) 2,4,6-Tri-tert-butylnitrobenzene (tBNB) |
0.00102 s−1 | n/a | n/a | 1 Mpa,b 3.5 MPac | 353 K,b 373 Kc | 31 | |
| Pt@MFI(0.8) | 100% |
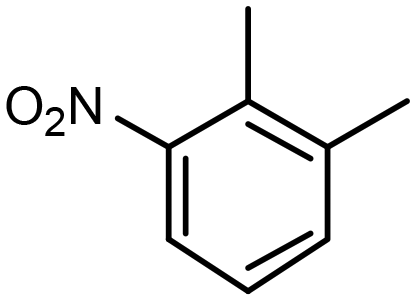 2,3-Dimethylnitrobenzene (DMNB) 2,3-Dimethylnitrobenzene (DMNB) |
∼50%d | 1 mmol NB + 1 mmol DMNB | 100 mg | 1 MPa | 353 K | 8 h | 46 |
| Si-Mod Pt@MFI(0.8) | ∼95%d | ∼18%d | |||||||
| Pt@HZSM-5(0.5) | 100%e | No molecule | n/a | 0.65 mmol | 100 mg | 1 MPa | 403 K | 3 h | 47 |
| Poisoned Pt@HZSM-5(0.5) | 100%f | ||||||||
| Pt–CeO2@SG-ZSM-5 | 100% |
 1,3-Dimethyl-2 nitrobenzene (DMNB) 1,3-Dimethyl-2 nitrobenzene (DMNB) |
7% | 0.25 mmol | 0.4 mol%g | 1 MPa | 393 K | 3 h | 49 |
| Pt@MFI-b | 86% |
 3-Nitrotolune (NT) 3-Nitrotolune (NT) |
58% | 0.1 mmol | 50 mg | 0.3 mmola | RT | 30 min | 48 |
| 0.8Pd0.2Ni(OH)2@S-1 | 100% | Negligible | 0.00146 mmol | 20.1 mg | 0.0438 mmola | 298 K | 5 min | 57 | |
Overall, it appears that in situ and core–shell synthesis routes frequently result in the effective encapsulation of the metallic phase. However, introducing NPs into zeolites using host–guest assembly often leads to incomplete encapsulation, and consequently, a decrease in selectivity. This phenomenon was demonstrated by impregnating nickel precursor on ZSM-5, leading to nickel oxide nanoparticles on the surface and in the ZSM-5 channels.52 This random distribution allowed hydrogenating both toluene and 1,3,5-tri-isopropyl benzene regardless of their different molecular size. Selectively extracting surface NPs by poly-4-styrenesulfonic acid decreased the conversion of 1,3,5-tri-isopropyl benzene considerably. Meanwhile, toluene hydrogenation was less affected and remained almost proportional to nickel content. Pt NPs encapsulated in hollow beta zeolite by dissolution–recrystallization method allowed the diffusion and hydrogenation of toluene with similar TOF to that obtained from the hydrogenation over Pt supported on silica (TOFPt@beta = 0.04 s−1, TOFPt/SiO2 = 0.06 s−1. On the other hand, diffusion of the bulkier mesitylene was hindered, leading to low conversions.53 After improving the catalytic system by decreasing the Pt particle size from 10–50 nm to 0.5–3 nm and removing the surface supported NPs, a 19-fold increase of activity towards hydrogenation of toluene was achieved. On the other hand, the slow and restricted diffusion of mesitylene prevented its interaction with the confined NPs; a limited conversion was reported over the traces of Pt available in the micropore region accessible by mesitylene.58 Using the reassembly-transformation method, Pt and Fe NPs were encapsulated in hollow ZSM-5 (Pt–Fe@ZSM-5) and the catalyst efficiently converted styrene into ethylbenzene. However, it was substantially less active in converting cis-cyclooctene due to mass transport limitations and the strong steric hindrance around the CC moiety.54 Rather than encapsulating the NPs in a central large hole, Pd NPs (∼9.5 nm) were placed in the mesopores of silicalite-1 (Pd@S-1). Owing to the limited size of the zeolite apertures, 3-methyl-2-butenal (0.38 × 0.62 nm) and cinnamaldehyde (0.54 × 0.92 nm) could diffuse into the NPs but not the larger 3,3-di-phenylacrylaldehyde (0.81 × 1.0 nm).59
The transformation of 2D zeolites into 3D ones also promotes a high intermolecular selectivity as demonstrated by several studies. For example, 3-nitrotoluene was hydrogenated over Pd@MCM-22,56 but not 1-nitronaphthalene, whose dimensions (0.73 × 0.66 nm) are larger than the zeolite pores (0.41 × 0.51 nm). Pd@FER39 (FER: ferrierite) promoted the excellent conversion of 1-hexene (90%) and benzaldehyde (90%) versus very low (∼10%) or no conversion of diphenylmethanone and 1-phenyl-1-cyclohexene, respectively. The selective behavior of both catalysts did not hold for their homologues with surface-supported NPs. A different behavior was reported for Pt@MCM-22 (ref. 55) due to incomplete encapsulation. The platinum SAs and ACs (0.2–0.7 nm) were mostly confined in the internal cages accessible uniquely through 10-membered ring windows, yet a part of the Pt species was located at the external cups on the surface of MCM-22. The small-sized propylene was able to diffuse through the zeolite 10 membered ring windows and reach the encapsulated metal clusters; consequently, a five times higher reaction rate was achieved compared to the impregnated homologue. On the other hand, the larger isobutene molecule could not diffuse through the 10-membered ring windows, yet it was converted over Pt species present in the external cups of with similar rate to that of the supported homologue. In this example, the higher activity of the confined NPs is explained by the higher turnover frequencies obtained by subnanometric Pt nanoparticles in the confined samples. It also highlights the importance of full confinement or the need to extract the eventual surface supported species to avoid drops in intermolecular selectivity.
Intermolecular selectivity effects were also investigated with structural and spatial isomers as substrates. A Pt/Al2O3 catalyst covered in a silicalite-1 shell exclusively hydrogenated para-xylene to 1,4-dimethylcyclohexane; meanwhile, the uncoated catalyst converted all xylene isomers (Fig. 3).41 Selectivity was attributed to the silicalite-1 sheath that allowed only para-xylene to diffuse through the micropores and interact with the metal functionality. The high selectivity was conserved even under the equimolar feed of para-/ortho- and para-/meta-xylene. In a follow-up study, the same workgroup introduced an additional H-ZSM-5 layer over the silicalite-1 shell; this modification promoted isomerization of meta- to para-xylene and consequently increased product yield by generating more para-xylene illegible to diffuse through the silicalite-1 molecular sieve towards the platinum catalyst.42
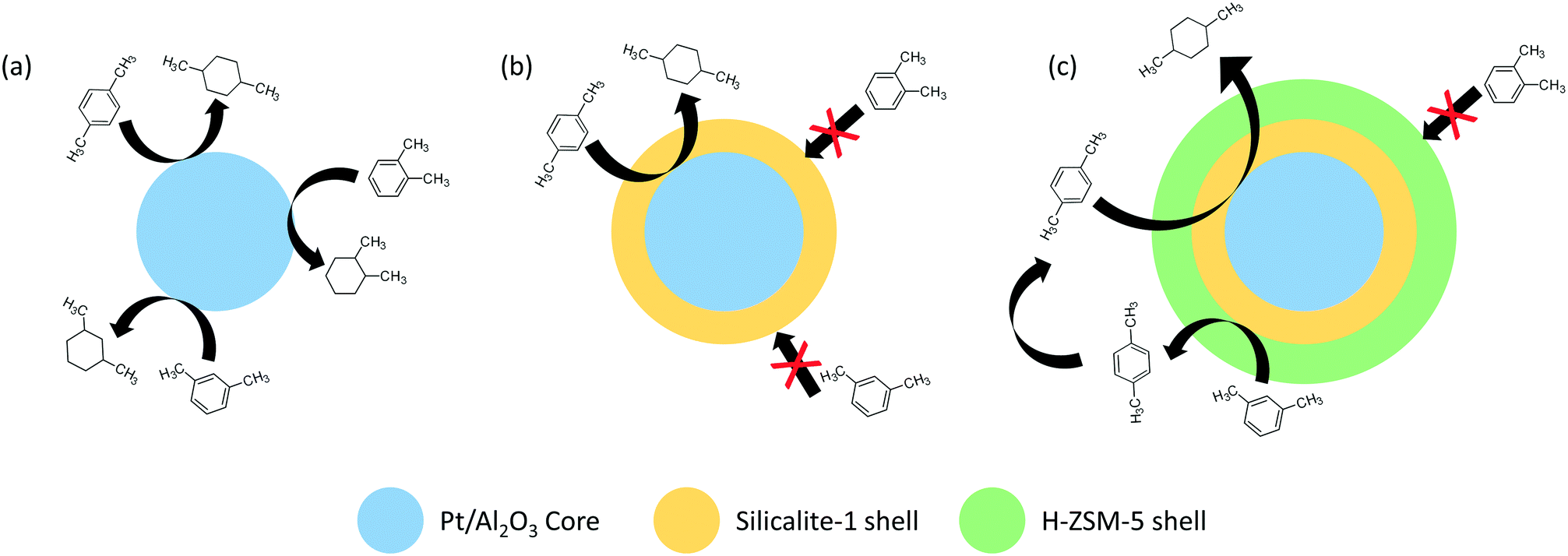 | ||
| Fig. 3 Illustration of the hydrogenation of ortho-, meta-, and para-xylene over (a) Pt/Al2O3, (b) Pt/Al2O3 core covered with silicalite-1 shell, and (c) Pt/Al2O3 core covered with silicalite-1 shell and H-ZSM-5 shell. | ||
In another example, Pt NPs encapsulated between two silicalite-1 layers, a core and a shell, selectively hydrogenated 1-hexene but not cyclohexene.43 Due to the difference in their molecular size, 1-hexene was able to cross the microporous shell and reach the Pt NPs, whereas the diffusion of cyclohexene was restricted. In the absence of a zeolite sheath, both substrates were fully converted with no selectivity.
An elegant illustration of discriminating the spatial isomers of triacylglycerol was reported by Sels and coworkers.50 In their work, the authors prepared a Pt encapsulated in ZSM-5 catalysts, by host–guest assembly, that selectively hydrogenates the CC unsaturations of trans-triacylglycerol. Due to its slimmer shape, the trans-triacylglycerol was significantly better adsorbed by the zeolite than the cis-isomer. Additionally, the hydrogenation of the central fatty acid chain (sn-2) was favored over the external ones (sn-1 and sn-3). The authors proposed that hydrogenation proceeded by adsorbing the substrate in fork conformation, as illustrated in Fig. 4, with the central chain protruding in the pore aperture and thus allowing the contact of the double bonds with the platinum particles placed in the pore opening, whereas the external chains (sn-1 and sn-3) were adsorbed on the external zeolite surface.
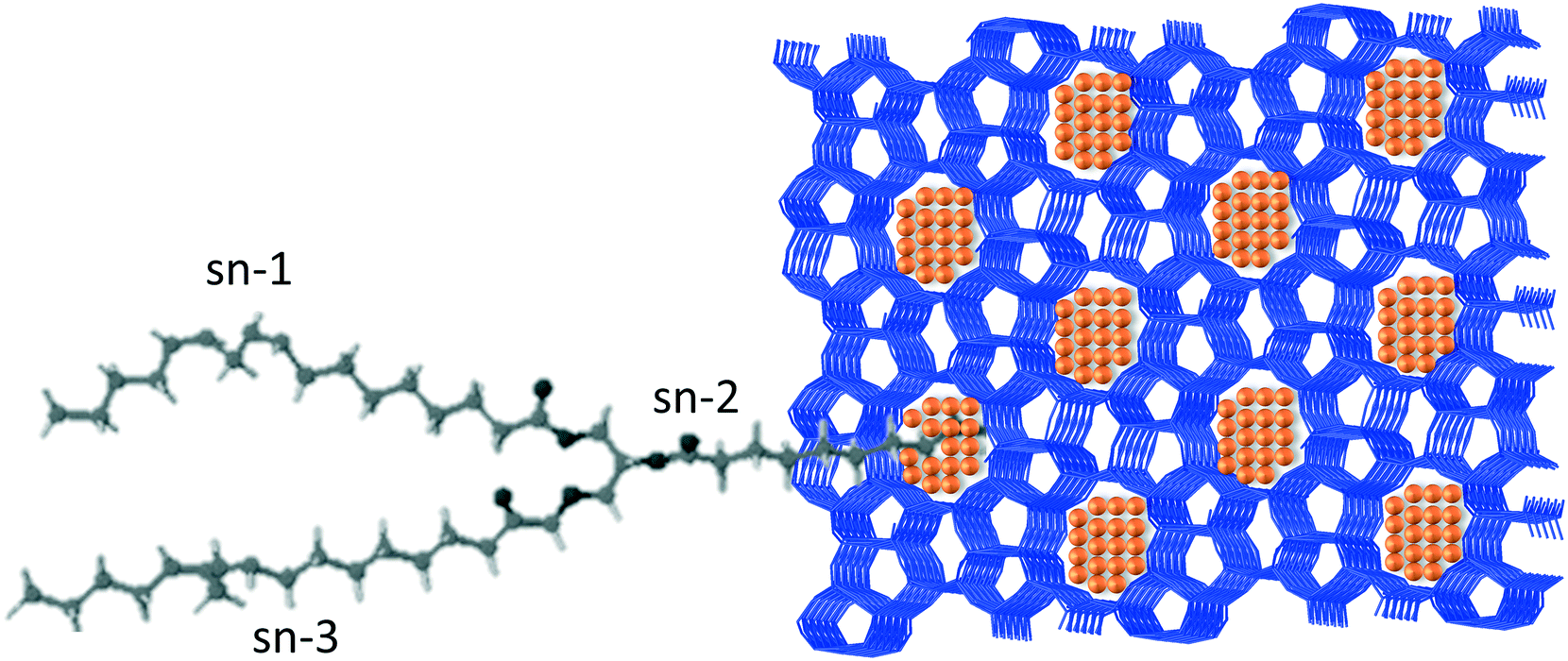 | ||
| Fig. 4 Sketch of the adsorption of triacylglycerol in turning fork conformation on a Pt nanoparticle encapsulated in a ZSM-5 zeolite, proposed by Sels and coworkers.50 This conformation explains the selective hydrogenation of the CC unsaturation in the central fatty acid chain. | ||
2.3. Intramolecular selectivity
Based on the available literature, intramolecular selectivity can be classified into four groups on the basis of the reagents considered: (a) semihydrogenation of acetylene (HCCH) to ethylene, (b) selective hydrogenation of a functional group in a molecule with multifunctional groups, (c) asymmetric hydrogenation, and (d) tandem reactions. Understanding intramolecular selectivity often requires detailed investigations on the interactions between the zeolite and the reagents, intermediates, and possible products. For such studies, spectroscopic measurements and theoretical calculations are often used.
Furfural is another interesting molecule to investigate intramolecular hydrogenation selectivity. It can be converted into distinct products based on the location of the active sites, the interactions of transition states and products with the internal zeolite environment, and/or the polarity of the zeolite encapsulant. For instance, this molecule was converted into furan in 98.7% selectivity over Pd NPs (7 nm) encapsulated in silicalite-1 by the core–shell method, versus 5.6% selectivity over the supported NPs of equal size.45 The selectivity was correlated to the fast desorption of furan from the zeolite compared to the other products. FTIR desorption studies revealed that the C–O band intensity of furan dropped faster than those of the starting material, intermediates, and side products. This result was further confirmed by the theoretical calculations that predicted the easier desorption of furan from the micropores due to their lower absolute value of adsorption energy (−6.06 eV) than the starting material (−6.18 eV) and the other possible side products, such as tetrahydrofuran (−7.05 eV), methylfuran (−7.05 eV), furfuryl alcohol (−9.34 eV), and tetrahydrofurfuryl alcohol (−9.75 eV). The selective conversion of furfural into furan can be also affected by the hydrophilicity of the zeolite host. Encapsulating Pd NPs in hydrophilic silicalite-1 (Pd@S-1-OH-10) crystals resulted in the full conversion to furan at 250 °C, versus 90% yield over Pd@S-1 catalyst with non-modified silanols and only 5.6% over the supported Pd/S-1.61 The improved catalytic performance of Pd@S-1-OH was attributed to the more hydrophilic zeolite sheath that promoted the adsorption of furfural on the one hand, and favored the desorption of furan rather than other byproducts on the other hand. It is noteworthy that the hydrogenation over supported NPs (Pd/S-1 and Pd/Al2O3) produced bulky molecules originating from the condensation of furfural; these were not detected upon using Pd@S-1-OH-10 or Pd@S-1 catalysts.
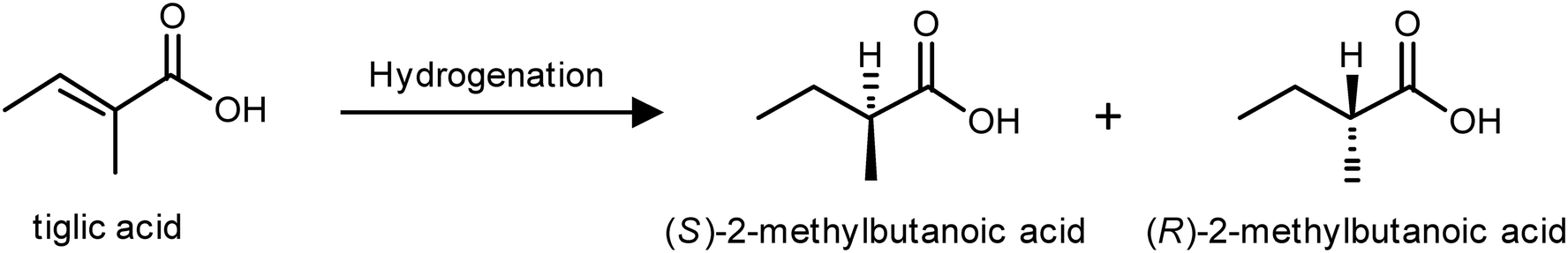 | ||
| Scheme 1 Hydrogenation products of tiglic acid. | ||
In another example, acetophenone was converted into to (R)-1-phenylethanol over the chiral Ru complex (1S,2S)-DPEN-Ru(TPP)2 encapsulated in the supercages of zeolite Y by host–guest assembly.63 In the absence of ligands, the Ru–NaY catalyst demonstrated very weak conversion (6.1%, Table 3) with no enantiomeric excess. On the other hand, the encapsulated chiral complexes achieved 37.7% to 100% conversion with larger values recorded for catalysts prepared from zeolites containing smaller cations. Additionally, DFT calculations unveiled that the adsorption energy of tetraphenylporphyrin (TPP) and 1,2-diphenyl-1,2-ethylenediamine (DPEN) ligands within the zeolite framework are the lowest in the presence of Li+ cation. These weak interactions render the complex and the substrate more free and less affected by the zeolite internal environment.
| Catalyst | Ru content (%) | Conversion (%) | ee (%) |
|---|---|---|---|
| Ru–NaY | 1 | 6.1 | 0 |
| (1S,2S)-DPEN-Ru(TPP)2/Li-Y | 0.66 | 100 | 75.3 |
| (1S,2S)-DPEN-Ru(TPP)2/Na-Y | 0.24 | 100 | 61.5 |
| (1S,2S)-DPEN-Ru(TPP)2/K-Y | 0.15 | 54.8 | 50.3 |
| (1S,2S)-DPEN-Ru(TPP)2/Cs-Y | 0.19 | 37.6 | 45.8 |
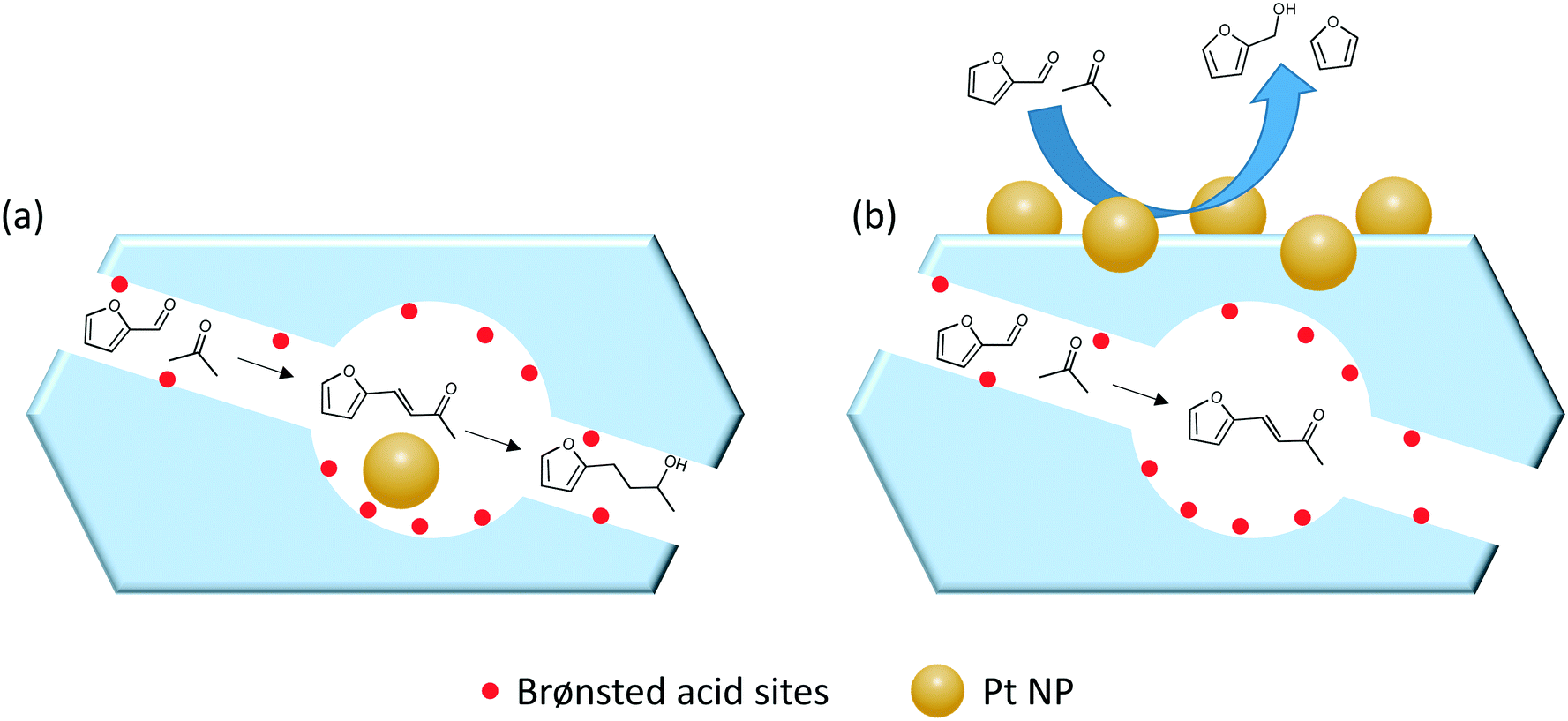 | ||
| Fig. 5 Illustration of the different product obtained during the tandem reaction of furfural with acetone over Pt NPs (a) encapsulated in H-ZSM-5 and (b) supported on the surface of H-ZSM-5. | ||
The relation between the internal zeolite environment and the product distribution was also investigated by converting furfural over Pt NPs confined, following the in situ preparation approach, in silicalite-1, Na-ZSM-5 and H-ZSM-5. The microenvironment significantly modify the reaction pathways and, consequently, the product distribution, as exemplified in Scheme 2.33 Both silicalite-1 and Na-ZSM-5 favor the adsorption of furfural in perpendicular configuration and hence promote the interaction with the aldehyde group. The inadequate activation of H2 on Pd@silicalite-1 resulted in the direct decarbonylation into furan (>70% selectivity). Meanwhile, the presence of Al in the framework of Pd@Na-ZSM-5 favored higher and stronger adsorption of H2 as exemplified by H2-TPD studies; this resulted in a more selective formation of furfural alcohol (>90% selectivity). In contrast, a non-selective behavior was reported for Pd@H-ZSM-5 that simultaneously produced furan via decarbonylation, furfural alcohol via hydrogenation, and 1,5-pentanediol (PDO) through hydrogenolysis.
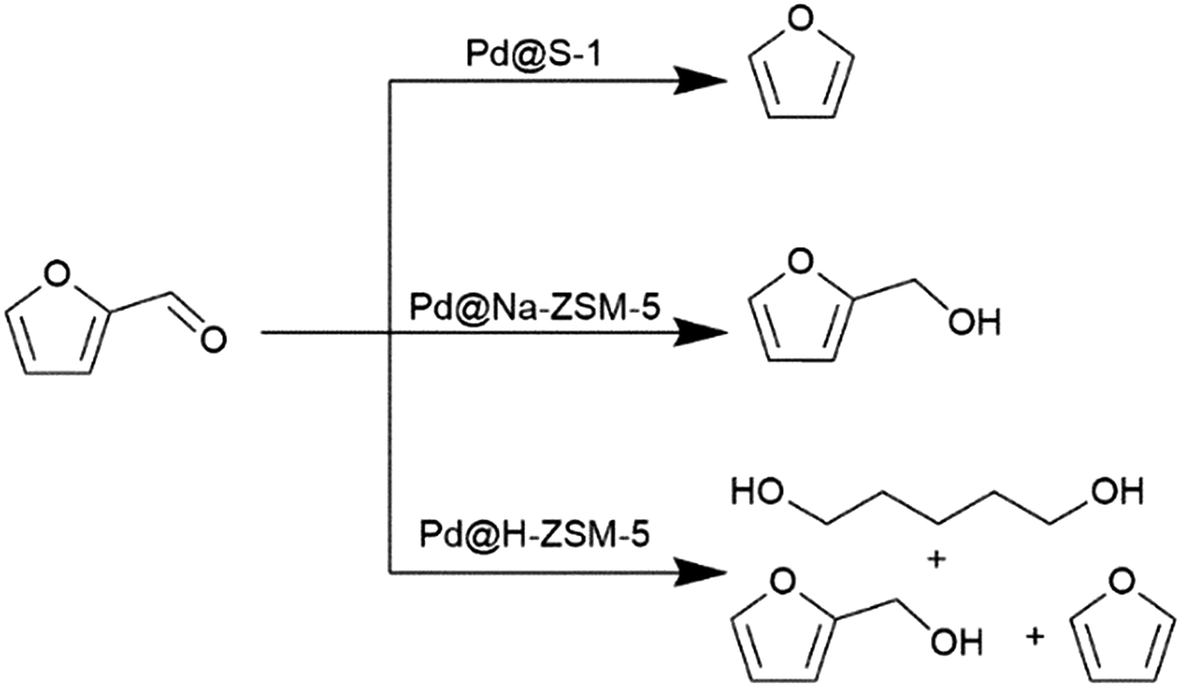 | ||
| Scheme 2 Product distribution of furfural hydrogenation over Pd NPs confined in MFI zeolite. | ||
3. Confinement effects in MOF based hydrogenation catalysts
Metal organic frameworks (MOFs) are well-ordered structures composed of metal ions or clusters connected to each other by coordinating organic linkers.64 Research on MOFs is a highly active field with currently over 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reported MOF crystal structures, owing to the large number of available metals and organic linkers.64 However, research on MOF-based catalysts is primarily aimed towards the synthesis of fine chemicals, while few MOFs are being developed for use in the synthesis of commodity chemicals.65,66
000 reported MOF crystal structures, owing to the large number of available metals and organic linkers.64 However, research on MOF-based catalysts is primarily aimed towards the synthesis of fine chemicals, while few MOFs are being developed for use in the synthesis of commodity chemicals.65,66
Compared to zeolites, MOFs present several advantages. While zeolites mostly possess a pore size below 1 nm, many MOFs have larger pores. This feature allows the diffusion of bulkier substrates that cannot be processed with zeolites. As a result, MOFs are developed for the production of larger and more complex compounds than commodity chemicals. Furthermore, predicting the structure and properties based on the linker and coordinating metal is sometimes possible for MOFs, thus enabling rational design for the conversion of complex molecules.65–67
On the other hand, MOFs suffer from several limitations for the synthesis of commodity chemicals which are often simpler and tend to be produced on a large scale. The production of MOFs is currently still expensive and on a relatively small scale, thus making it difficult to compete with current catalysts. Furthermore, despite the significant improvement in the thermal stability of MOFs, they still tend to decompose at temperatures above 400 °C. This limits their application to liquid phase reactions and gas phase reactions that do not employ high temperatures.65 Furthermore, it is improbable that MOFs are capable of withstanding long term corrosion and mechanical wear, which makes them unsuitable for use in fluidized bed reactors, where the chance of breaking apart is greater.65
Other prominent applications for MOFs are in membranes for separation due to their well-defined pore size and the large variations in composition and pore geometries.68,69 It is these properties that make MOFs also suitable for size-selective reactions. Metal active species are confined within the voids of MOFs for the use in several catalytic reactions such as hydrogenation and oxidation.
3.1. Synthesis methods for confining metal species within MOFs
MOF-Based size-selective catalysts are similar to zeolite catalysts in the sense that they are mostly composed of a catalytically active metal covered in a shell. As such, some preparation methods are comparable to the preparation methods observed in zeolite-based catalysts. Examples include the commonly used core–shell method,70–92 and host–guest methods (impregnation,93–96 ion exchange,97,98 and chemical vapor infiltration99). Preparation procedures that are different from those observed in zeolite synthesis generally take advantage of the unique metal and linker composition of MOFs. These include sacrificial approaches,100–109 partial decomposition of the MOF,110 and post-synthetic modification.111–113 These synthetic approaches are sketched in Fig. 6.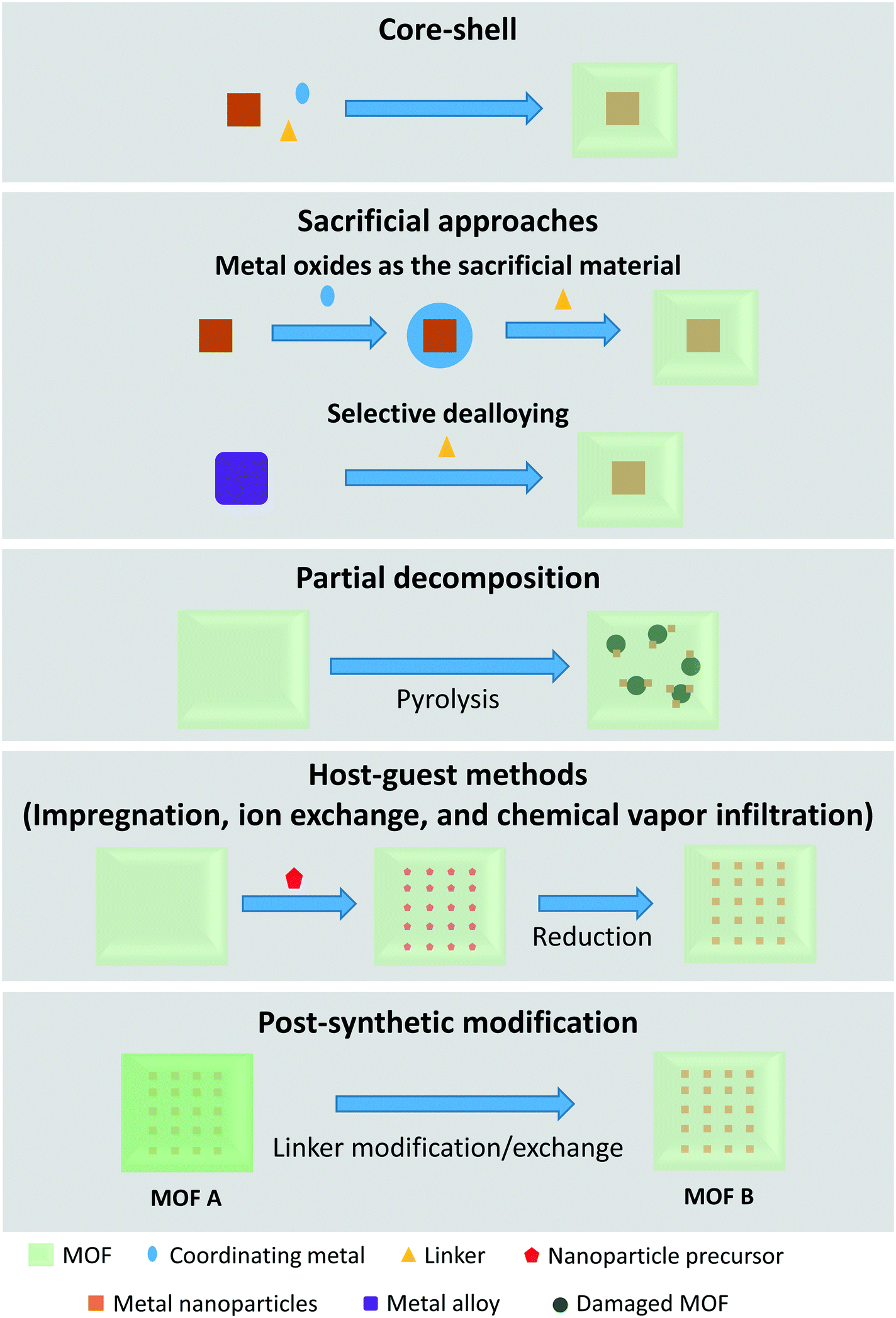 | ||
| Fig. 6 Methods of confining metal species in MOFs. | ||
The core–shell approach for encapsulating materials in MOFs is very similar to that described to encapsulate metal species in zeolites; pre-synthesized materials, such as freestanding or supported NPs, are coated with one or more MOF layers following a secondary growth synthesis step. However, catalysts that are prepared by these core–shell procedures often require chemicals to stabilize the NPs or enhance the affinity of the NPs with the MOF in order to promote the formation of a complete shell. Polyvinylpyrrolidone (PVP) is commonly used for these applications. Other materials have also been used to enhance the growth of a MOF shell and to achieve full protection of the particles. Some examples are polydopamine70 and poly(sodium-4-styrenesulfonate) (PSS).71
The mechanism by which NPs are encapsulated is suggested to be adsorption onto the growing MOF until all free particles are depleted.72 Accordingly, this also allows for manipulation of the spatial distribution of NPs by varying the time at which the nanoparticles are introduced during the MOF synthesis; immediate or early addition results in complete encapsulation of all particles, although they are often located deep inside the MOF crystal. By delaying the addition, the particles are located closer to the surface until at some point no encapsulation takes place. It is suggested that the synthesis of the MOF may have already been completed by the time the particles are added, which causes the lack of encapsulation.73,74
Nanoparticles can also be encapsulated using an emulsion synthesis approach where the aqueous phase contains the coordinating metal and the organic phase contains the linker. The formation of the MOF takes place on the water/organic interface and the shell thickness can be controlled by adjusting the crystallization time or by changing the amount of precursors. This approach is capable of producing both solid and hollow MOF shells.92
The synthesis of metal-encapsulated MOFs and zeolites by the core–shell method shares the same advantages: generally good encapsulation of the metal functionality and a fine size control of the predesigned nanoparticles. However, they also share the disadvantage of being a multistep synthesis method since the nanoparticles have to be prepared beforehand. Furthermore, the reagents that stabilize the NPs and ensure a good affinity between the particles and the MOF surface are generally hard to remove and can have negative influences on the catalytic properties of the final catalysts.75
A modified method to core–shell exists where metal precursors are added during the MOF synthesis instead of pre-synthesized materials.114–116 These metal precursors are reduced in situ to NPs or ACs before being incorporated in the MOF. This method, which can be considered as an in situ approach, does not require any chemicals to stabilize the NPs. The successful confinement relies on the faster reduction of precursor than the formation of the MOF. When the reduction of the metal precursor is much slower than the formation of the MOF instead, the formed metallic particles will be located primarily on the external surface. This problem has been avoided by adding additional reductants or by adding chemicals to slow down the formation of the MOF.114,115
The location of nanoparticles in a MOF prepared from metal oxides can be determined by adjusting the ligand concentrations, as exemplified in Fig. 7. The reaction follows a “dissolution–precipitation” mechanism at high ligand concentrations; a large amount of cations coordinate with the ligand and the complex is rapidly released into solution. MOF nucleation occurs in the oversaturated solution and deposits onto the surface of the metal oxides. Continued growth connects the MOF crystals resulting in a multi-crystalline layer. The deposition of the MOF from solution causes the nanoparticles to be located on the interface between the remaining metal oxide and the MOF.
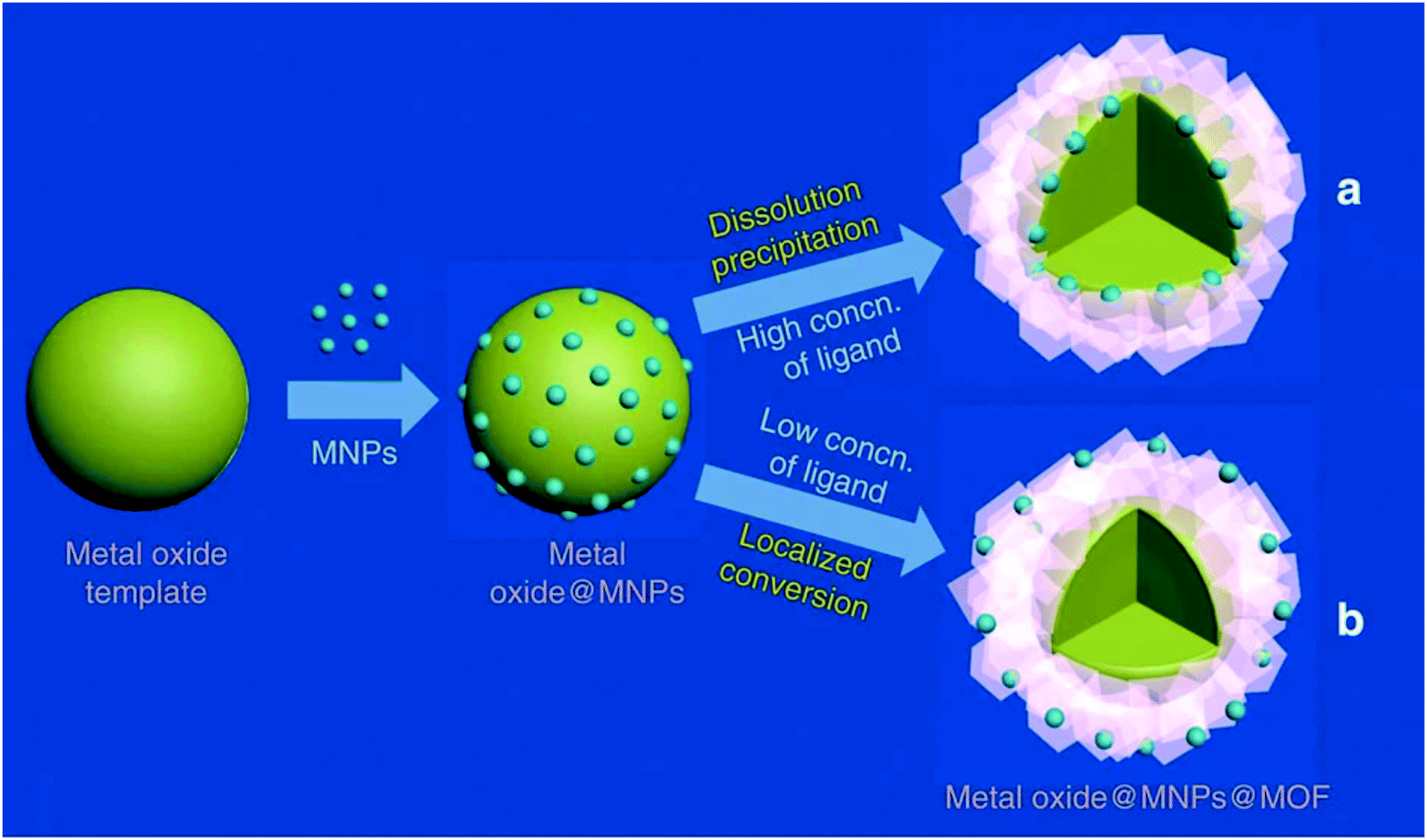 | ||
| Fig. 7 Illustration of the (a) dissolution precipitation mechanism which results in NPs at the interface of the MOF and the metal oxide, and (b) the localized conversion mechanism which results in NPs close to the MOF surface. Reproduced with permission from ref. 100. | ||
On the other hand, low ligand concentrations result in a “localized conversion” mechanism where the nucleation concentration cannot be reached during the synthesis. Instead, the MOF is directly produced on the metal oxide surface by means of a heterogeneous reaction. The growth pushes the NPs up and a material is obtained where the NPs are close to the surface of the MOF.100 Alloys are an alternative to metal oxides as sacrificial materials. The MOF shell is formed from alloys by selectively etching one metal in the presence of a linker.
In general, the size and structure of the particles loaded on the metal oxides are retained after the MOF synthesis. In the case of alloys, the etching process can result in porosity in the encapsulated metal.106,108 Furthermore, this method is widely applicable, with various types of NPs and sacrificial materials.
3.2. Intermolecular selectivity
The mechanism of intermolecular selectivity for MOFs-based catalysts is the same as that reported for zeolite-based ones. Interestingly, some MOFs possess a flexible structure that allows for the accommodation of substrates that are slightly larger than the pore openings. A significant amount of research focuses on developing intermolecular selectivity by confining NPs or ACs in a MOF. However, as of writing this review, applications of size selectivity are mostly limited to model substrates and the majority of the research is aimed towards developing and optimizing catalysts.Olefins and acetylenes are by far the most frequently used substrates for investigating the intermolecular selectivity of MOFs; the importance of these compounds lies in their facile hydrogenation with the rare formation of side products and their availability in a multitude of structures and molecular sizes. Furthermore, olefins are also used to prove the full confinement of the particles inside the MOF; reactants that are larger than the pore openings can only be converted when defects are present or when the particles are not fully encapsulated. 1-Hexene, cyclohexene, and cyclooctene are amongst the most frequently used substrates for demonstrating intermolecular selectivity (Table 4), since their molecular sizes are of comparable dimensions to the pores of a number of popular MOFs, such as ZIF-8. Surprisingly, the purely intermolecular hydrogenation of aldehydes is rare considering that unsaturated aldehydes are relevant substrates for catalysts displaying intramolecular selectivity. However, aldehydes are more prone to side reactions, which might explain their low interest.
| Preparation method | Catalyst | Conversion (%) | Ref. | ||
|---|---|---|---|---|---|
| 1-Hexene
|
Cyclohexene
|
Cyclooctene
|
|||
| a Values are estimated from bar graphs. | |||||
| Core–shell | Pd–mSiO2@ZIF-8 | n/a | 5.7 | n/a | 71 |
| Pt/ZIF-8 | 7.3 | n/a | 0 | 72 | |
| Pd@ZIF-8-1 | 70.5 | 7.5 | 0 | 73 | |
| meso-ZIF-8-Pt | 16.3 | n/a | n/a | 74 | |
| Pt0.74@ZIF-8 | 90.2 | n/a | 2.7 | 75 | |
| Pt-0.62@ZIF-8 | 76.4 | 0.13 | 0 | 76 | |
| Au@ZIF-8 | 10 | 0 | n/a | 77 | |
| Pd/SiO2@ZIF-8-II | 100 | 7.5 | 0 | 78 | |
| Pd/MSS@ZIF-8 | 100 | 13.8 | n/a | 79 | |
| Pd/ZSM-5@ZIF-8 II | 100 | 3.3 | n/a | 80 | |
| Pd NCs@ZIF-8 | ∼100 | 27 | 0 | 82 | |
| Pt–ZIF-8 hybrid thin film | 14.9 | n/a | 0 | 83 | |
| Pt/ZIF-8 hybrid thin films | 40 | n/a | 0 | 84 | |
| Pt–UiO-66 | 100 | n/a | 65.99 | 85 | |
| Pd@ZIF-L-1 | 75.7 | 32.6 | 7.4 | 86 | |
| 1:1 Pt/UiO-66 |
100 | n/a | n/a | 114 | |
| PtCo@UiO-66 | 100 | n/a | n/a | 115 | |
| Pd@NH2–UiO-66 | 100 | n/a | n/a | 116 | |
| Zn-MOF-74@(Pd@Fe2O3) | 100 | ∼40a | 5 | 120 | |
| Sacrificial approaches | Pt@ZIF-8-‘in’ | 7 | n/a | 0 | 100 |
| Pt@ZIF-8-‘sur’ | 21 | n/a | 0 | 100 | |
| Pd/ZnO@ZIF-8 | 100 | 13.9 | 0 | 101 | |
| Pd/F–ZnO@ZIF-8 | 100 | n/a | 0 | 102 | |
| PtAuDNP@HKUST-1 | 25 | n/a | n/a | 103 | |
| Pd@ZIF-8 | 98 | n/a | 0 | 104 | |
| RhCoNi@MOF-74(Ni) | 99.5 | 78.1 | n/a | 105 | |
| RhCoNi@ZIF-67(Co) | 98.6 | 9.9 | n/a | 105 | |
| Impregnation | 0.1 mmol Ag@HKUST-1 | ∼100a | ∼100a | ∼0a | 93 |
| 10.7 wt% Pt@UiO-66–NH2 | 13.4 | n/a | n/a | 94 | |
| Post-synthetic modifications | Pt@SALEM-2 | n/a | 7 | n/a | 111 |

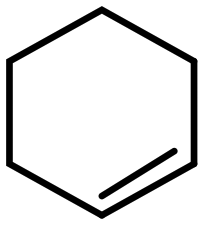
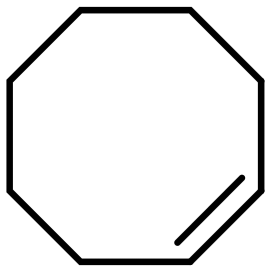
Numerous contributions have been devoted to investigate intermolecular selectivity over metals encapsulated in ZIF-8, a zeolitic imidazolate framework composed of a sodalite type structure with 11.6 Å cages connected through 3.4 Å six-membered windows.72,121 The majority of materials were synthesized by the core–shell method. For example, this synthesis route was used to grow the ZIF-8 shell around Pt NPs capped with PVP. The activity of the catalyst was demonstrated by the ability to hydrogenate 1-hexene, but conversions were low due to poor diffusion. No hydrogenation products were obtained when the same reaction was performed with cis-cyclooctene, which is larger than the pore openings of ZIF-8, demonstrating the size-selective nature of the Pt@ZIF-8 catalyst. Furthermore, excellent regioselectivity was obtained over this catalyst; 1-hexene was hydrogenated but not trans-2-hexene whose CC moiety could not reach the Pt NPs.72 Wang et al. similarly demonstrated that the hydrogenation rate of CC bonds over Pt@ZIF-8 decreases when the bond is located in the middle of the molecule using 3-methyl-2-butene-1-ol and 3-methyl-3-butene-1-ol. They attributed this observation to an easier adsorption of the terminal CC bond compared to the CC bond in the middle.76 This selectivity is not limited to Pt, Au@ZIF-8 was completely inactive towards 3-hexene but converted 1-hexene under the same conditions.77
The importance of proper encapsulation was demonstrated by Xing et al. who compared catalysts with all particles inside, all particles outside, and particles on the inside and outside in the hydrogenation of olefins. All catalysts are capable of hydrogenating cyclohexene and gave 7.5%, 46.7% and 13.4% conversion for inside, outside, and inside and outside, respectively. However, the hydrogenation of cyclooctene only proceeded in the presence of palladium on the surface.73
Confined metal particles often demonstrate lower apparent catalytic activity compared to surface supported particles due to the slow diffusion of substrates through the pores. Huo et al. alleviated this problem by selectively etching out nanoparticles that are embedded in MOFs. This process leaves behind highly defined mesopores while maintaining the integrity of the MOF, resulting in enhanced diffusion while the selectivity is unaffected. Mesoporous Pt@ZIF-8 was fabricated by etching out gold NPs while the Pt NPs remain intact; the presence of the mesopores resulted in an increased conversion in the hydrogenation of 1-pentene, 1-hexene, and 1-heptene, whereas the catalyst remained unable to convert the larger cyclooctene.74
As previously discussed, stabilizers are used to improve the affinity of the encapsulated materials with the MOF during core–shell synthesis. Modifying Pd/SiO2 with poly(diallyldimethylammonium chloride) (PDDA) and poly(sodium-4-styrenesulfonate) (PSS) introduced a negative charge on the particle surface. The negatively charged surface is compensated by Zn2+ ions thereby enhancing the formation of a continuous ZIF-8 shell. The sieving effect of Pd/SiO2@ZIF-8 catalyst was proven by a decrease in conversion with increasing substrate size in the hydrogenation of olefins. However, the ability to hydrogenate cyclooctene, albeit with a low conversion, was attributed to the presence of defects in the ZIF-8 membrane shell. A second ZIF-8 growth cycle increases the density and thickness of the MOF shell, which made the catalyst completely inactive in the hydrogenation of cyclooctene.78 Furthermore, the catalyst was also protected from bulky sulfur poisons such as triphenylmethyl mercaptan.78,79 PDDA and PSS have also been used to facilitate the growth of ZIF-8 on Pd/ZSM-5 particles. The advantage of the coating was demonstrated with a control experiment where ZIF-8 was grown on ZSM-5 without the PDDA and PSS; the resulting material only contained a few scattered MOF particles on the surface of the zeolite.80 Another alternative to a PDDA and PSS coating is a temporary coating of Cu2O, which was used during the synthesis of a Pd@ZIF-8 catalyst. The authors mention that the clean Cu2O surface assists in the formation of the ZIF-8 shell and it is etched off spontaneously and simultaneously during the formation of ZIF-8. The process leaves behind a cavity, resulting in a hollow ZIF-8 structure with a Pd nanocrystal inside. The good activity in the hydrogenation of ethylene and cyclohexene, and the inability to hydrogenate the larger cyclooctene proved that the layer was pristine.81 PVP-capped Pd nanocubes encapsulated in hollow ZIF-8 spheres prepared using an inverse emulsion similarly display excellent size selectivity. Pd@ZIF-8 catalysts were capable of reducing 1-hexene with good to excellent yields (92–58%) depending on the thickness of the MOF shell. trans-Stilbene, which is larger than the pore openings of ZIF-8, could be converted (4.3–1.3%) due to diffusion through the mesoporous voids between ZIF-8 nanocrystals. No conversion was observed for the larger tetraphenylethylene.92 Hollow ZIF-8 containing Pt nanoparticles have also been prepared by growing ZIF-8 around Pd supported on carboxylate-terminated polystyrene spheres and subsequently removing the polystyrene with DMF. The coordination ability of zinc ions with the carboxylate groups of the polystyrene ensured that a ZIF-8 shell is obtained. Full conversion of 1-hexene was obtained whereas no conversion was observed for cyclooctene, diphenylethylene and triphenylethylene.122
As mentioned earlier, residual stabilizing agents on the surface of NPs can also adversely affect the catalytic activity of a catalyst. To avoid their use, Li et al. reported the preparation of Pt@ZIF-8 starting from nanoparticles stabilized by 2-methyl imidazole, the linker of ZIF-8. Pt@ZIF-8 catalysts with various Pt loadings demonstrated higher turnover frequencies than Pt/C for the hydrogenation of 1-hexene. In contrast, very low conversion was observed during the hydrogenation of the larger cyclooctene over the confined NPs while Pt/C efficiently catalyzed the hydrogenation of this substrate.75 Well-encapsulated Pd nanoparticles could be obtained by the addition of [(5,5′-dicarboxy-2,2′-bipyridine-)palladium(II)] dichloride during the synthesis of UiO-67 followed by reduction. The 2,2′-bipyridne-5,5′-dicarboxylic acid ligand of this complex can act as linker for UiO-67. Furthermore, the use of this complex does not affect the crystallinity of the resulting MOF and the resulting Pd nanoparticles are uniformly distributed in the MOF. The catalyst achieved full conversion in the hydrogenation of styrene, while no conversion is observed in the hydrogenation of the much larger tetraphenylethylene. Under the same conditions a Pd0/UiO-67 catalyst prepared by impregnation achieved 24% conversion of tetraphenylethylene.123
ZIF-8 thin films containing nanoparticles can be produced by spin coating the desired NPs onto a glass substrate covered with ZIF-8, after which a new layer of MOF is grown on top. The steps of spin coating and MOF growth can be repeated until the desired thickness is reached. Furthermore, different nanoparticles can be combined in one layer or placed in separate layers. A Pt-ZIF-8 hybrid thin film produced in this manner was completely inactive in the hydrogenation of cis-cyclooctene; however, 1-hexene was successfully hydrogenated, albeit with a low conversion (14.9%) which was presumed to be due to the low loading and small aperture size of ZIF-8.83 Excellent regioselectivity can also be obtained over larger metal structures; for example, metal layers encapsulated in a MOF displayed regioselectivity in addition to size selectivity in the hydrogenation of olefins. These catalysts were prepared by sputter coating a layer of metal onto a MOF surface and subsequently growing a MOF shell around the metal. The process of MOF growing and sputtering can be repeated until the desired thickness is reached. The catalyst is finalized by adding several layers of MOF over the last metal layer to prevent it from peeling off. A Pt@ZIF-8 prepared by such method gave 40% conversion of 1-hexene; meanwhile trans-2-hexene and cis-2-hexene were converted in only 6.8% and 1.0%, respectively.84
The second most investigated structure as encapsulant is UiO-66, a MOF with a pore entrance of 6 Å. Several examples were prepared by the in situ method, a modification of the core–shell synthesis. Luque, Lin et al. prepared a Pt@UiO-66 catalyst that displayed size selectivity in the hydrogenation of alkenes. A sufficiently fast reduction of the metal precursor compared to the formation of the MOF was ensured by accelerating the reduction with hydrogen as additional reductant and by slowing down the formation of UiO-66 with the addition of acetic acid. Therefore, the formation of metal particles on the outside of the MOF could be prevented. The Pt@UiO-66 catalysts prepared in this manner achieved full conversion of 1-hexene, however, little to no hydrogenation of tetraphenylethylene was observed under identical experimental conditions.114In situ preparation is not limited to a single metal at a time; a combination of Pt and Co precursors was used for the synthesis of PtCo@UiO-66. Similarly, hydrogen is added to achieve a sufficiently fast reduction of the metals. The platinum precursor is capable of reducing on its own under the reaction conditions while the reduction of cobalt is enhanced by the presence of Pt–H species, resulting in encapsulated PtCo alloy particles. The bimetallic catalyst achieved full conversion of 1-hexene while a monometallic Pt@UiO-66 catalyst achieved 75% conversion under the same conditions. Both catalysts converted only a negligible amount of the very bulky tetraphenylethylene.115 The preparation of Pd@NH2–UiO-66 did not require the addition of hydrogen; the amine functionalities of NH2–UiO-66 grant it an excellent coordination capability for metal ions which enhances the encapsulation. XPS analysis show a strong interaction between Pd and the nitrogen atoms of the amine. The importance of the amine functionality was further demonstrated by preparing Pd@UiO-66 without amine functionality; a catalyst prepared under similar conditions suffered from aggregation and most of the Pd was located on the outside. The well encapsulated Pd@NH2–UiO-66 catalyst was capable of hydrogenating the smaller 1-hexene and styrene with excellent conversion, but was completely inactive in the hydrogenation of the much larger tetraphenylethylene.116
The selective substrate conversion is dependent on the size of the pore openings, therefore different MOFs were used to accommodate larger substrates. Pt@UiO-66 with pore openings of 6 Å successfully hydrogenated molecules up to the size of triphenylethylene but not the larger tetraphenylethylene.85 Similarly, ZIF-L (pore size: 6.6 Å) could also accommodate and hydrogenate substrates which do not fit into ZIF-8, such as cyclooctene.86
Luo et al. synthesized Zn-MOF-74@(Pd@Fe2O3) by using an in situ method, taking advantage of the reaction between the linkers of MOF-74 and Fe3+, and the favorable reduction potentials of iron and palladium. Pd0 and Fe3+ are spontaneously formed in solution by the combination of Pd2+ and Fe2+ species. The resulting catalyst demonstrated size selectivity in the hydrogenation of a wide range of olefins with full conversion for small olefins such as 1-pentene, cyclopentene and 1-hexene, but only reaching 5% in the case of the bulkier cyclooctene.120 The sacrificial synthesis approach has been explored for the encapsulation of supported nanoparticles in ZIF-8. Zhang et al. produced a Pd/ZnO@ZIF-8 catalyst using a sacrificial approach starting from Pd/ZnO and 2-methylimidazole. The use of the sacrificial material resulted in stronger interactions with the core and produced a more continuous layer of MOF which was also less liable to peel off. However, the continuity of the shell was dependent on the dissolution speed of the ZnO metal oxide: a continuous shell could only be formed when the dissolution kinetics were slower than the kinetics of ZIF-8 formation. The growth of the MOF shell was also limited to a certain thickness, after which the linker and Zn2+ could hardly pass through the continuous ZIF-8 layer. Well encapsulated Pd/ZnO@ZIF-8 fully converted 1-hexene with a lower rate than Pd/ZnO due to steric hindrance caused by the ZIF-8 layer. The hydrogenation of bulkier cyclohexene over the well encapsulated Pd/ZnO@ZIF-8 catalyst resulted in 13.9% conversion and no conversion of cyclooctene was observed under identical conditions.101 A Pt/ZnO@ZIF-8 catalyst prepared following the localized conversion mechanism by employing low linker concentrations was compared to a catalyst prepared following the dissolution–precipitation mechanism by employing high linker concentrations. The TEM images clearly show Pt deep inside the catalyst prepared under dissolution–precipitation conditions (Fig. 8a) whereas Pt is near the surface of the MOF prepared under localized conversion conditions (Fig. 8b). Neither catalysts was capable of hydrogenating cyclooctene. However, hydrogenation of 1-hexene was more favorable by the particles near the surface than those encapsulated deep inside. This behavior was attributed to the different diffusion distances.100
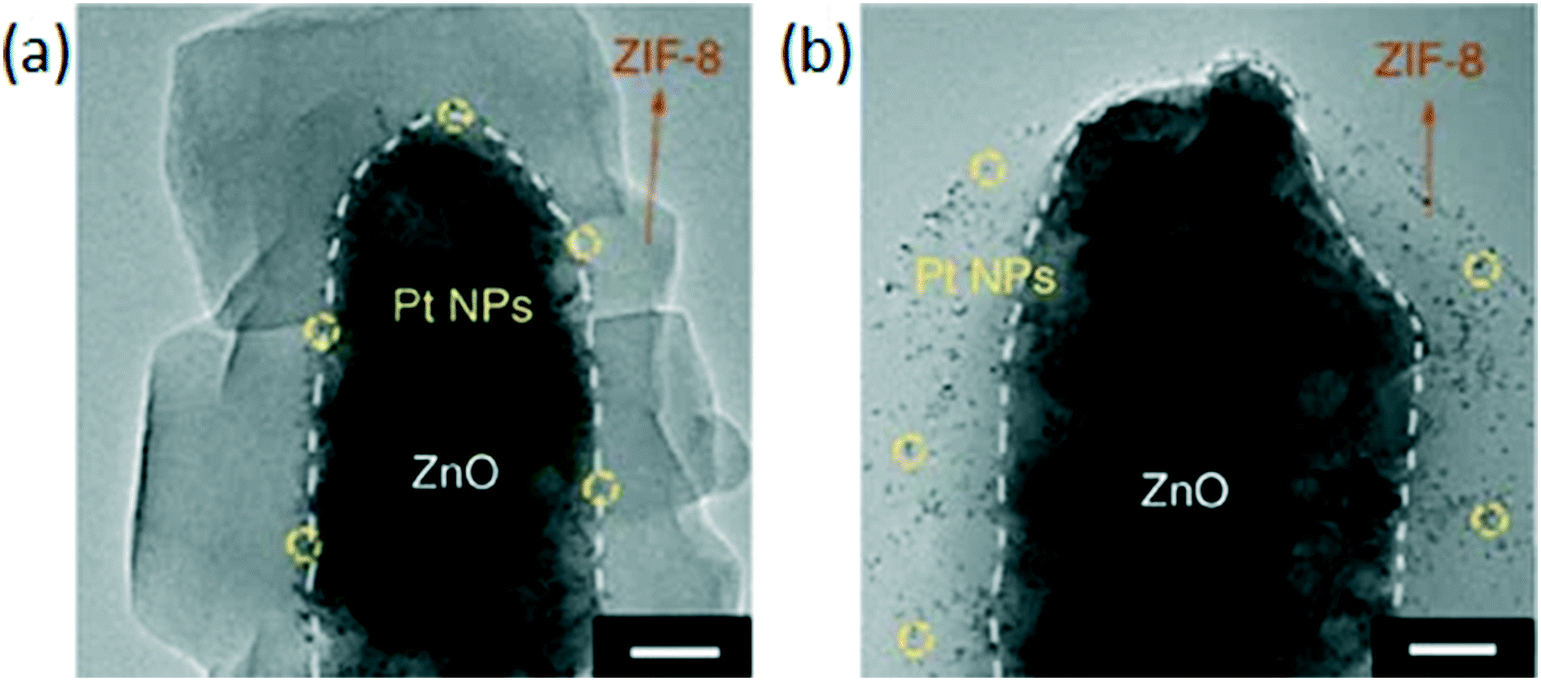 | ||
| Fig. 8 TEM images of Pt/ZnO@ZIF-8 catalysts (scale bar 20 nm), (a) Pt NPs are deep inside the catalyst prepared using high ligand concentrations, (b) Pt NPs near the surface of the MOF prepared with low ligand concentrations. Reproduced with permission from ref. 100. | ||
The morphology of the sacrificial metal oxide can also affect the encapsulation process. This was demonstrated by preparing Pd/ZnO@ZIF-8 catalysts from a Pd precursor and ZnO with a flower-like structure or a spherical structure. The use of flower-like structure resulted in a complete encapsulation of the particles and the authors speculated that the structure results in a stronger adsorption capacity for Pd and better anchoring during Pd nucleation, ZnO dissolution, and ZIF-8 encapsulation. Furthermore, the laminar structure of the ZnO can enhance the activity of the MOF growth. Meanwhile, naked ZnO could still be observed when spherical ZnO was employed. Fully encapsulated particles demonstrated size selectivity in the hydrogenation of olefins and offered protection against bulky catalyst poisons.102
The versatility of the sacrificial method enables the preparation of encapsulated catalysts with different structures. For example, hollow and core–shell structures can be synthesized by adjusting the crystallinity of the sacrificial agent. NPs can be confined in hollow HKUST-1 shells by coating the particles with polycrystalline Cu2O and subsequently converting this layer into the MOF. Continuous dissolution of the copper oxide resulted in expansion of the MOF outward while the hollow core is preserved. The shell was composed of multiple aggregated HKUST-1 crystals and the absence of freestanding MOF crystals suggested that the formation was restricted to the surface of the metal oxide. On the contrary, the use of monocrystalline Cu2O resulted in a core–shell structure. The formation of the different structures are governed by the kinetics of metal oxide dissolution and coordination of the ligands with the metal ions, as sketched in Fig. 9. Pd on Au dendritic nanoparticles encapsulated using this method were active in the catalytic hydrogenation of 1-hexene, but no products were observed in the catalytic hydrogenation of the much larger cis-stilbene.103
 | ||
| Fig. 9 Conversion of NPs@Cu2O to NPs@HKUST-1, (a) formation of a solid MOF shell around the NPs using monocrystalline Cu2O, (b) formation of a hollow MOF containing a NP using polycrystalline Cu2O. | ||
Recently, Chen et al. produced MOF-encapsulated nanoparticles by ball milling NPs supported on metal oxides in the presence of organic linkers. Preparation in this way greatly reduced the volume of the solvent and the NPs are located close to the surface of the MOF. The authors proposed that the MOF formation occurred through the localized conversion mechanism discussed earlier and exemplified in Fig. 7. The efficient encapsulation of Pd was proven by the lack of cyclooctene hydrogenation.104
The sacrificial method was also used to encapsulate Pt–Ni nanoparticles in Ni-MOF-74 by etching Ni from Pt–Ni nanocrystals. It's molecular sieving effect was proved by the trend of decreasing TOFs with increasing substrate size in the catalytic hydrogenation of styrene (9757 h−1), 2,4,6-trimethylstyrene (462 h−1), trans-stilbene (17 h−1) and 4,4′-dimethyl-trans-stilbene (9 h−1).106 Notably, more than one type of MOF can be formed from a single alloy, depending on which metal is leached out and what ligands are present. Starting from a RhCoNi alloy, Kuang et al. produced ZIF-67 and MOF-74 following the addition of 2-methylimidazole that leached out Co2+ and 2,5-dihydroxyterephtalic acid that removed Ni2+, respectively. RhCoNi@ZIF-67 has a smaller pore size and was able to fully hydrogenate 1-hexene but only partially converted cyclohexene diffusing though some MOF intercrystalline cracks. RhCoNi@MOF-74 on the other hand fully hydrogenated 1-hexene and converted most of the cyclohexene due to its larger pore openings.105
Other classical preparation methods, such as impregnation, have provided satisfactory metal encapsulation. Ag@HKUST-1 catalysts prepared by a double solvent impregnation demonstrated excellent size selectivity in the hydrogenation of olefins. For small olefins, such as 1-hexene, 1-octene, cyclohexene, and styrene, the encapsulated Ag particles achieved a higher conversion compared to the surface-supported NPs due to the smaller size of the encapsulated particles. Larger olefins on the other hand (1-decene, cyclooctene, diphenylethylene, and 1-dodecene) yielded almost no conversion, proving encapsulation of the NPs. However, high metal precursor concentrations lead to a small part of the NPs on the surface and such catalysts are capable of hydrogenating large olefins to some extent.93 Impregnation of a metal precursor that strongly interacts with the MOF has also shown good results in the encapsulation of metal functionalities; Huang et al. reported that Pt nanoclusters can be embedded inside UiO-66–NH2 by interactions of the Pt ions with the –NH2 groups of the MOF, followed by a reduction step. The catalyst demonstrated both inter- and intramolecular selectivity for the gas-phase catalytic hydrogenation of olefins. The intermolecular selectivity was demonstrated with the decreasing conversion for larger olefins; ethylene was hydrogenated with a 19.6% conversion while only 1.6% conversion for cyclooctadiene was obtained. In comparison, Pt/SiO2 achieved 12% conversion of 1,3-cyclooctadiene under identical conditions.94
Encapsulation of NPs was also successfully performed by chemical vapor infiltration under controlled conditions. In this way, palladium was introduced into UiO-66 and UiO-67 MOFs via chemical vapor infiltration with the Pd(η3-C3H5)(η5-C5H5) complex at different temperatures. The infiltration at 25 °C resulted in the formation of Pd NPs on the surface of UiO-66, most probably due to the uncontrollable decomposition of the precursor by the Lewis acidity of the Zr6O6 clusters on the MOF. However, performing chemical vapor infiltration at −15 °C decreases the uncontrolled decomposition. Accordingly, Pd@UiO-66 with a small fraction of Pd on the outside and Pd@UiO-67 with no particles on the outside were obtained. These catalysts displayed intermolecular selectivity in the selective hydrogenation of ketones with various molecular sizes.99
Pd was also simultaneously used as coordinating metal and active site for intermolecular hydrogenations. [Pd(2-pymo)2]n MOF was capable of hydrogenating 1-octene, although only 50% selectivity is obtained due to isomerization. Meanwhile, no conversion of the larger cyclododecene was reported. Unfortunately, the selectivity of this catalyst could only be maintained for a limited time due to a gradual degradation of the MOF and the formation of accessible Pd nanoparticles.117
Other synthetic approach involves the partial pyrolyzation of a Ni-MOF-74 to simultaneously produce Ni particles and create mesopores, resulting in Ni NP cores with carbon/Ni-MOF-74 composite shells. Size selectivity was reported for the catalytic hydrogenation of various olefins. Catalysts prepared with higher pyrolysis temperatures converted the large cyclooctene more efficiently. However, they exhibited lower conversion of the smaller 1-octene, which was attributed to aggregation of Ni NPs.110
The intermolecular selectivity of encapsulated catalysts can be tuned by post-synthetic methods, Farha et al. prepared Pt@SALEM-2 from Pt@ZIF-8 with solvent-assisted linker exchange without triggering leaching of the encapsulated material.111 Around 90% of the 2-methylimidazolate linkers in Pt@ZIF-8 were replaced with imidazole while the platinum particle size and location was retained. The larger apertures of SALEM-2 (6 Å) compared to ZIF-8 (3.4 Å) allowed the new catalyst to hydrogenate cyclohexene with 7% conversion while no conversion was obtained over the original Pt@ZIF-8. Huo et al. employed a post-synthetic method to decrease the pore apertures of Pt@UiO-66–NH2 by reacting the amino groups with anhydrides containing alkyl chains of various lengths. The modification preserves the crystal structure and tunes the size-selectivity; conversion of trans-stilbene decreased with increasing alkyl chain length. Hydrogenation of cyclooctene showed a similar trend, but with higher conversions due to its smaller size. The greatest ratio between the hydrogenation rates of trans-stilbene and cyclooctene was obtained over a catalyst that was modified with a C3 alkyl chain; at this size the diffusion of trans-stilbene is greatly reduced but cyclooctene faces less restrictions.112
3.3. Intramolecular selectivity
Compared to zeolites, MOFs provide additional means to enhance intramolecular selectivity by using linkers that interacts and modify the adsorption mode of substrates. However, as of writing this review, intramolecular selectivity over MOF based catalysts has been less reported than intermolecular selectivity. Investigations on intramolecular selectivity can be classified into four groups. (a) The hydrogenation of carbon–carbon double (CC) and triple bonds (CC). This includes the partial hydrogenation of dienes to alkenes, the selective partial hydrogenation of alkynes, and the suppression of isomerization during the hydrogenation of unsaturated alcohols. (b) The hydrogenation of CO in unsaturated aldehydes and ketones, (c) asymmetric hydrogenations, and (d) tandem reactions, in which a substrate or intermediate is selectively hydrogenated.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C) and triple bonds (C
C) and triple bonds (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C).
In addition to their uses as substrates in intermolecular selectivity, molecules containing carbon–carbon double and triple bonds are also convenient for investigating the intramolecular selectivity of catalysts. Various structures and sizes are readily available, allowing to choose substrates that match the pore dimensions closely.
C).
In addition to their uses as substrates in intermolecular selectivity, molecules containing carbon–carbon double and triple bonds are also convenient for investigating the intramolecular selectivity of catalysts. Various structures and sizes are readily available, allowing to choose substrates that match the pore dimensions closely.
Farha et al. demonstrated the excellent intramolecular selectivity of Pt@ZIF-8 prepared by a core–shell approach by selectively hydrogenating the CC double bond at the 1 position in 1,3-hexadiene, giving rise to 3-hexene with 95% selectivity.87 Pd NPs sandwich catalysts with UiO-67 core and UiO-66, UiO-67, or UiO-68 shell displayed excellent activity and selectivity in the semihydrogenation of alkynes. The catalysts were produced by the addition of a Pd precursor during the synthesis of UiO-67 followed by reduction with NaBH4 and a subsequent growth cycle of UiO MOF. The highest selectivity in the phenylacetylene hydrogenation was obtained with UiO-67@Pd@UiO-67. The authors attributed this selectivity to the electron transfer between Pd and the UiO supports which leads to easier desorption of the targeted styrene product.124 Rh–Ni encapsulated in MOF-74 displayed transition state selectivity in the partial hydrogenation of alkynes in order to preferentially obtain cis-products. The Rh–Ni@MOF-74 catalyst was produced by selectively removing a part of the nickel of a Rh–Ni alloy in the presence of a linker. The thickness of the MOF shell could be increased by increasing the Ni content of the initial alloy. The best performing Rh–Ni@MOF-74 catalyst was able to hydrogenate diphenyl acetylene to cis-stilbene with good selectivity (63.8%) while the best bare metal particles resulted in only 28.7% selectivity to the cis-product. The high stereoselectivity originated from the suppression of the isomerization from cis to trans; the isomerization goes through an intermediate which is too large to be accommodated in the pores of the MOF. This steric hindrance is lower for smaller substrates which results in a significantly lower cis/trans ratio. Hydrogenating 1-phenyl-1-pentyne resulted in 18.6% cis-1-phenyl-1-pentene whereas hydrogenating 1,4-dichloro-2-butyne did not give any cis product.107
Weller et al. confined an organometallic complex (Crabtree catalyst) in the voids of sulfonated MIL-101(Cr) by incorporating the cationic part of the complex via ion-exchange. The catalysts were tested in the hydrogenation of unsaturated alcohols, where the intramolecular selectivity of this catalyst was displayed; the confined complex achieved better selectivity for the hydrogenation of alcohols that are prone to isomerization. The authors proposed that suppression of the isomerization reaction resulted from extended coordination sphere interactions, such as H-bonding of the substrate with the sulfonated MOF.97 Rh NPs stabilized by a sulfonic acid functionalized MOF (Rh@S-MIL-101) were prepared by ion exchange followed by reduction with sodium borohydride. This Rh@S-MIL-101 catalyst was used for the partial hydrogenation of phenol to cyclohex-1-en-1-ol which then undergoes rapid tautomerization to cyclohexanone. High conversion (>95%) and selectivity (>92%) to cyclohexanone are obtained, which can be further increased to 96% at 91% conversion over a catalyst with less Rh on the surface. The selectivity of the catalysts was explained by the Cr(III) Lewis acidic sites of the MOF, which interact with the Lewis basic CO group of cyclohexanone and inhibit the overhydrogenation.98
C hydrogenation is more favored thermodynamically. Li et al. prepared Pt nanoparticles on the surface of MIL-101 while additional MIL-101 was grown over the surface via an epitaxial growth mechanism. Successive growth cycles afforded good control over the thickness of the MOF and allowed for the synthesis of catalysts with an optimum thickness. The hydrogenation of cinnamaldehyde over an optimized Pt@MIL-101 resulted in an excellent conversion and selectivity to cinnamyl alcohol. The high selectivity of CO hydrogenation versus CC hydrogenation was explained by both steric hindrance due to small pore openings and electronic repulsion between the phenyl moiety of the cinnamaldehyde and π-electrons from the linkers. As a result, cinnamaldehyde is preferentially adsorbed with the CO pointing forward. Additionally, the MOF linkers are capable of donating electron density to the Pt particles, which is known to enhance CO activation.88 Various MIL-101@Pt@MIL-101 sandwich catalysts, with Cr3+, Fe3+ or a combination of both as coordinating metal, have been used in the selective hydrogenation of α,β-unsaturated aldehydes to their corresponding unsaturated alcohols. These catalysts were prepared using a core–shell approach in which an additional layer of MIL-101 is grown over MIL-101@Pt. The selectivity in the hydrogenation of cinnamaldehyde to cinnamyl alcohol increases from Pt NPs < MIL-101(Cr)@Pt < MIL-101(Fe)@Pt. This increase was attributed to the activation of the CO bond by the Lewis acidic sites of MIL-101. A further increase in selectivity was obtained by coating these catalysts with another layer of MIL-101, with the best result being obtained over MIL-101(Fe)@Pt@MIL-101(Fe). Furfural, 3-methyl-2-butenal, and acrolein could similarly be hydrogenated with excellent selectivity.125
Wang et al. used Pt@ZIF-8 prepared by the core–shell method for the hydrogenation of 3-methylcrotonaldehyde. ZIF-8 has pore apertures of a comparable size to the 3-methylcrotonaldehyde, resulting in excellent selectivity to 3-methyl-2-buten-1-ol. The narrow pores force the substrate to approach in a linear fashion and prevents it from freely rotating, thus the CC bond cannot easily adsorb on the Pt particles while the CO moiety remains fully accessible.76 Similarly, Au@ZIF-8 was used in the catalytic hydrogenation of crotonaldehyde to crotyl alcohol with excellent selectivity. No butyraldehyde, which originates from CC hydrogenation, was detected. The observed selectivity was mostly attributed to the narrow pores of ZIF-8, which only allowed the CO moiety to adsorb on the active sites.77 Pt–SnOx@ZIF-8 demonstrated a good selectivity in converting 2-pentenal to 2-pentenol due to the synergetic effect that arises from combining the confinement effects of MOFs with the selectivity enhancement by SnOx, as shown in Fig. 10. Pt/SiO2 only achieved 4.3% selectivity to the unsaturated alcohol, Pt@ZIF-8 on the other hand achieved 31.0% selectivity to the targeted product. The authors used in situ FTIR to investigate the binding modes of the substrate in the catalyst. The FTIR investigation showed a lower ν2(CO) signal over Pt@ZIF-8 compared with Pt/SiO2. This indicates that the confinement enhances the η(CO) conformation. The selectivity was further increased by incorporating SnOx species, this increase went further for higher SnOx to Pt ratios, reaching 80.9% when SnOx/Pt = 1.5. This result was attributed to SnOx acting as electrophilic sites for adsorption and activation of the CO bond. Additionally, hydrogen spillover from Pt allows SnOx to hydrogenate CO, but not CC. The hydrogenation selectivity is highly dependent on the size of the substrate. Conversion of the bulkier 3-methylcrotonaldehyde over Pt–0.5SnOx@ZIF-8 resulted in 98.6% selectivity to the unsaturated alcohol due to the increased steric hindrance of the substrate.89
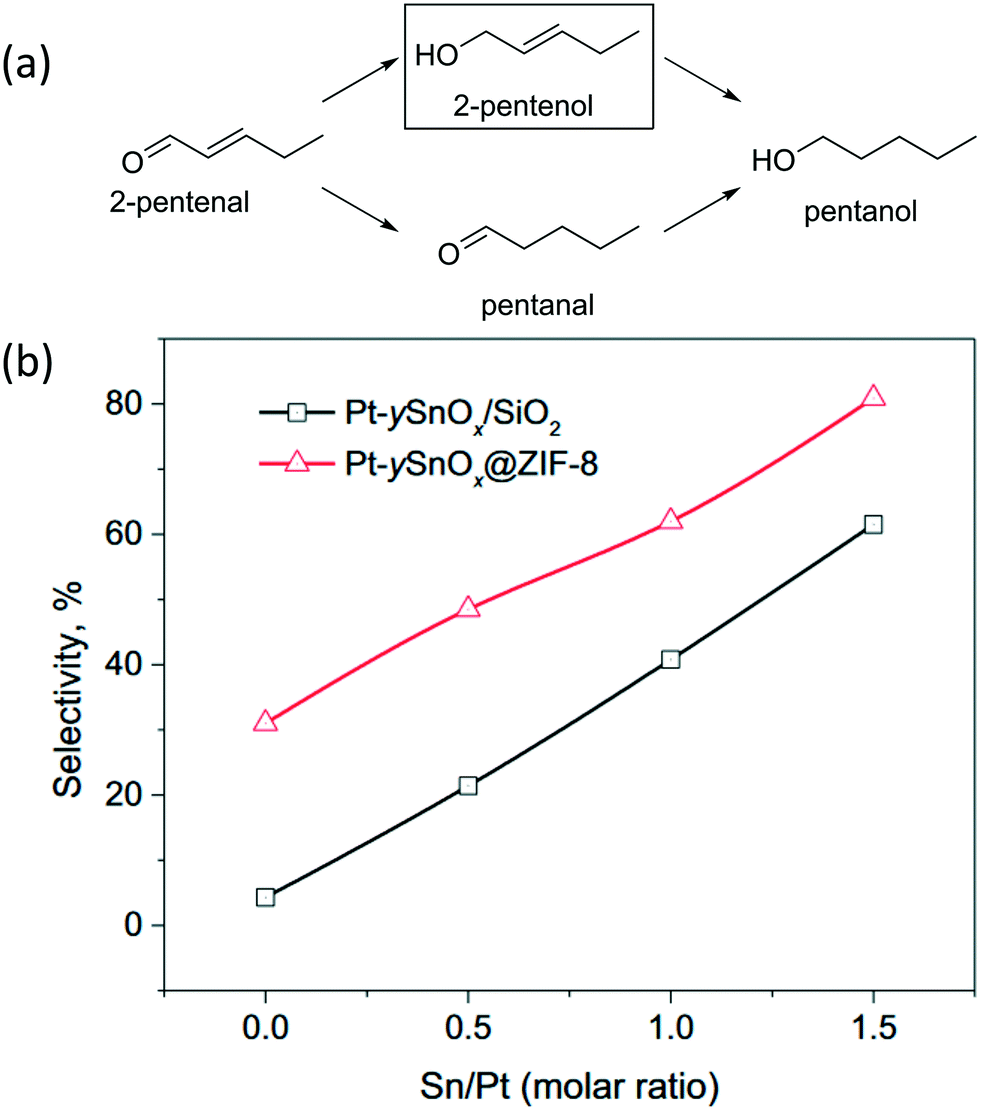 | ||
| Fig. 10 (a) Schematic presentation of 2-pentenal hydrogenation reaction. (b) Selectivity as a function of the Sn/Pt ratio in the conversion of 2-pentenal to 2-pentenol over encapsulated Pt–SnOx@ZIF-8 and supported Pt–SnOx/SiO2 catalysts. Reproduced with permission from ref. 89. | ||
Pt@UiO-66–NH2 prepared by impregnation demonstrated excellent intramolecular selectivity in the hydrogenation of cinnamaldehyde. The CC moiety in cinnamaldehyde could not easily adsorb onto the metal particles while the CO bond was able to easily reach and adsorb on the metal particle. As such only a small amount of hydrocinnamaldehyde and hydrocinnamyl alcohol were obtained and the selectivity to cinnamyl alcohol was over 90%.94 A recent computational study showed that the adsorption through the lone pairs of the cinnamaldehyde oxygen atom on Pt@UiO-66–NH2 is kinetically favorable, however, it is thermodynamically unfavorable compared to adsorption through CO and CC, as shown in Fig. 11. Furthermore, the hydrogenation energy barrier is much lower for cinnamaldehyde adsorbed through the O-atom compared to those for CC and CO adsorbed cinnamaldehyde.126
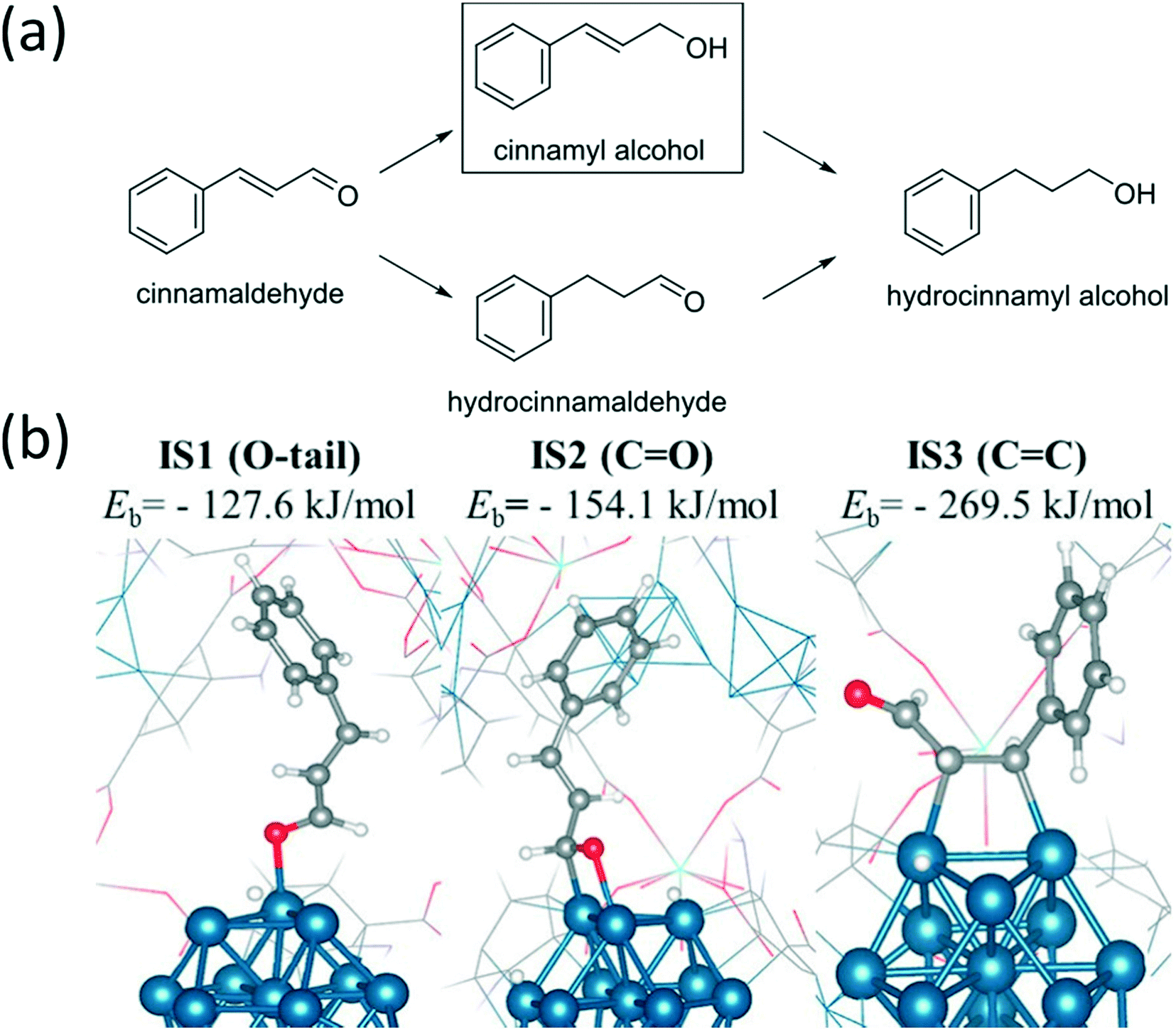 | ||
| Fig. 11 (a) Schematic presentation of cinnamaldehyde hydrogenation reaction. (b) The adsorption modes and binding energies of cinnamaldehyde on the metal surface of Pt28@UiO-66–NH2. Reproduced with permission from ref. 126. | ||
C bond of methyl α-acetamidoacrylate, methyl α-acetamidocinnamate, and dimethyl itaconate with full conversion and 85%, 70%, and 91% ee, respectively. However, the homogeneous catalyst used for comparison, Ru(Me2L)(DMF)2Cl2, achieved 88%, 81%, and 96% ee under the same conditions. The lower ee was attributed to residual achiral Ru complex that is trapped in the MOF framework. This was supported by a leaching test which showed that a significant amount of Ru leaches out during a reaction (3.6% vs. 0.1% of Zr).113 The Ru-BINAP-MOF could similarly be used for the enantioselective hydrogenation of β-keto ester compounds. The catalyst hydrogenated the carbonyl group of methyl-3-oxobutanoate, ethyl-3-oxopentanoate, and tert-butyl-3-oxobutanoate with quantitative yields and 97%, 94%, and 96% ee, respectively. Similar to the reaction with substituted alkenes, the homogeneous catalyst achieved better ee at full conversion; Ru(Me2L)(DMF)2Cl2 achieved >99% ee for all substrates.113
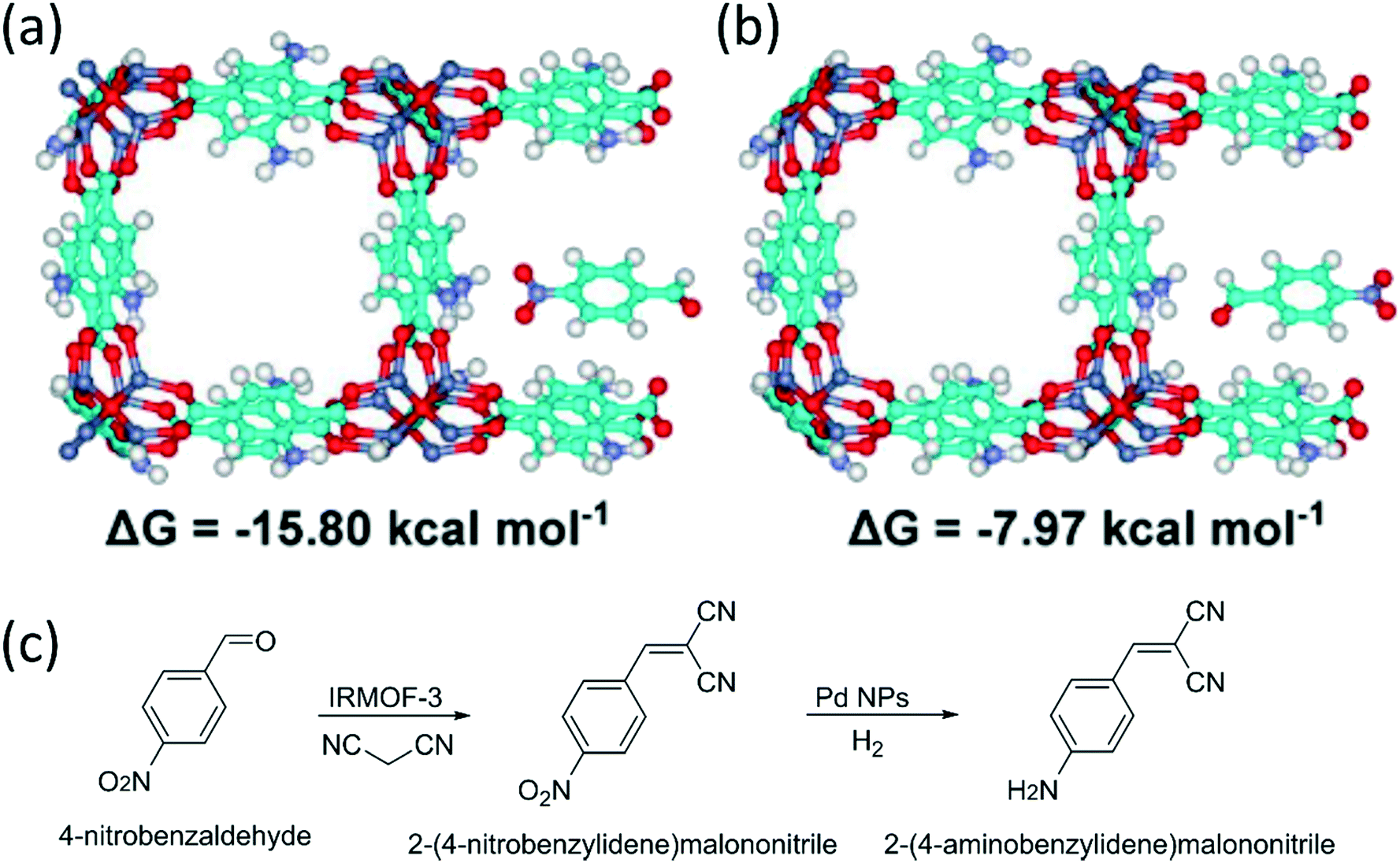 | ||
| Fig. 12 Theoretical models showing the preferred 4-nitrobenzaldehyde orientation over Pd@IRMOF-3. (a) NO2– pointing towards the MOF. (b) NO2– pointing away from the MOF. (c) Schematic presentation of converting 4-nitrobenzaldehyde to 2-(4-aminobenzylidene)malononitrile. Reproduced with permission from ref. 90. | ||
Li et al. used a sacrificial method to etch out part of the nickel of Pt–Ni alloy in the presence of linkers to form PtNi@Ni-MOF-74. The catalyst size-selectivity was beneficial for the tandem reduction of nitroarene and condensation to imines. Because the imine is larger than the pore opening of the MOF, diffusion to the Pt–Ni frame is prevented and over-reduction to the secondary amine is supressed.106
The sacrificial method was combined with aspects from the core–shell procedure by adding a polyoxometalate (phosphotungstic acid) during the initial phase of forming PtNi@MOF-74 from PtNi NPs. The resulting system was used for hydrogenation and esterification reactions. Encapsulating the metal reduced overhydrogenation whereas incorporating the acidic polyoxometalate promoted esterification reactions. PtNi encapsulated in MOF-74 converted p-nitrobenzoic acid predominately to p-aminobenzoic acid with less overhydrogenation products than those obtained from the unencapsulated PtNi branched alloy; this was attributed to a different adsorption configuration of the reactants on the catalyst. Phosphotungstic acid incorporated in the MOF promoted the esterification, resulting in the target product benzocaine in 81.4% yield.127 Pd@MIL-101, prepared by a double solvent impregnation was used for the tandem reaction to produce 2-(4-aminophenyl)-1H-benzimidazole. 4-Nitrobenzaldehyde and 1,2-phenylenediamine react over the Lewis acidic MIL-101 to form the 2-(4-nitrophenyl)-1H-benzimidazole, this intermediate subsequently passes over the encapsulated Pd and is reduced to the 2-(4-aminophenyl)-1H-benzimidazole target product with near quantitative yield. In contrast, Pd/C did not give any product due to the lack of acidity whereas MIL-101 only resulted in the intermediate and hydrogenation did not take place.96 A PdAg@MIL-101 catalyst was prepared in the same manner as the Pd@MIL-101 catalyst. Interestingly, the particle size of the PdAg alloy NPs (1.5 ± 0.3 nm) was smaller compared to the Pd NPs (2.5 ± 0.3) nm due to the presence of Ag that reduces aggregation. PdAg@MIL-101 was used in the one-pot reaction to produce n-benzylaniline. Nitrobenzene is hydrogenated to aniline followed by the acid catalyzed imine formation with benzaldehyde. Pd1Ag1@MIL-101 achieved 90% selectivity to the target product, while the Pd@MIL-101 catalyst only achieved 56% selectivity due to the high activity for the hydrogenation of benzaldehyde. The authors propose that the presence of Ag slows down the reduction rate of benzaldehyde. The importance of the Lewis acidity of MIL-101 was evaluated by the addition of pyridine, which poisons the acid sites and stops the reaction.96 Pt@UiO-66–NH2 synthesized by impregnation was used for the one-pot synthesis of nitrones starting from nitromethane and various benzaldehydes. The Pt NPs are responsible for the hydrogenation of nitromethane to N-methyl hydroxylamine, while the NH2 substituted MOF catalyzes the subsequent condensation with benzaldehydes. In the absence of NH2 groups (Pt@UiO-66) the selectivity is low and the main product is the undesired hydrogenation product of benzaldehyde. Furthermore, the activity and selectivity decrease significantly if a base probe molecule (triethylamine) is added, highlighting the importance of the Lewis acidity of UiO-66–NH2. The importance of encapsulation was demonstrated by a comparison of encapsulated Pt@UiO-66–NH2 and surface supported PVP-Pt/UiO-66–NH2 or Pt/UiO-66–NH2, which showed decreased activity and selectivity for the surface supported catalysts.128
4. Confining metal active species in carbon nanotubes
Metallic species confined in carbon nanotubes (CNTs) are prepared by several methods. Dispersed metallic species in the channels of CNTs can be achieved by loading metallic precursors using classical impregnation methods129–132 or chemical vapor deposition approaches.133 Other more complexes synthesis involve the thermal annealing of MOFs bearing the desired metal.134–136 N-Doped CNTs can also be used for improved metal dispersion, the synthesis methods include the pyrolysis of metal precursors137 or MOFs138 in the presence of N-containing material such as melamine. Depending on the synthetic procedure used and on the shape of the CNTs, the active species can be in the form of metal-centers (M–Nx), NPs, or even a co-activity between exposed NPs and M–Nx. The size of these species varies widely from <1 nm for the metal centers up to few hundreds of nanometers for metallic wires.Selective hydrogenation reactions over metals confined in CNTs
The strong capillarity forces and relatively large pore openings of CNTs result in the easy and fast diffusion of reagents into the channels. Consequently, the reagents are far more abundant in the proximity of the confined active species than on the surface (Fig. 13).129,131 Additionally, CNTs possess electron-deficient internal walls due to charge transfer from the internal walls towards the external ones. This deficiency is partially compensated through the interaction with the confined metallic species that, in turn, become electron deficient, and consequently, more active towards hydrogenation compared to surface supported ones.132 These factors explain the higher conversions reported over confined NPs compared to surface supported ones.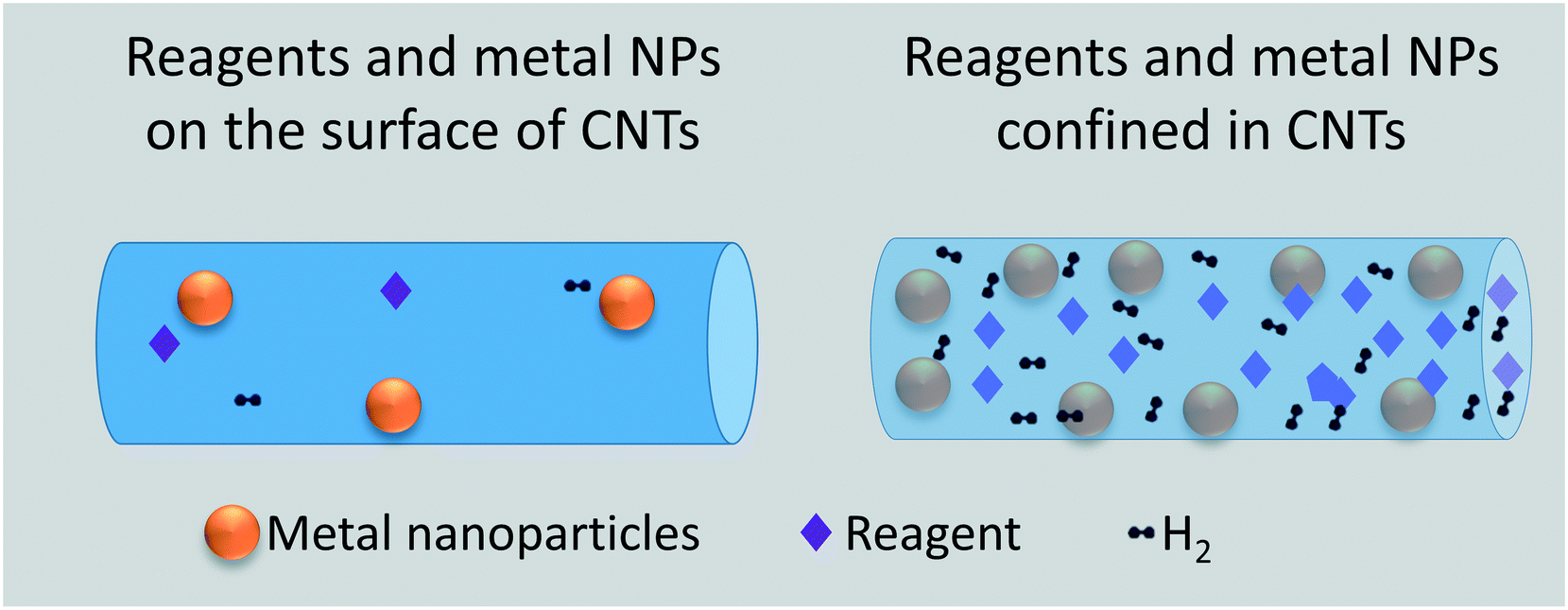 | ||
| Fig. 13 Distribution of metal NPs and reagents on the surface and in the cavities of CNTs. | ||
The capillary forces and the large pore openings of CNTs, during impregnation, favor the confinement of the metallic species in the internal cavities of CNTs. Nevertheless, some NPs inevitably appear on the external surface as well. To eliminate this undesired phenomenon, Serp and coworkers proposed a method to selectively confine NPs inside the CNTs.139 In this example, the external surface of CNTs was functionalized with amides having long alkyl chains to induce weak or repulsive interactions with the NPs. Ru–Pt bimetallic NPs active species were introduced by impregnation. These species were stabilized by a 4-(3-phenylpropyl)pyridine ligand which possesses a pyridyl ring with an affinity for the surface of the NPs and a phenyl ring with an affinity for the CNT graphene layers (π–π interaction). XPS and 3D TEM results revealed full confinement of the Ru–Pt NPs at 5–11% metal loading and an 80% confinement at the highest metal loading (23%). The prepared catalyst selectively hydrogenated the aldehyde group in cinnamaldehyde (CAL) with a linear increase of activity as the metal loading increased. Meanwhile, NPs anchored on the external surface of CNTs, functionalized with carboxylic acid instead of the alkyl amide, were less active and selective. The activity of the confined NPs was attributed to the passivated outer surface that directed CAL towards the channels; under high concentration, the CAL molecules are adsorbed perpendicular to the Pt surface with the aromatic ring in a parallel arrangement, thereby enhancing the selectivity towards cinnamyl alcohol.
In CNTs, the interaction of confined metal active species with the electron-deficient internal walls, and the N-doped structures of CNTs can create an electronic environment that allows the adsorption and activation of substrates in a specific conformation. This effect can promote the selective hydrogenation of molecules such as quinoline heterocycle and multisubstituted nitro arenes. The selective hydrogenation of the quinoline heterocycle was reported over Pd NPs incorporated in the tips of CNTs (Pd@CNTs)132 and over Co NPs encapsulated in N-doped CNTs dispersed on hollow N-doped carbon polyhedrons (Co@NCT/HNC).140 The high selectivity of Pd NPs was attributed to their interaction with the electron-deficient CNT internal walls. DFT calculations demonstrated that the inherent electron deficiency of these Pd NPs favors the preferential adsorption and activation of quinoline through the electron-rich nitrogen atom of its heterocycle rather than the benzene ring. In contrast, the electron-rich exterior surface reduces the tendency of Pd NPs to adsorb the nitrogen-containing ring, resulting in non-selective hydrogenation. On the other hand, the high performance of Co@NCT/HNC was attributed to the good Co dispersion, easy transfer of H2 to the surface of the reactant, and to the N-doped structures that weakened the coordination between N-heteroarenes and Co NPs. The combination of these factors resulted in the selective hydrogenation of the quinoline heterocycle in the presence of bromo, chloro, methoxy, and methyl substituents.
Metal NPs confined in CNTs selectively hydrogenated the nitro functional group of multi-substituted nitroarenes. Co3O4/NGr@CNT, comprising Co particles with Co3O4 oxidic shell confined in nitrogen-doped carbon layers, converted 1-iodo-4-nitrobenzene into iodoaniline with 93% yield both under batch and continuous flow conditions.141 In another example, a Fe/Fe3C catalyst encapsulated in N-doped CNTs promoted the selective reduction of the nitro functional group in multi-substituted arenes. In all cases, the nitro functional group was selectively hydrogenated over the Fe/Fe3C active sites of Fe/Fe3C@N6CT-900-1.138 Based on DFT calculations, the surface N-sites-that result from doping-efficiently trap nitrobenzene and aniline (used as models) by their phenyl groups with adsorption energies of −1.79 and −2.23 eV, respectively.138 Similar behavior was reported for the Ni NPs encapsulated in N-doped CNTs, the nitro substitution was selectively reduced in the presence of electron-donating or withdrawing groups except for aldehyde, which was converted into corresponding alcohol in 74.2% yield.142
Taking benefit of the capillarity forces of CNTs, several reports described the use of CNTs as micro-reactors for asymmetric hydrogenation reactions.129,131,143,144 Pt NPs confined in multi-walled (MW) CNTs with open channels were chirally modified by cinchonidine (CD), which was present in the internal cavities with a 2700 times higher concentration than in the bulk solution.131 This modification allowed for the asymmetric hydrogenation of ethyl pyruvate (α-ketoester) with a high TOF (105 h−1) and 96% ee. The high activity and selectivity were linked to the enrichment with the substrate and the chiral modifier in the proximity of confined NPs on the one hand, and to the higher affinity of CNTs with the reagent compared to the product on the other hand. In contrast, surface supported NPs of similar metal loading, size, and shape to the confined ones gave rise to much lower TOF values (∼1.5 × 104 h−1) and ee (75%). Later on, it was found that the presence of water traces in the solvent highly affects both the activity and the selectivity of the confined catalyst.143 When using anhydrous acetic acid as a solvent, the TOF and ee were calculated to be 6.2 × 104 h−1 and 86%. Introducing scarce amounts of H2O (acetic acid/H2O = 31) boosted the TOF up to 9.5 × 104 h−1 and the ee to 95%. H2O molecules were suspected to promote the hydrolysis of oxygen-containing functional groups (alcohols, aldehydes, ketones, and acids) from the CNT internal walls and thus reduced H-bonding interactions with the CD and the reagent. Therefore, the concentration of chiral modifier in channels increased to 3900 times the concentration in solution. The presence of water also enhanced the preferential adsorption of the reagents by CNTs over the product thus allowing further conversion of reagents and easier release of the product. Finally, generating cationic Ptn+ in the form of Pt(ONa)x was found to further increase the electronegativity of the Pt NPs and consequently strengthen the interaction of NPs with chiral modifier/reactant.129 This resulted in a better chiral environment on the surface of the Pt nanoparticles giving rise to high enantioselectivity.
5. Confining metal catalyst in other host materials
Enhancing the selectivity of metallic catalysts towards hydrogenation reactions was also achieved through confining the NPs in other materials such as polymers, silica, and free-standing metallic structures.Intermolecular selectivity has mostly been obtained over particles encapsulated in various polymers and mesoporous silica. Pt NPs encapsulated in polypyrrole prepared in a single-step exhibited size selectivity in the catalytic hydrogenation of substituted olefins. Hydrogenation of methyl acrylate, dimethyl maleate, diethyl maleate, and dibutyl maleate over this catalyst showed that the hydrogenation rate decreases with increasing size of the substrate.145 An alumina core surrounded by polyelectrolyte films containing Pd NPs was produced by alternating adsorption of poly(acrylic acid) and a polyethylenimine–Pd(II) complex followed by reduction of the metal complex with NaBH4. The film limits the aggregation of the metal particles while simultaneously imparting intermolecular size selectivity in the catalytic hydrogenation of unsaturated alcohols. The authors propose that the access to the NPs relies on specific paths through the film. Smaller substrates have more available paths and therefore diffuse faster to the palladium. The first-order reaction rates are also consistent with diffusion-limited kinetics. Selectivity could be improved, albeit at the cost of a decreased hydrogenation rate, by running the reaction in a water/methanol mixture instead of pure water. This observation is attributed to decreased swelling of the polymer in this solvent.146 Palladium NPs confined in the cavities of 1,2,3-triazolyl-containing porous organic polymers prepared with a click reaction (Pd@CPP-C) or Yamamoto coupling (Pd@CPP-Y) have been used in the catalytic hydrogenation of olefins to alkanes. Catalytic hydrogenation of various olefins such as 1-octene, 2-octene, cyclohexene, and (S)-(−)-limonene revealed a decreasing trend in catalytic activity with increasing size of the substrates which was attributed to steric hindrance.147
Intermolecular selectivity has also been obtained over particles encapsulated in dendrimers, an example of which are palladium nanoparticles confined in hydroxyl-terminated poly(amidoamine) (PAMAM) dendrimers. In this system, the accessibility to the palladium particles was controlled by the generation of the dendrimer (Fig. 14). A higher generation resulted in lower accessibility due to a higher packing density of the end groups. The hydrogenation of allylic alcohols showed a general trend of decreasing TOF with larger substrates (allyl alcohol > 3-buten-2-ol > 1-penten-3-ol > 2-methyl3-buten-2-ol > 3-methyl-1-penten-3-ol) and more crowded dendrimers (4th generation > 6th generation > 8th generation).148
 | ||
| Fig. 14 Illustration of size selectivity over NPs confined in a fourth generation dendrimer (left) and a sixth generation dendrimer (right). Reproduced with permission from ref. 148. | ||
PdAu alloy nanoparticles encapsulated in poly(propylene imine) (PPI) dendrimers grafted on MWCNTs similarly demonstrated intermolecular selectivity. This catalyst was prepared by introducing Pd and Au precursors into the dendrimers followed by reduction. An increase of the dendrimer generation lead to more amino groups and accordingly, a higher loading of nanoparticles. In the hydrogenation of a combination of a less and a more hindered alkene, the less hindered alkenes were preferentially converted at higher dendrimer generation. Additionally, the PdAu catalyst displayed negligible leaching and a good reusability over 6 runs.149
Nanoparticles encapsulated in mesoporous silica were used in the size-selective catalytic hydrogenation of olefins. Iron and noble metal NPs confined in mesoporous SiO2 tubular nanoreactors were produced by covering Fe3O4-MWCNTs in mesoporous silica, and subsequently etching away the CNTs. Reducing the iron oxide followed by a galvanic replacement reaction with noble metals yielded Pd–Fe@meso-SiO2, Pt–Fe@meso-SiO2, and Au–Fe@meso-SiO2 magnetic tubular nanoreactors. Excellent TOFs were obtained with Pd–Fe@meso-SiO2 in the catalytic hydrogenation of various olefins, such as styrene and 4-methylstyrene. However, a lower TOF was observed in the hydrogenation of (E)-1,2-diphenylethene, which was attributed to the larger size of this substrate and its lower reactivity. Furthermore, no dechlorination was observed when 1-chloro-4-vinylbenzene was hydrogenated. The authors attributed the excellent activity of the catalyst to the increased local concentration of the substrate.150
Intramolecular selectivity has been achieved over a wider range of materials compared to intermolecular selectivity and includes particles encapsulated in polymers, silica, and even free-standing metallic structures.
Dong et al. used a porous polymer made by polymerizing cyanuric chloride and benzidine to confine palladium nanoparticles. A magnetic core coated with dopamine inside the porous polymer facilitated recycling. The catalyst was used to hydrogenate substituted nitroarenes, alkynes and alkenes with excellent conversion and selectivity. Furthermore, no dehalogenation products were observed in the hydrogenation of halogenated nitroarenes.151
Pt and PtSn nanoparticles coated in porous silica by a polyol method converted crotonaldehyde into crotyl alcohol with a higher selectivity compared to particles supported on silica. The preferential hydrogenation of CO in the presence of CC was attributed to the confinement effect that reduced the interaction between the CC bond and nanoparticle. In contrast, the homolog catalyst with NPs anchored on the external surface demonstrated lower selectivity due to the equal chances to adsorb CC and CO on the surface of NPs.152 Similarly, Ni NPs were confined in a silica network with 4–10 nm and 20–60 nm interparticle pores. The hydrogenation of cinnamaldehyde over this catalyst resulted in 65% selectivity to cinnamyl alcohol at full conversion in 1.5 h; meanwhile, it took 10 h to reach full conversion for Ni supported on SiO2 and the selectivity to cinnamyl alcohol was near zero. The enhanced performance was attributed to Ni–support interactions and confinement effects.153
Ru nanoparticles supported on high surface area graphite (HSAG) and encapsulated in mesoporous silica were prepared by treating Ru/HSAG with a surfactant-directed sol–gel coating process, cetyl trimethylammonium bromide was used as the template and tetraethyl orthosilicate as the silica source. Graphite as a support by itself already enhances the CO hydrogenation over CC hydrogenation due to charge transfer from the support to the metal, thus decreasing the chance of CC activation. The encapsulation further increased the selectivity to the unsaturated alcohol from 50% over Ru/HSAG to 65% over Ru/HSAG@SiO2. This additional selectivity was attributed to a suppression of the adsorption of cinnamaldehyde in a flat configuration by the pores of the silica.154
MCM-41 was used for enantioselective hydrogenations by anchoring organometallic complexes inside the pores. Covalent bonding of (S)-(−)-2-aminomethyl-1-ethyl pyrrolidine and (1R,2R)-(+)-1,2-diphenylethylenediamine ligands in the pores of MCM-41 was achieved by first reacting MCM-41 with Cl3Si(CH2)3Br and subsequently reacting the ligands with the 3-bromopropyl tail. Immobilized Rh and Pd catalysts with these ligands outperformed their homogeneous counterparts in terms of conversion and ee in the enantioselective hydrogenation of E-α-phenylcinnamic acid to 2,3-diphenylpropionic acid, and the enantioselective hydrogenation of methyl benzoyl formate to methyl mandelate. The enhanced ee was attributed to the anchoring on the concave interior of the pores, which restricts the access of the substrate to the catalyst. Access of the substrates to the metal ions was only favored when the substrate approached the active sites along the axis of the pores.155,156
Catalysts for enantioselective hydrogenation were also prepared by adsorbing ionic liquids containing chiral organometallic complexes in MCM-41 or highly porous carbons. IrCl(COD)-(S,S)-BDPP and [IrCl-(S)-BINAP]2 confined in MCM-41 and porous carbon, and RuCl(p-cymene)[(S,S)-Ts-DPEN] confined in MCM-41 achieved a higher ee in the hydrogenation of trimethylindolenine compared to their homogeneous counterparts. Unfortunately, the ionic liquids and complexes were prone to leaching which resulted in a decreased ee and conversion in the recycling reactions.157 Dimethylitaconate was hydrogenated over [Rh(COD)(R,R)-Me-DuPHOS][BF4]/[BMIM][BF4]/MCM-41, which resulted in a greater ee compared to the homogeneous counterpart. Furthermore, the ee (>99%) did not decrease after recycling the catalyst, however, the conversion decreased from 22.5% to 7.5%.157
Enantioselectivity was reported by Liu et al. who encapsulated palladium on carbon (Pd/C) in a silica shell containing chiral ruthenium species. This encapsulation promoted the enantioselective carbonyl transfer hydrogenation of the various alkynone substrates. The Pd/C yolk subsequently catalyzed the hydrogenation of the alkyne moiety. Considering each part solely, the chiral Ru catalyst was only able to hydrogenate the carbonyl of (4-(phenylethynyl)phenyl)ethanone. Whereas in the absence of the Ru catalyst, the encapsulated Pd/C catalyzed the hydrogenation of the alkyne moiety, but also gave a small amount of non-chiral alcohol.158
Free-standing palladium network catalysts with unique morphologies were also capable of enhancing enantioselectivity. Hexagonally stacked Pd nanowires were produced with SBA-15 as template via a double solvent approach and ordered mesopores were created among the Pd wires due to this stacking. The catalyst was used in the hydrogenation of acetophenone in the presence of S-proline, producing enantioenriched 1-phenylethanol. A maximum ee of 28.8% was obtained, while an ultrafine palladium black catalyst only achieved a maximum ee of 16.8%. The authors explained that the mesopores form a restricted environment that confines the stereo-configuration of the adsorbed S-proline–acetophenone adduct. This confinement seemed to favor the adsorption of the adduct which results in the R-isomer, thereby enhancing the ee.159 This reaction was also performed on freestanding Pd gyroidal structures prepared with various KIT-6 mesoporous silicates as templates. The use of KIT-6 templates prepared at temperatures below 375 K resulted in palladium with a gyroidal morphology (Fig. 15a), meanwhile the use of KIT-6 prepared at 375 K or above resulted in a double gyroidal morphology (Fig. 15b).
 | ||
| Fig. 15 Graphical representation of freestanding Pd structures. (a) Skeletal model of a fragment of single gyroid with I4132 symmetry. (b) Skeletal model of a fragment of double gyroid with Ia3d symmetry. Reproduced with permission from ref. 160. | ||
The authors mentioned that the small pores present in the double gyroidal structures enhanced the selectivity by generating stronger steric constraints of the adsorbates and by improving interactions between the co-adsorbed reactant and the chiral modifier, which resulted in a maximum ee of 42% in the hydrogenation of acetophenone.160
6. Concluding remarks
Confining metal active species in solid matrices such as zeolites, MOFs, carbon nanotubes, and other host materials efficiently promotes their selectivity towards hydrogenation reactions. This effect extends from allowing the catalyst to discriminate reagents based on their size and shape, to selectively hydrogenating one functional group in multi-substituted molecules, and even to selectively produce one stereoisomer. The encapsulant also provides means to efficiently stabilize the active sites, thus minimizing deactivation pathways such as metal sintering and leaching. Advances in this field go hand in hand with the increasing sophistication on the synthesis of nanomaterials and understanding of reaction mechanisms.We foresee a great fundamental interest in metal-confined catalysts, not only for selective hydrogenation reactions but also for other metal-catalyzed reactions where selectivity is a must. Those include selective oxidations, hydroformulation, dehydrogenation and other organic reactions. Another fascinating application worth of further exploration is the encapsulation of several functionalities that act in a sequential or concerted fashion to perform cascade reactions. Despite the limited number of reported examples that mostly encompass a combination of acid/base functionalities with metal active sites, the results are promising and encourage the search for more challenging cascade reactions. Here, MOFs have a great potential owing to their remarkable versatility of linkers, which can bring tunable pore sizes and chemical functionalities.
Among the most widely used synthesis methods, the direct synthesis (or in situ preparation) is one of the easiest, fastest, versatile, and potentially more economical for the preparation of encapsulated catalysts. Core–shell methods generally allow for a higher metal loading and enable the engineering of distinct functionalities into the catalysts by introducing shells of different active sites, thickness and structure. As a downside, the complexity of synthesis is often translated into higher cost. Lastly, impregnation methods are facile and easy to implement, but encapsulation efficiencies are normally suboptimal. From a practical perspective, one could easily envisage that encapsulated catalysts may not be as cost efficient as traditional metal-supported catalysts. Hence, the synthesis premium can only be justified when the encapsulated catalyst provides outstanding selectivity and stability that outcompete other available catalytic technologies. Yet, it is fair to say that most of the investigations aim to provide an evidence of concept instead of a cost-effective catalyst.
Nevertheless, there is extensive room for catalyst improvements. For example, strategies to reduce diffusion pathlength through the encapsulant may be further investigated for reactions where the kinetics of mass transport are dominant. Solutions such as pore hierarchization, the use of nanosized encapsulants, or covering metal species with thinner shell should be further explored. Additionally, more efforts should be devoted on the use of cheaper hydrogenation metals and to metal atom efficiency. While most examples are based on encapsulated NPs, fewer exploit ACs and SAs. Such a decrease in particle size will promote higher activities and atom efficiencies, especially when expensive noble metals are used.
Diagnostic tools to evaluate the successful confinement of metal functionalities are highly important and deserve to be critically discussed. Catalytic studies can be conducted with probe molecules unable to diffuse inside of the porous structure of the encapsulant. The low or negligible conversion of such substrates is a conclusive sign of full encapsulation. Yet, the probe molecules must be carefully selected to avoid partial adsorption and reaction of the functional group in the catalyst pore mouth. Spectroscopic techniques, including XRD and TEM are often used as a diagnosis for metal encapsulation. However, they have several limitations and cannot be used as a definitive proof. XRD results are generally used to claim full confinement when large NPs are absent. The use of NP size as a descriptor for metal encapsulation is questionable as small NPs can also be present and stabilized in a non-encapsulated manner. When using TEM techniques, two-dimensional projections of catalyst particles should be avoided as a proof of confinement. Instead, electron tomography (ET) overcomes this limitation and offer reliable information of the metal encapsulation in three dimensions. As a drawback, ET is a time-consuming technique and only provides very local information, therefore obtaining statistically relevant results is challenging. Other techniques with certain surface sensitivity such as X-ray photoelectron spectroscopy (XPS) should be carefully interpreted considering that the information depth is a few nanometers. We consider that more surface specific tools, e.g., time-of-flight secondary ion mass spectrometry (TOF-SIMS)161 and specially high sensitivity low energy ion scattering spectroscopy (HS-LEIS)162 will be of high value for the detection and quantification of non-encapsulated NPs. Overall, it appears that several diagnostic tools are needed to give an unequivocal answer on the encapsulation of metal functionalities.
Lastly, we advocate for a more rational design of encapsulated catalysts, based on a deeper understanding of the catalytic event. Advanced electron microscopy studies will be crucial to investigate structure of the metal functionality in confined voids. In this field, seminal contributions by the groups of Corma,163 Han,164 and Terasaki165 are paving the way for studying metal active sites at the atomic level. Detailed spectroscopic studies are expected to bring complementary structural information about the active confined void, and to unravel the structure of the adsorbed reactants and transition states. These methods, combined with theoretical calculations will provide guidance for the synthesis of novel encapsulated catalysts. Also monitoring the assembly of encapsulated materials under synthesis conditions will help to understand the parameters governing the location and encapsulation of the metal active species.
Conflicts of interest
There are no conflicts to declare.References
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001 Search PubMed.
- R. A. Sheldon and H. Van Bekkum, Fine Chemicals through Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2001 Search PubMed.
- M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS.
- I. McManus, H. Daly, J. M. Thompson, E. Connor, C. Hardacre, S. K. Wilkinson, N. Sedaie Bonab, J. ten Dam, M. J. H. Simmons, E. H. Stitt, C. D'Agostino, J. McGregor, L. F. Gladden and J. J. Delgado, J. Catal., 2015, 330, 344–353 CrossRef CAS.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- C. Louis and L. Delannoy, in Advances in Catalysis, Elsevier Inc., 1st edn, 2019, vol. 64, pp. 1–88 Search PubMed.
- F. Alonso, P. Riente, F. Rodríguez-Reinoso, J. Ruiz-Martínez, A. Sepúlveda-Escribano and M. Yus, J. Catal., 2008, 260, 113–118 CrossRef CAS.
- J. Ruiz-Martínez, A. Sepúlveda-Escribano, J. A. Anderson and F. Rodríguez-Reinoso, Catal. Today, 2007, 123, 235–244 CrossRef.
- J. Ruiz-Martínez, F. Coloma, A. Sepúlveda-Escribano, J. A. Anderson and F. Rodríguez-Reinoso, Catal. Today, 2008, 133–135, 35–41 CrossRef.
- P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2005, 292, 1–49 CrossRef.
- Z. Zhang, Y. Li, J. Gu, L. Ding, N. Xue, L. Peng, X. Guo, Y. Zhu, J. Ma and W. Ding, Catal. Sci. Technol., 2018, 8, 6384–6395 RSC.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS.
- S. T. Marshall, M. O'Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwartz and J. W. Medlin, Nat. Mater., 2010, 9, 853–858 CrossRef CAS.
- K. R. Kahsar, D. K. Schwartz and J. W. Medlin, J. Am. Chem. Soc., 2014, 136, 520–526 CrossRef CAS.
- P. T. Witte, WO Pat., 096783Al, 2009 Search PubMed.
- P. T. Witte, P. H. Berben, S. Boland, E. H. Boymans, D. Vogt, J. W. Geus and J. G. Donkervoort, Top. Catal., 2012, 55, 505–511 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- G. Vilé, D. Albani, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 21–33 CrossRef.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS.
- S.-M. Wu, X.-Y. Yang and C. Janiak, Angew. Chem., Int. Ed., 2019, 58, 12340–12354 CrossRef CAS.
- M. Shamzhy, M. Opanasenko, P. Concepción and A. Martínez, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- D. Farrusseng and A. Tuel, New J. Chem., 2016, 40, 3933–3949 RSC.
- J. Meng, X. Liu, C. Niu, Q. Pang, J. Li, F. Liu, Z. Liu and L. Mai, Chem. Soc. Rev., 2020, 49, 3142–3186 RSC.
- S. Yang, L. Peng, S. Bulut and W. L. Queen, Chem. – Eur. J., 2019, 25, 2161–2178 CrossRef CAS.
- S. A. Miners, G. A. Rance and A. N. Khlobystov, Chem. Soc. Rev., 2016, 45, 4727–4746 RSC.
- F. Alonso, P. Riente and M. Yus, Acc. Chem. Res., 2011, 44, 379–391 CrossRef CAS.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang, C. Yang, T. Chen, M. Lin, P. Wang, N. Xue, L. Peng, X. Guo, Y. Zhu, Z. Chen and W. Ding, Chem, 2020, 6, 2673–2689 Search PubMed.
- S. Goel, Z. Wu, S. I. Zones and E. Iglesia, J. Am. Chem. Soc., 2012, 134, 17688–17695 CrossRef CAS.
- Q. Chen, M. Wang, C. Zhang, K. Ren, Y. Xin, M. Zhao and E. Xing, Chem. - Asian J., 2018, 13, 2077–2084 CrossRef CAS.
- S. Wang, Z.-J. Zhao, X. Chang, J. Zhao, H. Tian, C. Yang, M. Li, Q. Fu, R. Mu and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 7668–7672 CrossRef CAS.
- Y. Chai, S. Liu, Z.-J. Zhao, J. Gong, W. Dai, G. Wu, N. Guan and L. Li, ACS Catal., 2018, 8, 8578–8589 CrossRef CAS.
- T.-L. Cui, W.-Y. Ke, W.-B. Zhang, H.-H. Wang, X.-H. Li and J.-S. Chen, Angew. Chem., Int. Ed., 2016, 55, 9178–9182 CrossRef CAS.
- M. Choi, Z. Wu and E. Iglesia, J. Am. Chem. Soc., 2010, 132, 9129–9137 CrossRef CAS.
- Z. Wu, S. Goel, M. Choi and E. Iglesia, J. Catal., 2014, 311, 458–468 CrossRef CAS.
- H. J. Cho, D. Kim, J. Li, D. Su and B. Xu, J. Am. Chem. Soc., 2018, 140, 13514–13520 CrossRef CAS.
- M. Moliner, J. E. Gabay, C. E. Kliewer, R. T. Carr, J. Guzman, G. L. Casty, P. Serna and A. Corma, J. Am. Chem. Soc., 2016, 138, 15743–15750 CrossRef CAS.
- Z. Zhao, Y. Li, M. Feyen, R. McGuire, U. Müller and W. Zhang, ChemCatChem, 2018, 10, 2254–2259 CrossRef CAS.
- F. Wang, J. Ren, Y. Cai, L. Sun, C. Chen, S. Liang and X. Jiang, Chem. Eng. J., 2016, 283, 922–928 CrossRef CAS.
- Y. Wu, Y. Chai, J. Li, H. Guo, L. Wen and C. Liu, Catal. Commun., 2015, 64, 110–113 CrossRef CAS.
- Y. Wu, J. Li, Y. Chai, H. Guo and C. Liu, J. Membr. Sci., 2015, 496, 70–77 CrossRef CAS.
- K. Miyake, R. Inoue, M. Nakai, Y. Hirota, Y. Uchida, S. Tanaka, M. Miyamoto and N. Nishiyama, Microporous Mesoporous Mater., 2018, 271, 156–159 CrossRef CAS.
- J. Zhang, L. Wang, Y. Shao, Y. Wang, B. C. Gates and F.-S. Xiao, Angew. Chem., 2017, 129, 9879–9883 CrossRef.
- C. Wang, L. Wang, J. Zhang, H. Wang, J. P. Lewis and F.-S. Xiao, J. Am. Chem. Soc., 2016, 138, 7880–7883 CrossRef CAS.
- J. Gu, Z. Zhang, P. Hu, L. Ding, N. Xue, L. Peng, X. Guo, M. Lin and W. Ding, ACS Catal., 2015, 5, 6893–6901 CrossRef CAS.
- J. Gu, Z. Zhang, L. Ding, K. Huang, N. Xue, L. Peng, X. Guo and W. Ding, Catal. Commun., 2017, 97, 98–101 CrossRef CAS.
- X. Yang, Q. Liu, Y. Zhang, X. Su, Y. Huang and T. Zhang, Nanoscale, 2018, 10, 11320–11327 RSC.
- J. Wang, L. Liu, X. Dong, L. Alfilfil, C.-E. Hsiung, Z. Liu and Y. Han, Chem. Mater., 2018, 30, 6361–6369 CrossRef CAS.
- A. Philippaerts, S. Paulussen, A. Breesch, S. Turner, O. I. Lebedev, G. Van Tendeloo, B. Sels and P. Jacobs, Angew. Chem., Int. Ed., 2011, 50, 3947–3949 CrossRef CAS.
- T. Iida, D. Zanchet, K. Ohara, T. Wakihara and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2018, 57, 6454–6458 CrossRef CAS.
- D. V. Peron, V. L. Zholobenko, M. R. de la Rocha, M. Oberson de Souza, L. A. Feris, N. R. Marcilio, V. V. Ordomsky and A. Y. Khodakov, J. Mater. Sci., 2019, 54, 5399–5411 CrossRef CAS.
- A. R. Morgado Prates, C. Pagis, F. C. Meunier, L. Burel, T. Epicier, L. Roiban, S. Koneti, N. Bats, D. Farrusseng and A. Tuel, Cryst. Growth Des., 2018, 18, 592–596 CrossRef CAS.
- K. M. Kwok, S. W. D. Ong, L. Chen and H. C. Zeng, ACS Appl. Mater. Interfaces, 2019, 11, 14774–14785 CrossRef CAS.
- L. Liu, U. Díaz, R. Arenal, G. Agostini, P. Concepción and A. Corma, Nat. Mater., 2017, 16, 132–138 CrossRef CAS.
- Y. Zhang, K. Fulajtárová, M. Kubů, M. Mazur, M. Shamzhy, M. Hronec and J. Čejka, Mater. Today Nano, 2019, 8, 100056 CrossRef.
- Q. Sun, N. Wang, Q. Bing, R. Si, J. Liu, R. Bai, P. Zhang, M. Jia and J. Yu, Chem, 2017, 3, 477–493 CAS.
- A. R. Morgado Prates, F. Meunier, M. Dodin, R. Martinez Franco, D. Farrusseng and A. Tuel, Chem. – Eur. J., 2019, 25, 2972–2977 CrossRef CAS.
- C. Liu, J. Liu, S. Yang, C. Cao and W. Song, ChemCatChem, 2016, 8, 1279–1282 CrossRef CAS.
- Y. Chai, G. Wu, X. Liu, Y. Ren, W. Dai, C. Wang, Z. Xie, N. Guan and L. Li, J. Am. Chem. Soc., 2019, 141, 9920–9927 CrossRef CAS.
- C. Wang, Z. Liu, L. Wang, X. Dong, J. Zhang, G. Wang, S. Han, X. Meng, A. Zheng and F.-S. Xiao, ACS Catal., 2018, 8, 474–481 CrossRef CAS.
- Q.-H. Xia, S.-C. Shen, J. Song, S. Kawi and K. Hidajat, J. Catal., 2003, 219, 74–84 CrossRef CAS.
- X. Shi, B. Fan, B. Xing, G. Liu, X. Pang, R. Ren and R. Li, J. Mol. Catal. A: Chem., 2014, 385, 85–90 CrossRef CAS.
- Y. Luo, M. Ahmad, A. Schug and M. Tsotsalas, Adv. Mater., 2019, 31, 1901744 CrossRef.
- C. Wang, B. An and W. Lin, ACS Catal., 2019, 9, 130–146 CrossRef CAS.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Catal. Sci. Technol., 2013, 3, 2509–2940 RSC.
- F.-X. Coudert and A. H. Fuchs, Coord. Chem. Rev., 2016, 307, 211–236 CrossRef CAS.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef.
- J. Zhou, P. Wang, C. Wang, Y. T. Goh, Z. Fang, P. B. Messersmith and H. Duan, ACS Nano, 2015, 9, 6951–6960 CrossRef CAS.
- B. Xi, Y. C. Tan and H. C. Zeng, Chem. Mater., 2016, 28, 326–336 CrossRef CAS.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS.
- S. Ding, Q. Yan, H. Jiang, Z. Zhong, R. Chen and W. Xing, Chem. Eng. J., 2016, 296, 146–153 CrossRef CAS.
- W. Zhang, Y. Liu, G. Lu, Y. Wang, S. Li, C. Cui, J. Wu, Z. Xu, D. Tian, W. Huang, J. S. Ducheneu, W. D. Wei, H. Chen, Y. Yang and F. Huo, Adv. Mater., 2015, 27, 2923–2929 CrossRef CAS.
- P. Wang, J. Zhao, X. Li, Y. Yang, Q. Yang and C. Li, Chem. Commun., 2013, 49, 3330–3332 RSC.
- X. Lan, N. Huang, J. Wang and T. Wang, Catal. Sci. Technol., 2017, 7, 2601–2608 RSC.
- C. J. Stephenson, C. L. Whitford, P. C. Stair, O. K. Farha and J. T. Hupp, ChemCatChem, 2016, 8, 855–860 CrossRef CAS.
- L. Lin, T. Zhang, X. Zhang, H. Liu, K. L. Yeung and J. Qiu, Ind. Eng. Chem. Res., 2014, 53, 10906–10913 CrossRef CAS.
- T. Zhang, B. Li, X. Zhang, J. Qiu, W. Han and K. L. Yeung, Microporous Mesoporous Mater., 2014, 197, 324–330 CrossRef CAS.
- T. Zhang, X. Zhang, X. Yan, L. Lin, H. Liu, J. Qiu and K. L. Yeung, Catal. Today, 2014, 236, 41–48 CrossRef CAS.
- C.-H. Kuo, Y. Tang, L.-Y. Chou, B. T. Sneed, C. N. Brodsky, Z. Zhao and C.-K. Tsung, J. Am. Chem. Soc., 2012, 134, 14345–14348 CrossRef CAS.
- Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685–3689 CrossRef CAS.
- W. Zhang, G. Lu, S. Li, Y. Liu, H. Xu, C. Cui, W. Yan, Y. Yang and F. Huo, Chem. Commun., 2014, 50, 4296–4298 RSC.
- Z. Xu, W. Zhang, J. Weng, W. Huang, D. Tian and F. Huo, Nano Res., 2016, 9, 158–164 CrossRef CAS.
- W. Zhang, G. Lu, C. Cui, Y. Liu, S. Li, W. Yan, C. Xing, Y. R. Chi, Y. Yang and F. Huo, Adv. Mater., 2014, 26, 4056–4060 CrossRef CAS.
- S. Xue, H. Jiang, Z. Zhong, Z.-X. Low, R. Chen and W. Xing, Microporous Mesoporous Mater., 2016, 221, 220–227 CrossRef CAS.
- C. J. Stephenson, J. T. Hupp and O. K. Farha, Inorg. Chem. Front., 2015, 2, 448–452 RSC.
- H. Liu, L. Chang, L. Chen and Y. Li, ChemCatChem, 2016, 8, 946–951 CrossRef CAS.
- X. Lan, K. Xue and T. Wang, J. Catal., 2019, 372, 49–60 CrossRef CAS.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS.
- L. Chen, W. Zhan, H. Fang, Z. Cao, C. Yuan, Z. Xie, Q. Kuang and L. Zheng, Chem. – Eur. J., 2017, 23, 11397–11403 CrossRef CAS.
- Y. Yang, F. Wang, Q. Yang, Y. Hu, H. Yan, Y.-Z. Chen, H. Liu, G. Zhang, J. Lu, H.-L. Jiang and H. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 18163–18171 CrossRef CAS.
- W.-J. Chen, B.-H. Cheng, Q.-T. Sun and H. Jiang, ChemCatChem, 2018, 10, 3659–3665 CrossRef CAS.
- Z. Guo, C. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. Li, D. Tesfagaber, A. Thiel and W. Huang, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS.
- T. W. Goh, C.-K. Tsung and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 23254–23260 CrossRef CAS.
- Y.-Z. Chen, Y.-X. Zhou, H. Wang, J. Lu, T. Uchida, Q. Xu, S.-H. Yu and H.-L. Jiang, ACS Catal., 2015, 5, 2062–2069 CrossRef CAS.
- A. Grigoropoulos, A. I. McKay, A. P. Katsoulidis, R. P. Davies, A. Haynes, L. Brammer, J. Xiao, A. S. Weller and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2018, 57, 4532–4537 CrossRef CAS.
- I. E. Ertas, M. Gulcan, A. Bulut, M. Yurderi and M. Zahmakiran, J. Mol. Catal. A: Chem., 2015, 410, 209–220 CrossRef CAS.
- I. Luz, C. Rösler, K. Epp, F. X. L. i Xamena and R. A. Fischer, Eur. J. Inorg. Chem., 2015, 2015, 3904–3912 CrossRef CAS.
- Q. Yang, W. Liu, B. Wang, W. Zhang, X. Zeng, C. Zhang, Y. Qin, X. Sun, T. Wu, J. Liu, F. Huo and J. Lu, Nat. Commun., 2017, 8, 14429 CrossRef CAS.
- L. Lin, T. Zhang, H. Liu, J. Qiu and X. Zhang, Nanoscale, 2015, 7, 7615–7623 RSC.
- L. Lin, H. Liu and X. Zhang, Appl. Surf. Sci., 2018, 433, 602–609 CrossRef CAS.
- Y. Liu, W. Zhang, S. Li, C. Cui, J. Wu, H. Chen and F. Huo, Chem. Mater., 2014, 26, 1119–1125 CrossRef CAS.
- X. Li, Z. Zhang, W. Xiao, S. Deng, C. Chen and N. Zhang, J. Mater. Chem. A, 2019, 7, 14504–14509 RSC.
- L.-N. Chen, H.-Q. Li, M.-W. Yan, C.-F. Yuan, W.-W. Zhan, Y.-Q. Jiang, Z.-X. Xie, Q. Kuang and L.-S. Zheng, Small, 2017, 13, 1700683 CrossRef.
- Z. Li, R. Yu, J. Huang, Y. Shi, D. Zhang, X. Zhong, D. Wang, Y. Wu and Y. Li, Nat. Commun., 2015, 6, 8248 CrossRef CAS.
- L. Chen, H. Li, W. Zhan, Z. Cao, J. Chen, Q. Jiang, Y. Jiang, Z. Xie, Q. Kuang and L. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 31059–31066 CrossRef CAS.
- N. Zhang, Q. Shao, P. Wang, X. Zhu and X. Huang, Small, 2018, 14, 1704318 CrossRef.
- T. Xu, K. Sun, D. Gao, C. Li, X. Hu and G. Chen, Chem. Commun., 2019, 55, 7651–7654 RSC.
- K. Nakatsuka, T. Yoshii, Y. Kuwahara, K. Mori and H. Yamashita, Chem. – Eur. J., 2018, 24, 898–905 CrossRef CAS.
- C. J. Stephenson, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 1361–1363 CrossRef CAS.
- Y. Liu, Y. Shen, W. Zhang, J. Weng, M. Zhao, T. Zhu, Y. R. Chi, Y. Yang, H. Zhang and F. Huo, Chem. Commun., 2019, 55, 11770–11773 RSC.
- J. M. Falkowski, T. Sawano, T. Zhang, G. Tsun, Y. Chen, J. V. Lockard and W. Lin, J. Am. Chem. Soc., 2014, 136, 5213–5216 CrossRef CAS.
- H. Liu, L. Chang, C. Bai, L. Chen, R. Luque and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 5019–5023 CrossRef CAS.
- L. Chang and Y. Li, Mol. Catal., 2017, 433, 77–83 CrossRef CAS.
- S. Zheng, P. Yang, F. Zhang, D. L. Chen and W. Zhu, Chem. Eng. J., 2017, 328, 977–987 CrossRef CAS.
- S. Opelt, V. Krug, J. Sonntag, M. Hunger and E. Klemm, Microporous Mesoporous Mater., 2012, 147, 327–333 CrossRef.
- A. Corma, H. García and F. X. L. i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC.
- Y. Tao, H. Q. Wu, J. Q. Li, L. X. Yang, W. H. Yin, M. B. Luo and F. Luo, Dalton Trans., 2018, 47, 14889–14892 RSC.
- Y.-R. Lee, M.-S. Jang, H.-Y. Cho, H.-J. Kwon, S. Kim and W.-S. Ahn, Chem. Eng. J., 2015, 271, 276–280 CrossRef CAS.
- X. Wang, M. Li, C. Cao, C. Liu, J. Liu, Y. Zhu, S. Zhang and W. Song, ChemCatChem, 2016, 8, 3224–3228 CrossRef CAS.
- L. Chen, H. Chen, R. Luque and Y. Li, Chem. Sci., 2014, 5, 3708–3714 RSC.
- K. Choe, F. Zheng, H. Wang, Y. Yuan, W. Zhao, G. Xue, X. Qiu, M. Ri, X. Shi, Y. Wang, G. Li and Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 3650–3657 CrossRef CAS.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS.
- Z. Tian, D.-L. Chen, T. He, P. Yang, F.-F. Wang, Y. Zhong and W. Zhu, J. Phys. Chem. C, 2019, 123, 22114–22122 CrossRef CAS.
- L. Chen, X. Zhang, J. Zhou, Z. Xie, Q. Kuang and L. Zheng, Nanoscale, 2019, 11, 3292–3299 RSC.
- X. Li, B. Zhang, L. Tang, T. W. Goh, S. Qi, A. Volkov, Y. Pei, Z. Qi, C.-K. Tsung, L. Stanley and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 16371–16375 CrossRef CAS.
- Z. Guan, S. Lu and C. Li, Chin. J. Catal., 2015, 36, 1535–1542 CrossRef CAS.
- J. Zheng, X. Duan, H. Lin, Z. Gu, H. Fang, J. Li and Y. Yuan, Nanoscale, 2016, 8, 5959–5967 RSC.
- Z. Chen, Z. Guan, M. Li, Q. Yang and C. Li, Angew. Chem., Int. Ed., 2011, 50, 4913–4917 CrossRef CAS.
- K. Li, B. Hao, M. Xiao, Y. Kuang, H. Shang, J. Ma, Y. Liao and H. Mao, Appl. Surf. Sci., 2019, 478, 176–182 CrossRef CAS.
- H. Ma, T. Yu, X. Pan and X. Bao, Chin. J. Catal., 2017, 38, 1315–1321 CrossRef CAS.
- W. Gong, Q. Yuan, C. Chen, Y. Lv, Y. Lin, C. Liang, G. Wang, H. Zhang and H. Zhao, Adv. Mater., 2019, 31, 1906051 CrossRef CAS.
- L. Ning, S. Liao, H. Li, R. Tong, C. Dong, M. Zhang, W. Gu and X. Liu, Carbon, 2019, 154, 48–57 CrossRef CAS.
- W. Gong, Y. Lin, C. Chen, M. Al-Mamun, H.-S. Lu, G. Wang, H. Zhang and H. Zhao, Adv. Mater., 2019, 31, 1808341 CrossRef.
- W. Gong, C. Chen, H. Zhang, G. Wang and H. Zhao, Catal. Sci. Technol., 2018, 8, 5506–5514 RSC.
- R. Yun, S. Zhang, W. Ma, X. Lv, S. Liu, T. Sheng and S. Wang, Inorg. Chem., 2019, 58, 9469–9475 CrossRef CAS.
- E. Castillejos, P.-J. Debouttière, L. Roiban, A. Solhy, V. Martinez, Y. Kihn, O. Ersen, K. Philippot, B. Chaudret and P. Serp, Angew. Chem., Int. Ed., 2009, 48, 2529–2533 CrossRef CAS.
- R. Yun, L. Hong, W. Ma, R. Zhang, F. Zhan, J. Duan, B. Zheng and S. Wang, ChemCatChem, 2020, 12, 129–134 CrossRef CAS.
- T. Baramov, P. Loos, J. Hassfeld, H. Alex, M. Beller, T. Stemmler, G. Meier, M. Gottfried and S. Roggan, Adv. Synth. Catal., 2016, 358, 2903–2911 CrossRef CAS.
- Y. Sun, X. Li, Z. Cai, H. Bai, G. Tang and Z. Hou, Catal. Sci. Technol., 2018, 8, 4858–4863 RSC.
- Z. Guan, S. Lu, Z. Chen and C. Li, J. Catal., 2013, 305, 19–26 CrossRef CAS.
- Z. Guan, S. Lu and C. Li, J. Catal., 2014, 311, 1–5 CrossRef CAS.
- M. Atobe, M. Okamoto, T. Fuchigami and J.-E. Park, Ultrason. Sonochem., 2010, 17, 26–29 CrossRef CAS.
- S. Kidambi, J. Dai, J. Li and M. L. Bruening, J. Am. Chem. Soc., 2004, 126, 2658–2659 CrossRef CAS.
- L. Li, H. Zhao and R. Wang, ACS Catal., 2015, 5, 948–955 CrossRef CAS.
- Y. Niu, L. K. Yeung and R. M. Crooks, J. Am. Chem. Soc., 2001, 123, 6840–6846 CrossRef CAS.
- A. Shaabani and M. Mahyari, Catal. Lett., 2013, 143, 1277–1284 CrossRef CAS.
- S. Yang, L. Peng, C. Cao, F. Wei, J. Liu, Y.-N. Zhu, C. Liu, X. Wang and W. Song, Chem. - Asian J., 2016, 11, 2797–2801 CrossRef CAS.
- J. Yang, Y. Zhu, M. Fan, X. Sun, W. D. Wang and Z. Dong, J. Colloid Interface Sci., 2019, 554, 157–165 CrossRef CAS.
- K. Taniya, C. H. Yu, S. C. Tsang, Y. Ichihashi and S. Nishiyama, Catal. Commun., 2011, 14, 6–9 CrossRef CAS.
- F. Wang, Y. Bi, N. Chen, K. Hu and X. Wei, Chem. Phys. Lett., 2018, 711, 152–155 CrossRef CAS.
- H. Shen, H. Tang, H. Yan, W. Han, Y. Li and J. Ni, RSC Adv., 2014, 4, 30180–30185 RSC.
- M. D. Jones, R. Raja, J. M. Thomas, B. F. G. Johnson, D. W. Lewis, J. Rouzaud and K. D. M. Harris, Angew. Chem., Int. Ed., 2003, 42, 4326–4331 CrossRef CAS.
- M. D. Jones, R. Raja, J. Meurig Thomas and B. F. G. Johnson, Top. Catal., 2003, 25, 71–79 CrossRef CAS.
- I. Podolean, C. Hardacre, P. Goodrich, N. Brun, R. Backov, S. M. Coman and V. I. Parvulescu, Catal. Today, 2013, 200, 63–73 CrossRef CAS.
- Y. Su, F. Chang, R. Jin, R. Liu and G. Liu, Green Chem., 2018, 20, 5397–5404 RSC.
- X. Chen, Z. Lou, M. Qiao, K. Fan, S. C. Tsang and H. He, J. Phys. Chem. C, 2008, 112, 1316–1320 CrossRef CAS.
- Y. Wang, N. Su, L. Ye, Y. Ren, X. Chen, Y. Du, Z. Li, B. Yue, S. C. E. Tsang, Q. Chen and H. He, J. Catal., 2014, 313, 113–126 CrossRef CAS.
- L.-T. Weng, Appl. Catal., A, 2014, 474, 203–210 CrossRef CAS.
- H. R. J. ter Veen, T. Kim, I. E. Wachs and H. H. Brongersma, Catal. Today, 2009, 140, 197–201 CrossRef CAS.
- L. Liu, M. Lopez-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Nat. Mater., 2019, 18, 866–873 CrossRef CAS.
- L. Liu, N. Wang, C. Zhu, X. Liu, Y. Zhu, P. Guo, L. Alfilfil, X. Dong, D. Zhang and Y. Han, Angew. Chem., Int. Ed., 2020, 59, 819–825 CrossRef CAS.
- Q. Zhang, A. Mayoral, J. Li, J. Ruan, V. Alfredsson, Y. Ma, J. Yu and O. Terasaki, Angew. Chem., Int. Ed., 2020, 59, 19403–19413 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01709d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |