
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon microfabricated reactor for operando XAS/DRIFTS studies of heterogeneous catalytic reactions†
B.
Venezia‡
 a,
E.
Cao‡
a,
S. K.
Matam
bc,
C.
Waldron
a,
G.
Cibin
d,
E. K.
Gibson
be,
S.
Golunski
c,
P. P.
Wells
bf,
I.
Silverwood
g,
C. R. A.
Catlow
bch,
G.
Sankar
h and
A.
Gavriilidis
*a
a,
E.
Cao‡
a,
S. K.
Matam
bc,
C.
Waldron
a,
G.
Cibin
d,
E. K.
Gibson
be,
S.
Golunski
c,
P. P.
Wells
bf,
I.
Silverwood
g,
C. R. A.
Catlow
bch,
G.
Sankar
h and
A.
Gavriilidis
*a
aDepartment of Chemical Engineering, University College London, London WC1E 7JE, UK. E-mail: a.gavriilidis@ucl.ac.uk
bThe UK Catalysis Hub, Research Complex at Harwell, Harwell, OX11 0FA, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK
dDiamond Light Source Ltd., Harwell Science and Innovation Campus, Didcot OX11 0DE, UK
eSchool of Chemistry, The University of Glasgow, Glasgow G12 8QQ, UK
fSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK
gISIS Pulsed Neutron and Muon Facility, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Oxon, UK
hDepartment of Chemistry, University College London, London WC1H 0AJ, UK
First published on 16th October 2020
Abstract
Operando X-ray absorption spectroscopy (XAS), diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and mass spectrometry (MS) provide complementary information on the catalyst structure, surface reaction mechanisms and activity relationships. The powerful combination of the techniques has been the driving force to design and engineer suitable spectroscopic operando reactors that can mitigate limitations inherent to conventional reaction cells and facilitate experiments under kinetic regimes. Microreactors have recently emerged as effective spectroscopic operando cells due to their plug-flow type operation with no dead volume and negligible mass and heat transfer resistances. Here we present a novel microfabricated reactor that can be used for both operando XAS and DRIFTS studies. The reactor has a glass–silicon–glass sandwich-like structure with a reaction channel (3000 μm × 600 μm; width × depth) packed with a catalyst bed (ca. 25 mg) and placed sideways to the X-ray beam, while the infrared beam illuminates the catalyst bed from the top. The outlet of the reactor is connected to MS for continuous monitoring of the reactor effluent. The feasibility of the microreactor is demonstrated by conducting two reactions: i) combustion of methane over 2 wt% Pd/Al2O3 studied by operando XAS at the Pd K-edge and ii) CO oxidation over 1 wt% Pt/Al2O3 catalyst studied by operando DRIFTS. The former shows that palladium is in an oxidised state at all studied temperatures, 250, 300, 350, 400 °C and the latter shows the presence of linearly adsorbed CO on the platinum surface. Furthermore, temperature-resolved reduction of palladium catalyst with methane and CO oxidation over platinum catalyst are also studied. Based on these results, the catalyst structure and surface reaction dynamics are discussed, which demonstrate not only the applicability and versatility of the microreactor for combined operando XAS and DRIFTS studies, but also illustrate the unique advantages of the microreactor for high space velocity and transient response experiments.
1. Introduction
The relationship between catalyst structure, activity and product selectivity for a given reaction has been extensively studied to design and develop catalytic processes.1–4 Catalyst structure and activity relationships can be explored using a variety of spectroscopic and microscopic techniques and combination of two or more spectroscopic methods in a single reactor can offer complementary information.2,4 X-ray absorption spectroscopy (XAS) is an element-specific technique, which provides information on the geometric, electronic and chemical state of an atom in the catalyst.1,5–9 One of the most commonly used techniques to study catalysts is infrared (IR) spectroscopy, which interrogates the vibrational state of the catalyst and the adsorbed molecules on the catalyst surface, providing useful information on the reaction and deactivation mechanisms.10,11 In particular, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) is suitable for studying catalysts in the form of a powder under reaction conditions.12 Therefore, the combination of XAS, DRIFTS and MS (mass spectrometry) is proven to offer a holistic approach to study catalyst structure and activity relationships.2,13–18In the last two decades, there has been a surge in the application of operando techniques.19–23 However, operando cells often suffer from a range of issues including poor sealing, contamination, large dead volume, and temperature and concentration gradients along the catalyst bed, which can lead to biased conclusions.19,24,25 Furthermore, in order to reduce mass transfer resistances across the catalyst bed and to derive reliable intrinsic kinetic data, the operando cells must be capable of handling high space velocities.26,27 Different configurations of operando and in situ reactor cells have been reported.28–34 Among these, it is common to find modified Harrick cells, which have been used for combined DRIFTS and XAS studies.32–34 However, these cells have a large dead volume that can lead to a broad residence time distribution, by-pass and inhomogeneous reaction conditions. Agostini et al. proposed a spectroscopic cell with a body-dome configuration, which was optimised for time-resolved combined XAS/DRIFTS/MS studies.35 This consisted of a body containing the sample holder and the heater, and a dome including the X-ray and IR windows. The catalyst in powder form was hosted in a crucible, placed in between two carbon-glass windows transparent to X-rays and below a CaF2 window for IR radiation. The reactor could be operated at up to 600 °C and 5 bar. The dead volume in the reaction chamber was around 0.5 cm3 which increased to 1 cm3 when a dome for higher pressure experiments had to be employed. In other studies, different configurations were reported as alternatives.27,36,37 Among these, Chiarello et al. demonstrated a spectroscopic cell for in situ transmission XAS with DRIFTS.36 In this design, the X-ray and IR beams were focussed on the same optical window and were perpendicular to the catalyst bed. Furthermore, the gas inlet and the outlet shared the same direction and the dead volume was reduced compared to a modified Harrick33 or SpectraTech cell.38 However, the gas flow was replaced in the cell in 5 s, due to the dead volume of the inlet and outlet tubes and for combined IR-XAS studies the authors had to drill a hole in the CaF2 window for IR and fill it with carbon based glue to allow X-ray passing through. Dann et al., developed a new spectroscopic packed-bed reactor to study the kinetic oscillations of CO oxidation over a Pd/Al2O3 catalyst with combined energy dispersive EXAFS (EDE) and DRIFTS.37 The reactor cell was made of pure aluminium with a square cross sectional channel (5 mm × 5 mm) that hosted the catalyst in the form of pellets (250–335 μm). It had thin walls on two opposite sides, to allow X-ray spectroscopy analysis to be performed in transmission mode, while on top a rectangular CaF2 window was installed for IR transmission. The heating plate was placed underneath the reactor and the temperature was measured near the outlet region. This cell was tested up to 140 °C and allowed to perform simultaneous operando EDE/DRIFTS at 8 different positions along the packed bed.
The use of microfluidic and microfabricated reactors which can offer accurate control of process conditions, homogeneous temperature distribution and a distinct flow pattern, has been reported for operando spectroscopic studies under realistic reaction conditions.39–47 Such microreactors are valuable and compact tools for spectroscopic studies24,39 including Raman,40,48,49 IR,50–53 X-ray diffraction (XRD)45,54,55 and XAS.42,45,55,56 Among such reactors, silicon microfabricated reactors have unique advantages due to their high thermal conductivity, chemical inertness and mechanical stability. Micromachining allows fabricating microreactors with accurate and versatile microchannel geometries. Silicon microreactors, sealed with glass through an anodic bonding process, can be operated up to 500 °C. In particular, the transparency of silicon to X-rays and IR beams (also glass for Raman) makes the glass-bonded silicon microreactor ideal for combined operando XAS and DRIFTS studies.55 In our previous work, a silver sputter-coated microchannel of a silicon-glass microreactor was employed for operando Raman spectroscopic studies of continuous oxidative dehydrogenation of methanol to formaldehyde.40 The silver sputter-coated microchannel of the microreactor served itself as a catalyst surface and it enabled us to evaluate the effect of temperature and reaction feed composition on the catalyst structure and activity, and to identify reaction intermediates. The same reactor was also employed for in situ XAS studies at the silver K-edge in fluorescence mode.42 This work demonstrated the applicability and versatility of the microchannel reactor for multi technique operando spectroscopic studies. However, the application of the sputter coated microchannel reactor is limited to unsupported metal catalysts. Therefore, here we report a novel silicon–glass microreactor for supported catalysts that can be used for both operando XAS and DRIFTS, addressing many of the limitations imposed by conventional operando cells. The applicability and versatility of the microreactor are demonstrated by investigating the Pd K-edge during methane combustion over 2 wt% Pd/Al2O3 and surface adsorbed species during carbon monoxide oxidation on 1 wt% Pt/Al2O3. The experiments provide new information on catalyst structure and dynamics for these widely studied systems.
2. Materials and methods
2.1. Microreactor cell design and fabrication
The silicon microreactor developed in this work has a glass–silicon–glass sandwich-like structure that can be employed for various spectroscopic studies including XAS, IR and Raman spectroscopy. On the two sides of the silicon layer, microchannels were designed to host a packed-bed of catalyst, inlet and outlet flow channels and slits for XAS measurements. Fig. 1 illustrates the technical drawing of the silicon layer. On the back side of the silicon layer, a serpentine-shaped gas inlet channel (1000 μm × 600 μm; width × depth) leads to the reaction channel (3000 μm × 600 μm; width × depth) that hosts the catalyst bed. A set of small pillars (500 μm × 40 μm; length × width; arranged in 20 μm intervals) is placed at the outlet of the reaction channel acting as catalyst retainer. Perpendicular to and on either side of the reaction channel, six dead-end slits (2000 μm × 600 μm; width × depth) allow the X-ray beam to probe the catalyst bed in transmission mode at six different axial locations along the bed. The silicon wall between the six dead-end X-ray slits and the reaction channel is 250 μm thick. Alternating with the six X-ray slits, seven thermocouple wells (600 μm × 600 μm; width × depth) enable monitoring of the axial temperature profile of the catalyst bed using 0.25 mm-diameter thermocouples (Omega Engineering). Beyond the set of small pillars, the back side shares a common hole with the front side (see top of Fig. 1). This allows the gas coming from the catalyst bed to flow into the outlet channel in the front side of the silicon layer (600 μm × 200 μm; width × depth). The microstructured channels on both sides (front and back) of the silicon layer are sealed with two 0.8 mm thick glass wafers (Corning 7740). The front glass cover has a hollow window (36 mm × 6 mm; length × depth) to allow the IR beam to probe the catalyst bed through the silicon wall, and the back-side glass cover has two pre-drilled holes for the gas inlet and outlet connections. Fig. 2 shows a schematic representation of the two sides (back and front) of the microreactor with inlet and outlet flows and X-ray and IR beam paths. A representation of a 15 mm long catalyst bed, preceded by glass beads used to distribute the gas flow and pillars for retaining the catalyst particles at the outlet, is shown in the back side view. The overall thickness of the fabricated reactor is 2.6 mm, with a length and width of 72 mm and 20 mm, respectively. Pictures of the reactor are shown in Fig. S1 (see ESI†).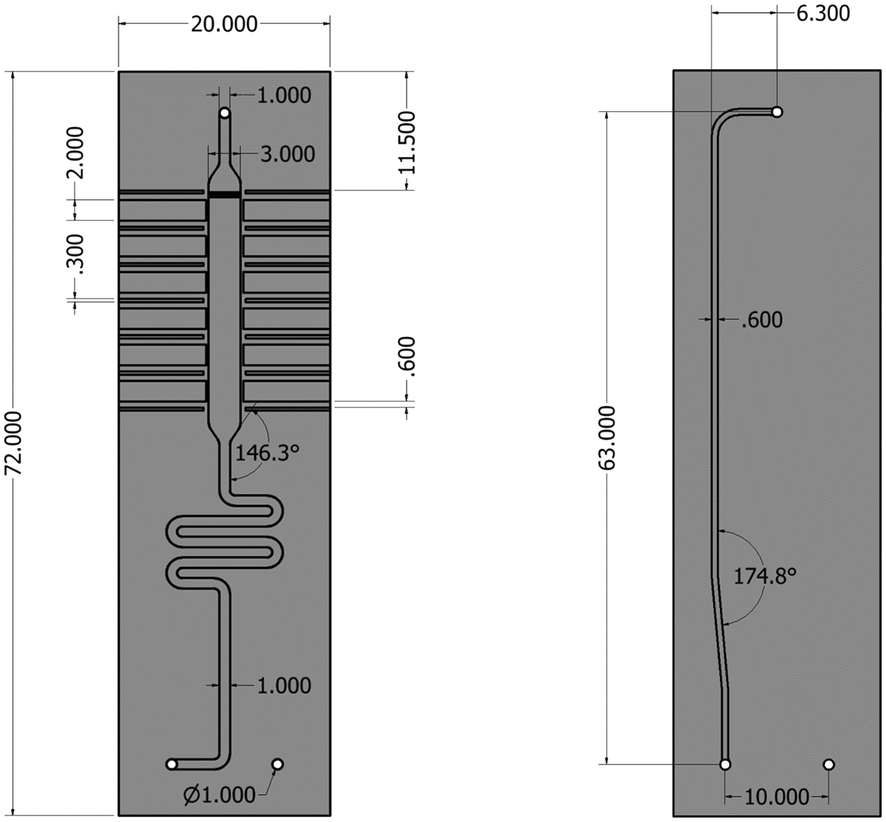 | ||
| Fig. 1 Technical drawing of the silicon microreactor. Back side (left), front side (right). Dimensions are in millimetres. | ||
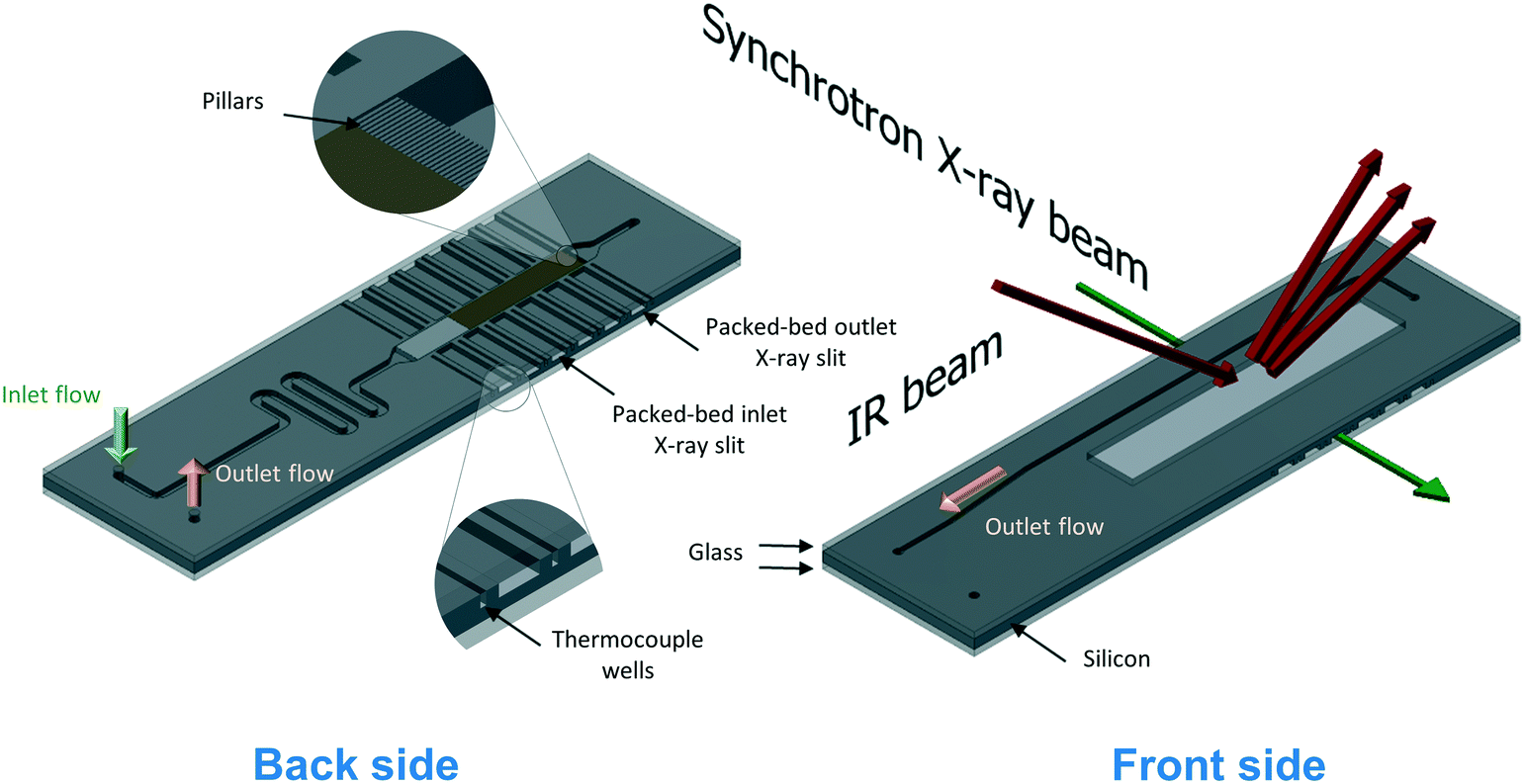 | ||
| Fig. 2 Schematic representation of the back and front side of the microreactor for operando XAS/MS and DRIFTS/MS studies. A packed-bed catalyst (brown) and glass beads (light grey) upstream of the catalyst bed are depicted on the back side image. Insets show detailed view of the pillars used for retaining the catalyst particles and the thermocouple wells. | ||
The microfabrication process was performed via double-sided photolithography (Q4000-6, Quintel) followed by deep reactive-ion etching (DRIE, STS ASE) of each side of a double-sided polished 1 mm thick and 100 mm diameter silicon wafer (undoped float zone grown, CRYSTRAN UK). For the wafer, three microfabrication steps were carried out. The front side of the reactor with its gas channel was processed first, patterned and etched to ca. 200 μm. Except for the inlet/outlet holes, the etched channel was then covered with a photoresist (SPR-220-7, Rohm and Haas) which was applied manually using a small pipette tip. This was followed by a soft baking at 90 °C for 10 min. The front side was further etched by ca. 200 μm for the holes. Afterwards, the wafer with the processed front side was cleaned thoroughly with Piranha solution (H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2 = 3:1) at 100 °C for 15 min, followed by another lithography step. The back side of the wafer was patterned with the help of infrared beam alignment. Subsequently, DRIE was used to etch for 600 μm to create the reactor channel. The holes on the silicon layer were etched through to form connections between the gas inlet channel and the reaction channel. The micro-fabricated silicon wafer was then cut into three individual reactor chips. Double-sided anodic bonding followed to seal the two sides of the silicon layer with the two glass layers by performing two steps of anodic bonding. The first step was to assemble the front glass layer to the front surface of the silicon layer on a hotplate (Stuart SD162) at a temperature of 420 °C while applying a DC voltage of 500 V across the assembly (cathode attached to the silicon and anode to the glass). After the first anodic bonding, the assembled silicon–glass chip was removed from the hot plate, cooled down and cleaned with Piranha solution. The second step was performed by placing the silicon–glass chip onto the hotplate (when the hot plate was cool enough) with the back surface of the silicon layer facing up and then putting the back glass cover on the top of the silicon chip by aligning the holes on the glass cover to the inlet and outlet holes on the silicon layer. After heating the hotplate to 420 °C, a DC voltage of 500–700 V was applied across the sandwich assembly.
:H2O2 = 3:1) at 100 °C for 15 min, followed by another lithography step. The back side of the wafer was patterned with the help of infrared beam alignment. Subsequently, DRIE was used to etch for 600 μm to create the reactor channel. The holes on the silicon layer were etched through to form connections between the gas inlet channel and the reaction channel. The micro-fabricated silicon wafer was then cut into three individual reactor chips. Double-sided anodic bonding followed to seal the two sides of the silicon layer with the two glass layers by performing two steps of anodic bonding. The first step was to assemble the front glass layer to the front surface of the silicon layer on a hotplate (Stuart SD162) at a temperature of 420 °C while applying a DC voltage of 500 V across the assembly (cathode attached to the silicon and anode to the glass). After the first anodic bonding, the assembled silicon–glass chip was removed from the hot plate, cooled down and cleaned with Piranha solution. The second step was performed by placing the silicon–glass chip onto the hotplate (when the hot plate was cool enough) with the back surface of the silicon layer facing up and then putting the back glass cover on the top of the silicon chip by aligning the holes on the glass cover to the inlet and outlet holes on the silicon layer. After heating the hotplate to 420 °C, a DC voltage of 500–700 V was applied across the sandwich assembly.
The microreactor was incorporated in an in-house made heating unit (see ESI†). The heating was achieved using a ceramic heater (25 mm × 50 mm × 2.5 mm, ULTRAMIC® ceramic heaters, Watlow) which was fitted into a stainless steel holder. High temperature O-rings (Perlast G80A, O Rings Ltd) were used to seal the inlet and outlet connections to the microreactor, which was placed onto the heater holder and fixed firmly in position using the top clamp of the holder. The microreactor can be operated up to a maximum temperature of 400 °C due to the material used for insulating the electrical connections of the thermocouple embedded in the ceramic heater, which can be changed to achieve a higher reaction temperature if required.
2.2. Catalytic combustion of methane
Prior to the operando spectroscopic studies, the applicability of the microreactor was tested for the catalytic combustion of methane at atmospheric pressure over a catalyst with a nominal composition of 2 wt% Pd/Al2O3 prepared by a modified impregnation method. The synthesis procedure for this catalyst and its characterisation have been reported elsewhere.57 The microreactor was loaded with the catalyst (28.5 mg and 53–63 μm sieve fraction), by applying vacuum at the outlet port, which formed a catalyst bed length of ca. 15 mm. Glass beads (75 μm, Sigma Aldrich) were placed at the inlet of the catalyst bed in order to ensure flow equi-distribution. The catalyst was heated up at 10 °C min−1 under 10% O2/He and calcined for 30 min at 400 °C. After calcination, the catalyst was cooled down to 200 °C and the gas was switched from 10% O2/He to a mixture of 1% CH4 and 4% of O2 in He with a flowrate of 40 NmL min−1, corresponding to a space velocity of around 84250 cm3 gcat−1 h−1. The temperature was increased from 200 °C to 400 °C (at 10 °C min−1) with a steady state dwell of around 45 min every 50 °C increment. The outlet gas composition was analysed by an online gas chromatograph (GC, Agilent 7890A) equipped with a thermal conductivity detector and two columns (HP-PLOT Molesieve and GS-CarbonPLOT). The temperature profile along the catalyst bed was measured in order to assess isothermality of the bed during reaction.
2.3. Operando XAS/MS
Operando XAS/MS experiments for the catalytic combustion of methane over 2 wt% Pd/Al2O3 were conducted at the B18 beamline of the Diamond Light Source (DLS), Didcot, UK. A picture of the setup is shown in Fig. 3, while the schematic of the experimental rig is presented in ESI.† XAS measurements were performed at the Pd K-edge in transmission mode using the quick EXAFS (QEXAFS) setup with a fast-scanning Si(311) double crystal monochromator.58 The data acquisition setup consisted of three detectors measuring the incident beam intensity, I0, the transmitted beam through the sample, It, and the one through the reference, Iref. A standard Pd foil reference was placed between It and Iref and each spectrum was acquired with an exposure time of 180 s. The X-ray beam size was optimised to around 200 μm × 100 μm for the measurement through the narrow X-ray access slits of the microreactor, which was placed on an adjustable stage in order to allow the alignment of the beam inside the X-ray slits. XAS measurements were conducted at the inlet and the outlet positions of the catalyst bed (see back side in Fig. 2). The reaction mixture from the outlet of the microreactor was monitored using an online MKS mass spectrometer. | ||
| Fig. 3 Microreactor assembly (a) at the beamline B18 at the Diamond Light Source for the operando XAS/MS study and (b) at the Research Complex at Harwell for the operando DRIFTS/MS study. | ||
Operando XAS studies were conducted similarly to the reaction test in section 2.2. The 2 wt% Pd/Al2O3 catalyst particles (27.7 mg of 53–63 μm sieve fraction) were loaded into the microreactor. The catalyst was heated up at 10 °C min−1 under 10% O2/He flow and calcined at 400 °C for 30 min. The temperature was then decreased to 250 °C and the gas flow was switched from 10% O2/He to 1% CH4, 4% O2 and 5% Ar in He (total flow rate of 40 NmL min−1). The combustion reaction was carried out at 250, 300, 350 and 400 °C (at 10 °C min−1) with a dwell time of around 45 min at each temperature to achieve steady state conditions. Under steady state, the catalyst bed at the inlet region was probed with the X-ray beam in transmission mode.
In another experiment, a temperature-resolved reduction of 2 wt% Pd/Al2O3 was performed with methane in flow. After calcination at 400 °C, the catalyst was cooled down to 225 °C under 10% O2/He flow. Then the gas mixture was switched from 10% O2/He to 1% CH4/He at 40 NmL min−1 and the temperature of the reactor was gradually increased from 225 to 300 °C at a rate of 1 °C min−1 while monitoring the catalyst at the bed inlet by XAS. After the experiment, the gas mixture was switched back to 10% O2/He, the temperature increased up to 400 °C and the catalyst was re-calcined. This time the X-ray beam was directed at the outlet bed position and a temperature-resolved reduction was performed again under 1% CH4/He at 40 NmL min−1 from 225 to 305 °C (1 °C min−1). During the two temperature-resolved reductions, XAS scans were taken every 3.46 min, generating 21 and 24 spectra respectively.
The raw XAS spectra were processed using the software Athena.59 The X-ray absorption near edge structure (XANES) spectra were pre-edge subtracted and normalised to the post-edge background. The linear combination fitting (LCF) of the normalised XANES spectra was then performed around the Pd K-edge, 24150–24986 eV, using the software Athena.59 The XANES spectra of the reference Pd metal foil were used as standard for the palladium metal (Pd0) in the LCF, while for the PdO component the spectra at the inlet during calcination were taken as reference. Their weightings were constrained to be between 0 and 1 and their sum to be equal to 1. The fitting quality was assessed using R-factors, which were consistently low for all the fittings (R-factor = 0.00083 ± 0.00021). An example of a LCF of a XANES spectrum for the catalyst at 250 °C is presented in Fig. S4 (see ESI†). The Fourier transform (FT) of the EXAFS data was performed on the k3-weighted functions between 2.7 and 10 Å−1.
2.4. Operando DRIFTS/MS
The applicability of the microreactor for operando DRIFTS studies was evaluated for the catalytic combustion of methane over the 2 wt% Pd/Al2O3 catalyst and for the oxidation of carbon monoxide over a commercial 1 wt% Pt/Al2O3, taking DRIFTS spectra in the middle of the catalytic bed, approximately between the inlet and the outlet region. Methane combustion was performed between 250 and 400 °C, which is directly comparable with the operando XAS studies. In fact, the same reaction conditions described in section 2.2 were applied in the operando DRIFTS study of methane combustion over 2 wt% Pd/Al2O3 (28 mg and 53–63 μm sieve fraction). Moreover, the same reaction was also conducted using a commercial high temperature Harrick DRIFTS cell for comparative purposes.Oxidation of carbon monoxide was performed using a typical amount of 29.6 mg (sieve fraction of 38–53 μm) of a commercial 1 wt% Pt/Al2O3 catalyst (Sigma Aldrich). The catalyst was heated up at 10 °C min−1 under 10% O2/He flow and calcined at 400 °C for 60 min. Then the flow was switched from 10% O2/He to pure He, and then to 10% H2/He at 400 °C for 30 min in order to reduce the catalyst. Subsequently, the hydrogen flow was replaced with pure helium at the same temperature to flush the catalyst bed for 15 min and the reactor was cooled to 30 °C under the same helium flow. Temperature-resolved CO oxidation over the reduced Pt/Al2O3 catalyst was then conducted between 30 and 400 °C using 10% O2/He (at 30 NmL min−1) and 10% CO/He (at 10 NmL min−1) reaction mixture. The reaction temperature was ramped at 5 °C/min from 30 to 400 °C and DRIFTS spectra were collected approximately every 30 s, while reaction products were simultaneously monitored by MS. At 400 °C, the reaction mixture was switched to 10% O2/He for 30 min to calcine the catalyst. Afterwards, the catalyst was cooled to 30 °C (10 °C min−1) under the same O2/He flow. A temperature-resolved CO oxidation over the oxidised 1 wt% Pt/Al2O3 catalyst was carried out using the same reaction mixture and temperature ramp as described above for the CO oxidation with the pre-reduced catalyst.
After the reaction at 400 °C, the temperature of the reactor was decreased to 310 °C and the gas was switched to a stoichiometric composition of 10% O2/He (5 NmL min−1), 10% CO/He (10 NmL min−1), balance He (25 NmL min−1) to study possible concentration gradients of adsorbed species along the catalyst bed by DRIFTS. For this experiment, the catalyst bed was probed under steady-state conditions with DRIFTS from the inlet to the outlet of the catalyst bed, by moving (at 2 mm steps) the reactor stage.
All experiments were conducted using an Agilent Cary 680 series spectrometer equipped with a Harrick DaVinci arm (see Fig. 3). The DaVinci arm was fitted with Praying Mantis optics, which focussed the IR beam onto the catalyst bed of the microreactor. The microreactor outlet was connected to a Hiden QGA mass spectrometer for analysis of the reactor effluent. As in the operando XAS experiments, the microreactor was placed onto an adjustable stage that was located under the DaVinci arm. The experimental rig schematic is shown in the ESI.† The extended IR focal length of the Praying Mantis optics allowed the microreactor to be located at about 4.7 mm below the DaVinci arm. Prior to DRIFTS experiments, alignment of the IR beam along the central line of the catalyst bed was required. DRIFTS spectra were acquired by taking 64 scans with a resolution of 4 cm−1 using a liquid nitrogen cooled mercury cadmium telluride (MCT) detector.
In all the experiments, the catalyst performance was assessed using the reactant consumption rate, r, according to eqn (1), where F is the inlet molar flowrate, yin and yout are the gas inlet and outlet concentrations (vol%, methane or carbon monoxide) respectively and ncat is the molar amount of metal catalyst.
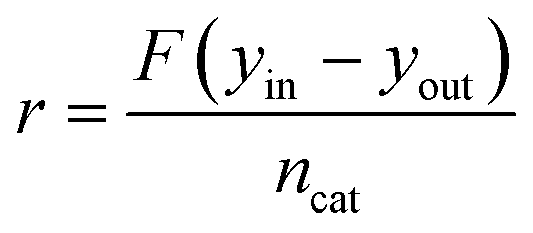 | (1) |
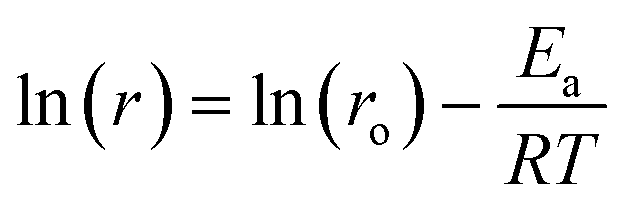 | (2) |
3. Results and discussion
3.1. Catalytic combustion of methane
The combustion of methane over 2 wt% Pd/Al2O3 was performed using the microreactor, as described in section 2.2, and the methane outlet concentration is shown in Fig. 4 at four different set temperatures: 250, 300, 350 and 400 °C. The combustion of methane starts at 250 °C with an outlet methane concentration of 0.99% which decreases to 0.48% at 400 °C, equivalent to a methane consumption rate of 107 molCH4 molPd−1 h−1. Similar methane consumption rates over Pd/Al2O3 catalysts at 400 °C and atmospheric pressure can be found in previous studies (see Table 1).60–64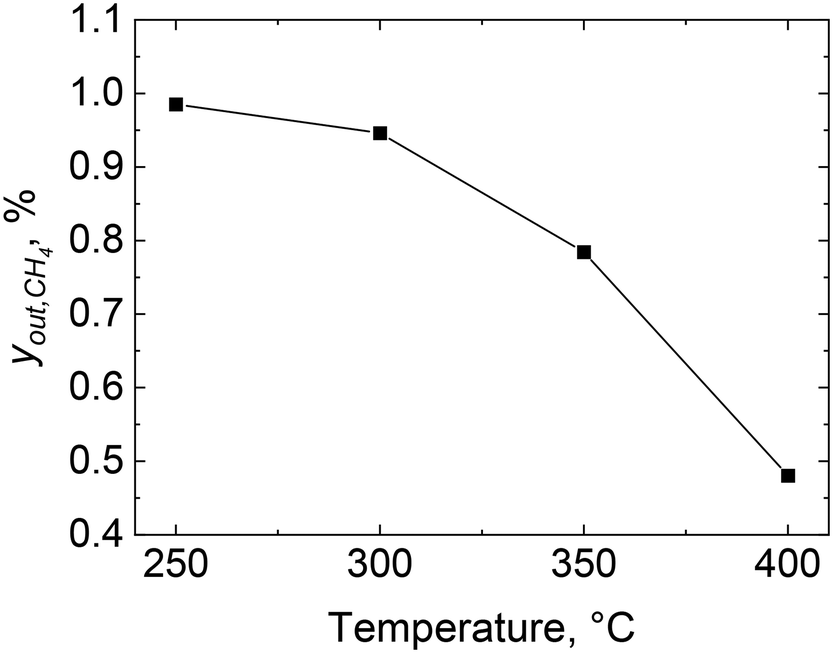 | ||
| Fig. 4 Methane outlet concentration, yout, CH4, during combustion over 2 wt% Pd/Al2O3 catalyst at different reactor set temperatures: 250, 300, 350 and 400 °C. Gas inlet composition: 1% CH4 and 4% of O2 in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 28.5 mg (53–63 μm). | ||
| Reference | wt% Pd | Catalyst pretreatment | y in,CH4, % | O2/CH4, − | r CH4, molCH4 molPd−1 h−1 |
|---|---|---|---|---|---|
| Burch et al.60 | 4 | Calcination | 0.8 | 20 | 91 |
| Briot et al.61 | 1.95 | Reduction | 1 | 4 | 10 |
| Briot et al.61 | 1.95 | Ageing | 1 | 4 | 65 |
| Burch et al.62 | 4 | Calcination | 0.3 | 5 | 35 |
| Mouaddib et al.63 | 1.93 | Reduction | 1 | 4 | 5 |
| Mouaddib et al.63 | 1.93 | Ageing | 1 | 4 | 70 |
| This work | 2 | Calcination | 1 | 4 | 107 |
Fig. 5 shows the temperature values along the reactor bed during methane combustion. It is evident that no large temperature gradient is present along the glass beads and the catalyst bed, especially below 400 °C. At 400 °C, the temperature profile along the bed shows the highest variation (±1 °C from the set temperature). Based on the conversion and temperature profiles obtained, the microreactor design and performance was judged to be satisfactory to be employed for operando XAS/MS studies for the combustion of methane.
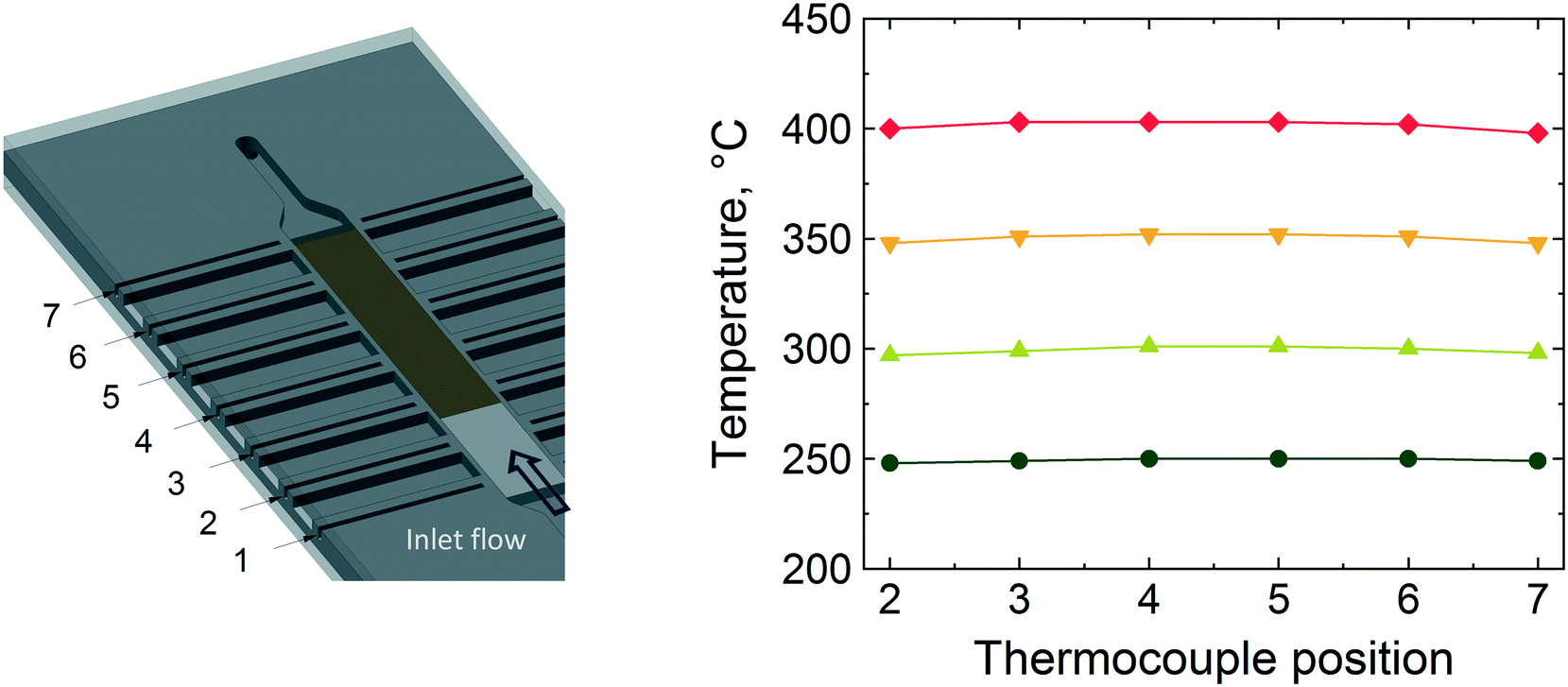 | ||
| Fig. 5 Temperature profiles along the reactor bed during methane combustion over 2 wt% Pd/Al2O3 catalyst at different reactor set temperatures: 250, 300, 350 and 400 °C. The thermocouple positions are at the six of the seven thermowells shown in the figure on the left. | ||
From the observed reaction rate (107 molCH4 molPd−1 h−1), the catalyst particle size (53–63 μm sieve fraction) and the gas physical properties at 400 °C, the Péclet (Pe), Mears (MR) and Weisz–Prater (CWP) numbers can be calculated (see Table 2). The large Péclet number suggests that axial dispersion within the packed bed can be neglected, hence the microreactor can be regarded as an ideal plug-flow reactor. The low Mears and Weisz–Prater numbers indicate that the reactor operates under no external or internal mass transfer resistances, respectively. The details on the calculation of these numbers are reported in the ESI.†
| Pe | MR | C WP |
|---|---|---|
| 61 (>6) | 0.07 (<0.15) | 0.003 (≪1) |
3.2. Operando XAS/MS
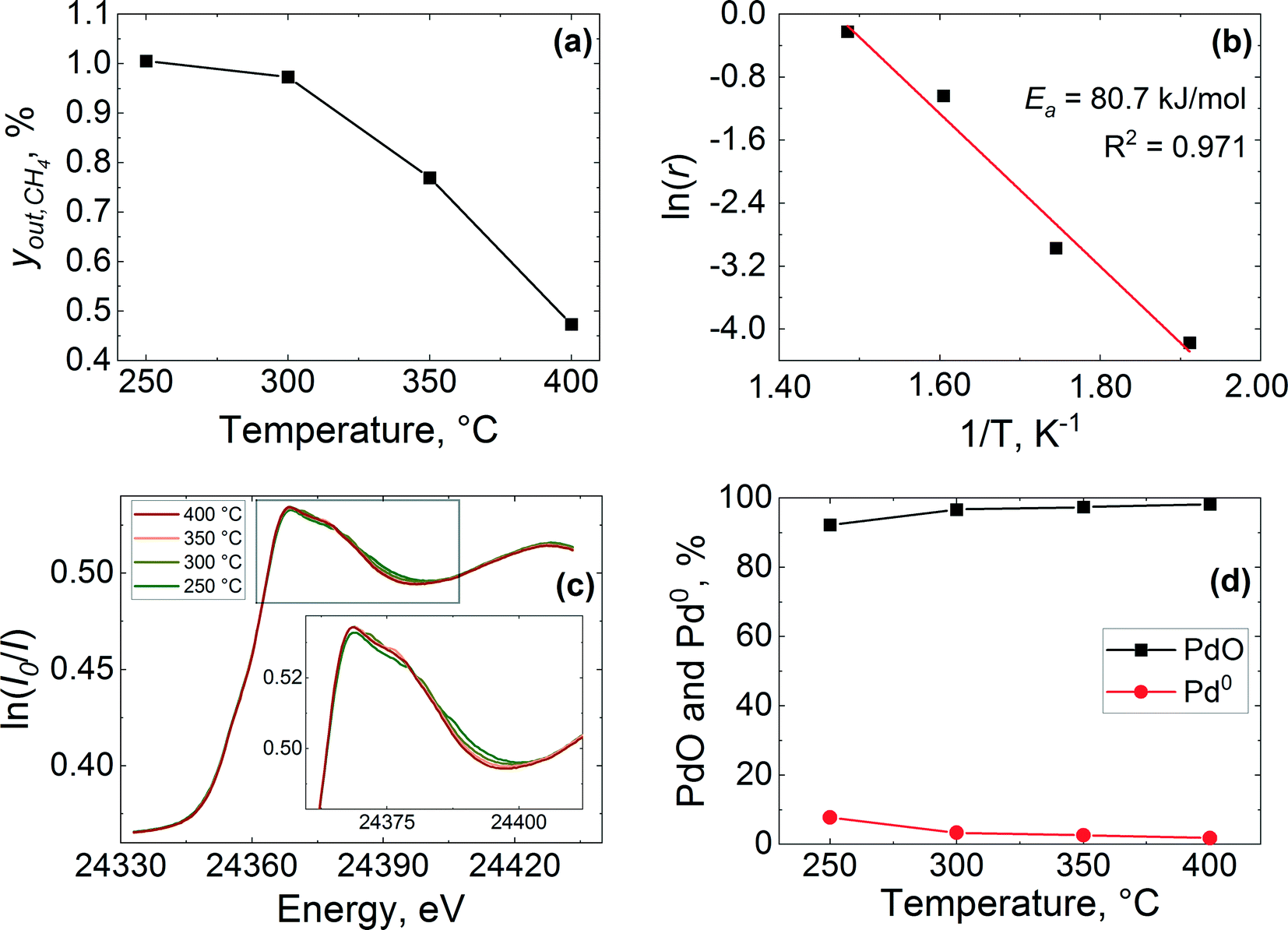 | ||
| Fig. 6 Methane combustion over 2 wt% Pd/Al2O3 catalyst: (a) methane outlet concentration, yout,CH4, (b) Arrhenius plot of the reaction rate, (c) normalised operando XANES spectra at the Pd K-edge of the inlet of the catalyst bed and (d) PdO and Pd0 and concentrations obtained by LCF of the spectra. Gas inlet composition: 1% CH4 and 4% of O2 in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 27.7 mg (53–63 μm). | ||
In agreement with XANES and LCF data (Fig. 6(c) and (d)), the Fourier transform (FT) of the corresponding EXAFS data shows a contribution from oxygen in the first coordination shell of the PdO like structure at ca. 1.5 Å (phase shift uncorrected) and another contribution from the corresponding Pd–Pd pair in the second coordination shell at around 3 Å (see Fig. 7). No other peaks attributable to metallic palladium are apparent except for a small contribution from Pd–Pd metal (ca. 2.4 Å) at 250 °C. These results further indicate that the palladium was predominantly in oxidised state in the catalyst during reaction at all studied temperatures.
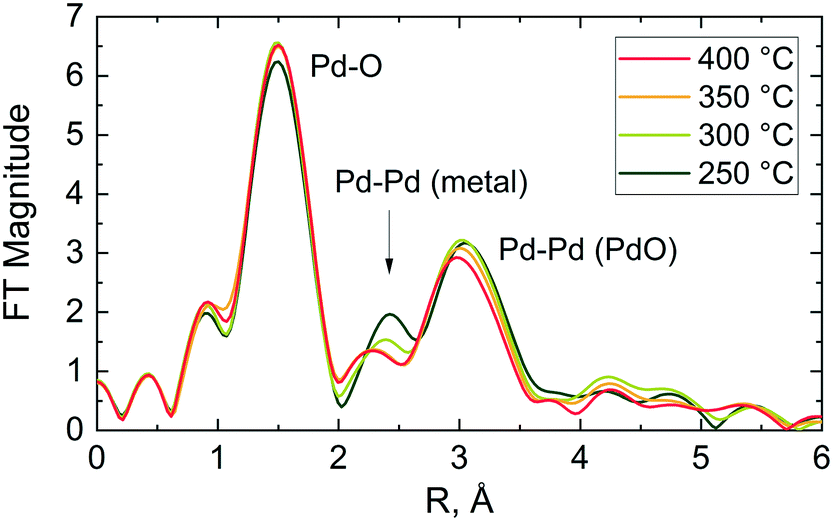 | ||
| Fig. 7 Phase shift uncorrected Fourier transform magnitude of the Pd K-edge EXAFS of the 2 wt% Pd/Al2O3 catalyst during methane combustion. Gas inlet composition: 1% CH4 and 4% of O2 in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 27.7 mg (53–63 μm). | ||
The stacked XANES, PdO and Pd metal component extracted from LCF analysis of XANES data and the FT of the EXAFS data collected for the catalyst at the reactor inlet are plotted as function of reaction temperature in Fig. 8. It is evident from the change in the white line of the XANES spectra that the palladium reduction occurred already at around 240 °C. The palladium speciation at the catalyst bed inlet is determined in more detail from XANES spectra by application of LCF and it can be readily seen from Fig. 8(b) that the PdO and Pd0 components change with the increase in temperature. At 225 °C, immediately after calcination at 400 °C for 30 min and at the beginning of the reduction process, it appears that the inlet of the catalyst bed was almost completely oxidised as evident from the low presence of a metallic Pd phase fraction (2.1%). This was determined by LCF and indicates that the calcination was effective. During the temperature-resolved reduction, the isosbestic point (i.e., the point at which the phase composition of two species is equal) is observed at 246 °C. These results reveal that the reduction of palladium oxide at the inlet of the catalyst bed was gradual.
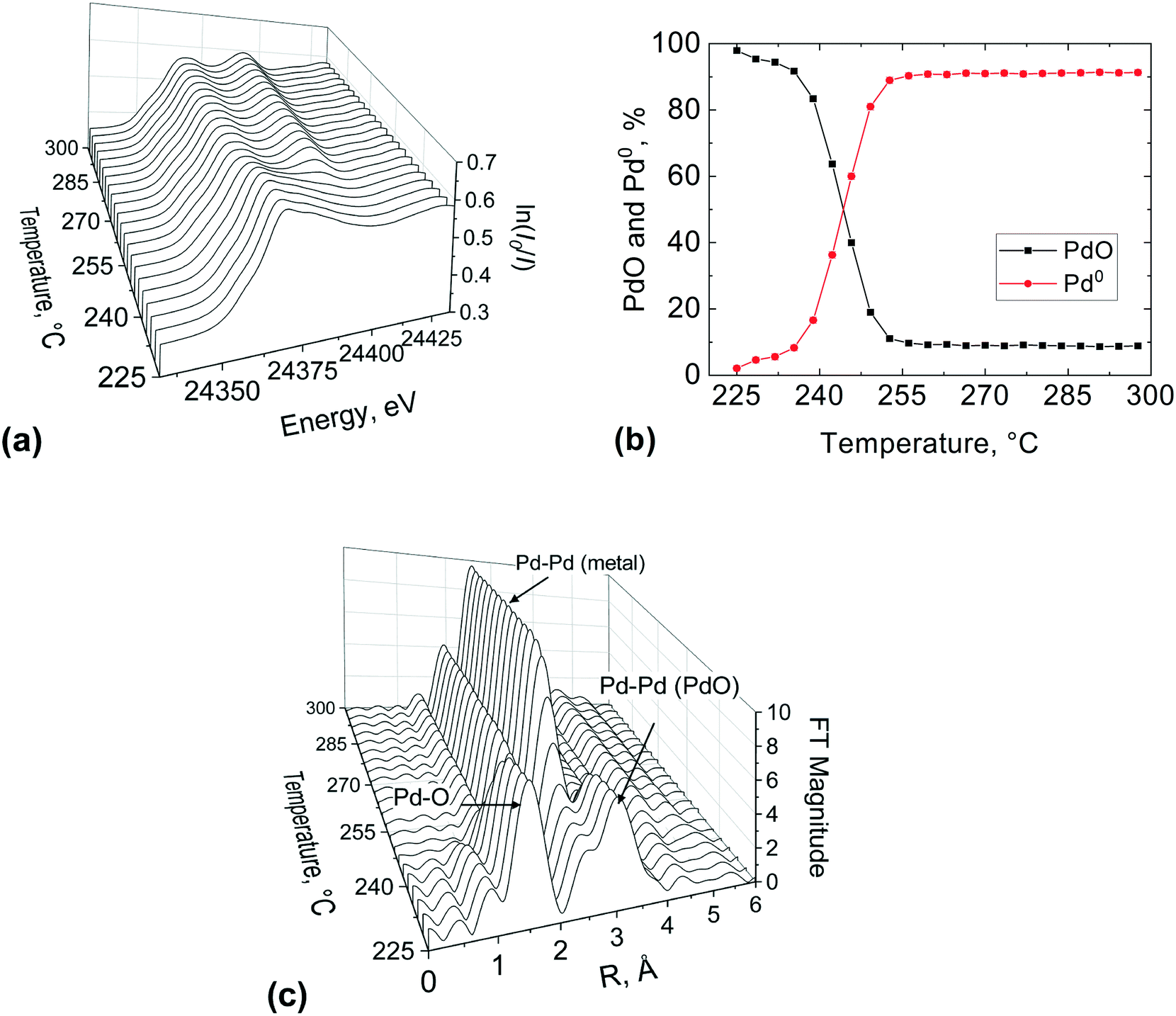 | ||
| Fig. 8 XAS study at the Pd K-edge of the 2 wt% Pd/Al2O3 catalyst at the bed inlet position during the first temperature-resolved reduction with methane: (a) normalised XANES spectra, (b) PdO and Pd0 concentrations obtained by LCF of the spectra and (c) phase shift uncorrected Fourier transform of EXAFS. Gas inlet composition: 1% CH4 and 4% of O2 in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 27.7 mg (53–63 μm). | ||
These observations are further corroborated by the FT of the EXAFS (Fig. 8(c)). At 225 °C the catalyst at the bed inlet presents two peaks at ca. 1.5 Å (phase shift uncorrected) in the first and at ca. 3 Å in the second coordination shells, respectively, attributable to Pd–O and Pd–Pd bonds of PdO like species. The presence of these two peaks at 225 °C suggests that the catalyst was mainly in the oxidised state, consistent with XANES. During the temperature-resolved reduction of the catalyst, the characteristic peak of the Pd–Pd in the first coordination shell of metallic Pd emerges at around 2.4 Å (phase shift uncorrected), with the concomitant progressive decrease of the PdO peaks in the first coordination shell at around 1.5 Å (phase shift uncorrected), which indicates the reduction of palladium oxide to metallic Pd. The amplitude of the metallic Pd–Pd peak in the first coordination shell establishes at around 250 °C, which is in excellent agreement with XANES and LCF.
The outlet region of the catalyst bed was investigated in a similar way after re-calcination of the catalyst, and results are reported in Fig. 9. It is evident from the change in the white line of the XANES spectra (Fig. 9(a)) that the palladium reduction occurred at around 280 °C, which is at higher temperature compared to the bed inlet (Fig. 8). This could be ascribed to the re-calcination before the second temperature-resolved reduction, which might have sintered the catalyst. The metal palladium speciation at the outlet can be seen in Fig. 9(b). At the beginning of the second reduction, it appears that the catalyst bed was not completely oxidised as evident from the presence of a metallic Pd phase fraction (9.9%). This suggests that the first calcination was more effective than the second one.
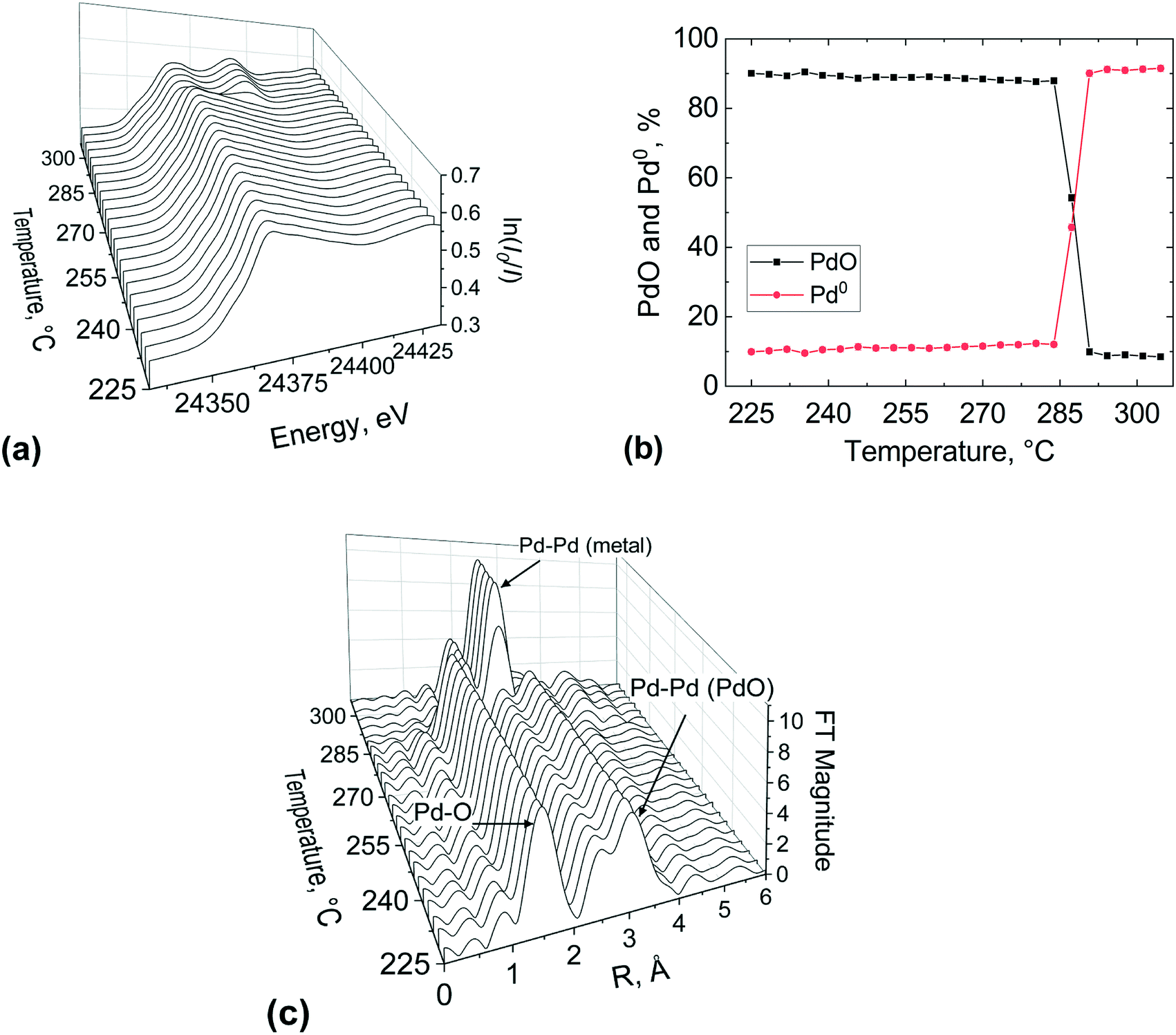 | ||
| Fig. 9 XAS study at the Pd K-edge of the 2 wt% Pd/Al2O3 catalyst at the bed outlet position during the second temperature-resolved reduction with methane: (a) normalised XANES spectra, (b) PdO and Pd0 concentrations obtained by LCF of the spectra and (c) phase shift uncorrected Fourier transform of EXAFS. Gas inlet composition: 1% CH4 and 4% of O2 in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 27.7 mg (53–63 μm). | ||
The isosbestic point is observed at 285 °C, unlike for the inlet bed location (246 °C). However, it is important to note that the occurrence of metallic Pd above 300 °C was the same (ca. 92%) at the inlet and outlet of the catalyst bed, for the first and second calcination respectively. The FT of the EXAFS (Fig. 9(c)) shows the characteristic peak of the Pd–Pd in the first coordination shell of (2.4 Å) emerging at around 290 °C and at 300 °C the Pd phase dominated at the outlet bed position. These results indicate that spatial information along the catalyst bed could be derived employing this novel microreactor, enabling the user to avoid averaging out the information along the catalyst bed, which is typically the case with conventional spectroscopic cells used for XAS measurements.
3.3. Operando DRIFTS/MS studies
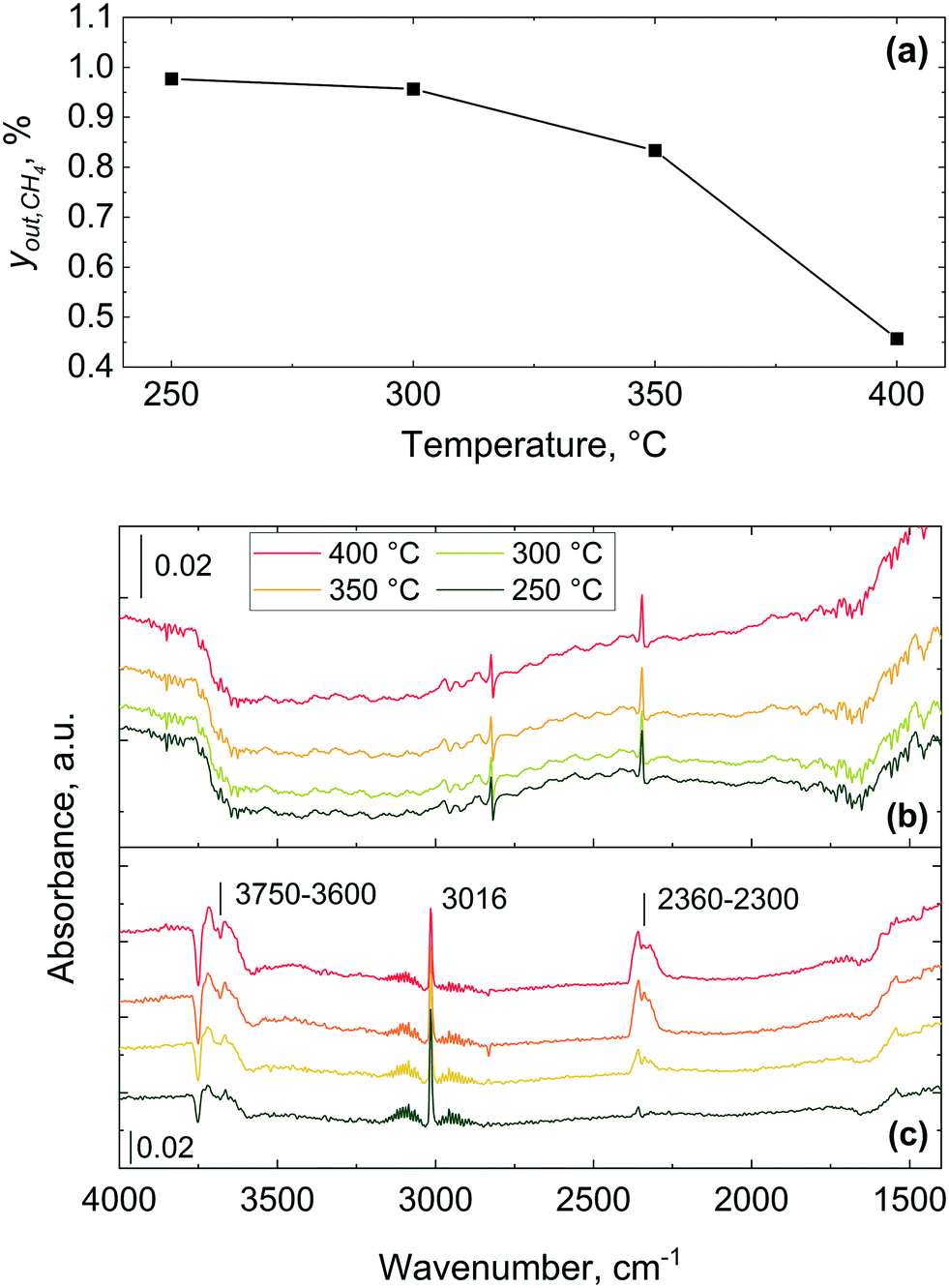 | ||
| Fig. 10 Methane combustion over 2 wt% Pd/Al2O3 catalyst: (a) methane outlet concentration, yout,CH4, (b) infrared spectra recorded using the microreactor and (c) spectra obtained using a commercial Harrick cell. Gas inlet composition: 1% CH4 and 4% of O2 in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 28 mg (53–63 μm). | ||
The gas phase features significantly dominate the spectra collected using the commercial Harrick DRIFTS cell (Fig. 10(c)) and the gas phase fingerprints of methane and carbon dioxide bands appear at 3016 cm−1 and 2300–2360 cm−1, respectively. As also shown in previous IR studies of methane combustion, methane and carbon dioxide are predominantly detected in their gaseous forms.67–69 The absence of strong gas phase bands in the infrared spectra collected with the microreactor is attributed to the lack of dead volume in the microreactor, unlike in the commercial DRIFTS cell. The lack of a gas-filled head space (i.e. dead volume) above the catalyst bed effectively minimises the interference of gas phase bands from the bands arising from the surface adsorbed species, making the microreactor ideal for transient response operando DRIFTS studies.36
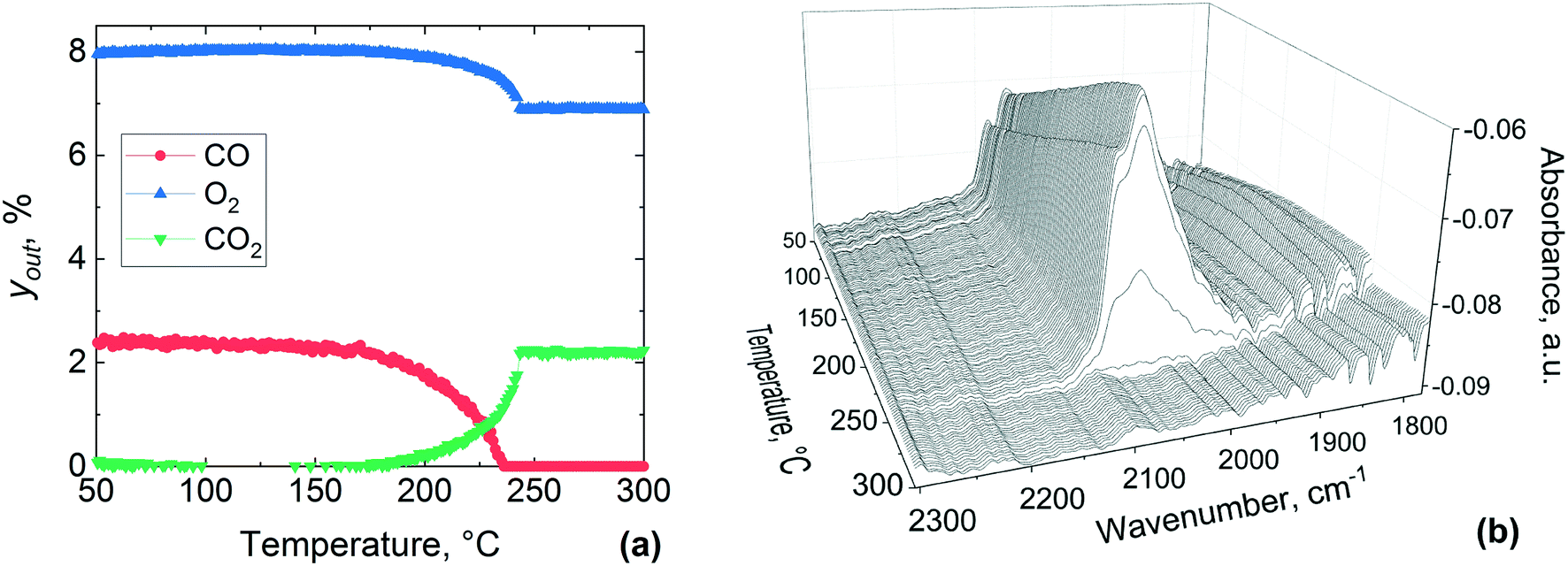 | ||
| Fig. 11 Temperature-resolved CO oxidation over pre-reduced 1 wt% Pt/Al2O3 catalyst: (a) concentrations of the gas phase species, yout, and (b) infrared spectra in the middle of the catalyst bed during the temperature-resolved CO oxidation. Gas inlet composition: 7.5% O2 and 2.5% of CO in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 29.6 mg (38–53 μm). | ||
Bowker elucidated the CO “light-off”, using X-ray photoelectron spectroscopy over Pd catalyst during CO oxidation.73 At low temperature the surface coverage is dominated by adsorbed CO, which poisons the catalyst and prevents oxidation. However, at a certain temperature molecular oxygen starts adsorbing and dissociating into atoms, which react with adsorbed CO to produce CO2. This process frees four free sites for further oxygen adsorption, making the reaction self-accelerating. Therefore, the abrupt disappearance of the CO band can be ascribed to the complete conversion of adsorbed CO, which at high temperature could not be detected by DRIFTS due to its fast reaction with oxygen. Based on these results, CO adsorbed on the catalyst surface hinders the activation of oxygen below 180 °C and above this temperature oxygen activation starts to take place, with CO oxidation light-off observed in the MS results.
Fig. 12 shows reactor outlet concentrations and DRIFTS spectra collected between 50 and 300 °C during CO oxidation over the pre-oxidised catalyst. From Fig. 12(a) it is clear that the gas phase CO2 appears at around 200 °C, which coincides with a drop in the CO and O2 concentration in the gas phase, indicating the occurrence of CO oxidation. At around 250 °C, CO disappears from the gas phase, while O2 and CO2 concentrations stabilise. From these results, it is evident that the gas phase conversion profiles of CO, O2 and CO2 shift to ca. 20 °C higher temperatures over the pre-oxidised catalyst as compared to the pre-reduced one reported in Fig. 11. The DRIFTS data show a band at 2086 cm−1 appearing between 50 and 120 °C, which is different from that on the pre-reduced platinum surface (Fig. 11). Above 120 °C, the band at 2086 cm−1 starts to redshift to 2078 cm−1 and a new band appears to emerge at 2060 cm−1. These two bands at 2078 and 2060 cm−1 become prominent at around 135 °C and grow in intensity up to 260 °C during the temperature-resolved oxidation. Above 260 °C the bands disappear abruptly, similar to that observed for the pre-reduced catalyst (Fig. 11), which coincides with the MS results that show complete conversion of CO at around 250 °C.
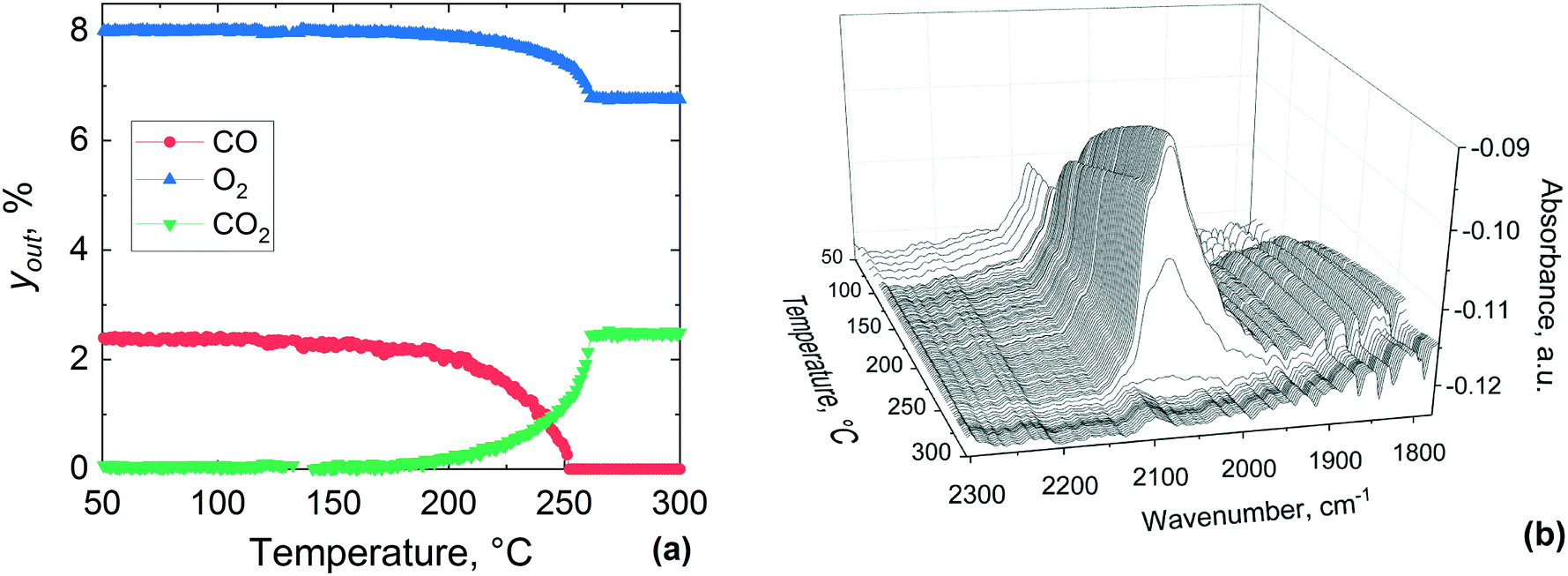 | ||
| Fig. 12 Temperature-resolved CO oxidation over pre-oxidised 1 wt% Pt/Al2O3 catalyst: (a) concentrations of the gas phase species, yout, and (b) infrared spectra in the middle of the catalyst bed during the temperature-resolved CO oxidation. Gas inlet composition: 7.5% O2 and 2.5% of CO in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 29.6 mg (38–53 μm). | ||
The logarithmic values of CO consumption rates on the pre-reduced and pre-oxidised catalysts are plotted against the inverse of temperature in Fig. 13. The apparent activation energy is derived from the CO conversion range between 7 and 30%. It is evident from Fig. 13 that the reaction rates and activation energies are of similar magnitudes for both the pre-reduced and pre-oxidised Pt/Al2O3 catalysts. In particular, the activation energy for the pre-reduced catalyst, determined to be ca. 46.6 kJ mol−1, is slightly lower than that observed for the pre-oxidised catalyst (48.4 kJ mol−1). In general, the activation energies are consistent with earlier studies. Different global or apparent activation energies for CO oxidation over Pt catalysts have been reported previously.74–77 An apparent activation energy of 56 kJ mol−1 was reported for the CO oxidation on a pre-reduced 5 wt% Pt/SiO2,77 while a value of 84 kJ mol−1 was reported for calcined 1–2 wt% Pt/Al2O3 catalysts, which was found to be independent of the platinum metal dispersion.74
 | ||
| Fig. 13 Arrhenius plots of the CO consumption rate for (a) pre-reduced and (b) pre-oxidised 1 wt% Pt/Al2O3 catalyst. | ||
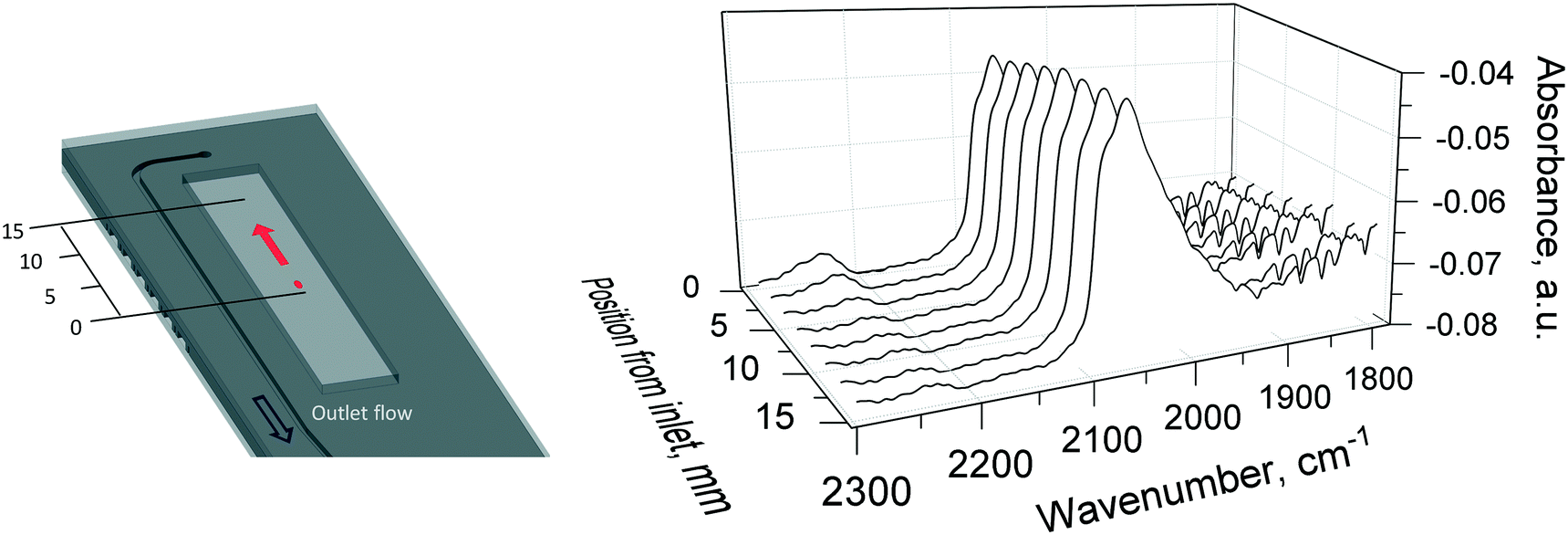 | ||
| Fig. 14 Infrared spectra of CO adsorbed over pre-oxidised 1 wt% Pt/Al2O3 catalyst during CO oxidation at 310 °C. The red dot and arrow on the left indicate the DRIFTS beam and the scanning direction along the catalyst bed. Gas inlet composition: 1.25% O2 and 2.5% of CO in He, flowrate: 40 NmL min−1, atmospheric pressure, catalyst amount: 29.6 mg (38–53 μm). | ||
4. Summary and conclusions
We have developed a novel silicon-glass plug-flow microreactor operating under no external and internal mass transfer resistances, suitable for operando X-ray absorption spectroscopy (XAS), diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and mass spectrometry (MS) studies. The applicability and versatility of the microreactor for operando XAS and DRIFTS studies is demonstrated in the catalytic combustion of methane and carbon monoxide oxidation. The reactor design and materials used resulted in an isothermal temperature profile along the catalyst bed. Operando XAS at the Pd K-edge conducted in transmission mode during methane combustion over Pd/Al2O3 showed that the palladium was in oxidised state at all studied temperatures at the inlet of the catalyst bed. Linear combination fitting showed a percentage of PdO larger than 90% for all the temperatures. The corresponding operando DRIFTS presented very weak features attributable to gas phase methane and CO2 due to the absence of dead volume. Temperature-resolved operando XAS during the reduction of the palladium catalyst with methane showed that the inlet and the outlet regions of the catalyst exhibited a different reduction temperature possibly due to ageing of the catalyst after re-calcination. Operando DRIFTS during the temperature-resolved CO oxidation indicated the presence of linearly adsorbed CO on the pre-reduced and pre-oxidised platinum surface. Spatially-resolved DRIFTS has been performed during the CO oxidation at 310 °C, by probing 9 different axial positions along the catalyst bed. Activation energies have been calculated for both methane combustion over 2 wt% Pd/Al2O3 (80.7 kJ mol−1) and CO oxidation over a reduced (46.6 kJ mol−1) and oxidised (48.4 kJ mol−1) 1 wt% Pt/Al2O3 surface, and were found to be in the same order of magnitude of those reported in previous studies. This novel microreactor with no dead volume facilitates experiments with high space velocities under true kinetic regimes and is particularly suited for transient response experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The UK Catalysis Hub is thanked for resources and support provided via membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/I038748/1, EP/I019693/1, EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1, EP/L003279/1 and EP/M013219/1). We thank the London Centre for Nanotechnology, in particular Steve Etienne for his support in the double-sided photolithography and deep reactive ion etching, and the Diamond Light Source for provision of beam time and support facilities at the beamline B18 (Experiment: SP19359-1).References
- J. M. Thomas, C. R. A. Catlow and G. Sankar, Chem. Commun., 2002, 2921–2925 RSC
.
- M. A. Newton and W. van Beek, Chem. Soc. Rev., 2010, 39, 4845–4863 RSC
- M. A. Bañares, Catal. Today, 2005, 100, 71–77 CrossRef
- B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4557–4559 RSC
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850 CrossRef CAS
- G. Sankar and J. M. Thomas, Top. Catal., 1999, 8, 1–21 CrossRef CAS
- G. Sankar, J. M. Thomas and C. R. A. Catlow, Top. Catal., 2000, 10, 255–264 CrossRef CAS
- J. M. Thomas and G. Sankar, J. Synchrotron Radiat., 2001, 8, 55–60 CrossRef CAS
- J. M. Thomas and G. Sankar, Acc. Chem. Res., 2001, 34, 571–581 CrossRef CAS
- N.-Y. Topsøe, Catal. Today, 2006, 113, 58–64 CrossRef
- J. Ryczkowski, Catal. Today, 2001, 68, 263–381 CrossRef CAS
- T. Armaroli, T. Bécue and S. Gautier, Oil Gas Sci. Technol., 2004, 59, 215–237 CrossRef CAS
- C. M. A. Parlett, C. V. Gaskell, J. N. Naughton, M. A. Newton, K. Wilson and A. F. Lee, Catal. Today, 2013, 205, 76–85 CrossRef CAS
- Y. Zhou, D. E. Doronkin, M. Chen, S. Wei and J.-D. Grunwaldt, ACS Catal., 2016, 6, 7799–7809 CrossRef CAS
- A. M. Abdel-Mageed, G. Kučerová, J. Bansmann and R. J. Behm, ACS Catal., 2017, 7, 6471–6484 CrossRef CAS
- E. K. Gibson, E. M. Crabb, D. Gianolio, A. E. Russell, D. Thompsett and P. P. Wells, Catal., Struct. React., 2017, 3, 5–12 CrossRef CAS
- E. K. Dann, E. K. Gibson, R. A. Catlow, P. Collier, T. Eralp Erden, D. Gianolio, C. Hardacre, A. Kroner, A. Raj, A. Goguet and P. P. Wells, Chem. Mater., 2017, 29, 7515–7523 CrossRef CAS
- D. Ferri, M. S. Kumar, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC
- A. Chakrabarti, M. E. Ford, D. Gregory, R. Hu, C. J. Keturakis, S. Lwin, Y. Tang, Z. Yang, M. Zhu, M. A. Bañares and I. E. Wachs, Catal. Today, 2017, 283, 27–53 CrossRef CAS
- A. M. Beale, A. M. J. van der Eerden, K. Kervinen, M. A. Newton and B. M. Weckhuysen, Chem. Commun., 2005, 3015–3017 RSC
- Y. Li, D. Zakharov, S. Zhao, R. Tappero, U. Jung, A. Elsen, P. Baumann, R. G. Nuzzo, E. A. Stach and A. I. Frenkel, Nat. Commun., 2015, 6, 7583 CrossRef CAS
- B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4351–4360 RSC
- H. Topsøe, J. Catal., 2003, 216, 155–164 CrossRef
- N. Al-Rifai, E. Cao, V. Dua and A. Gavriilidis, Curr. Opin. Chem. Eng., 2013, 2, 338–345 CrossRef
- F. C. Meunier, Chem. Soc. Rev., 2010, 39, 4602–4614 RSC
- J. D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037–3047 RSC
- S. K. Matam, O. Korsak, L. Bocher, D. Logvinovich, P. Hug, A. Weidenkaff and D. Ferri, Top. Catal., 2011, 54, 1213 CrossRef CAS
- C. Lamberti, C. Prestipino, S. Bordiga, G. Berlier, G. Spoto, A. Zecchina, A. Laloni, F. La Manna, F. D'Anca, R. Felici, F. D'Acapito and P. Roy, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 200, 196–201 CrossRef CAS
- N. Weiher, E. Bus, B. Gorzolnik, M. Moller, R. Prins and J. A. van Bokhoven, J. Synchrotron Radiat., 2005, 12, 675–679 CrossRef CAS
- A. Longo, A. Balerna, F. d'Acapito, F. D'Anca, F. Giannici, L. F. Liotta, G. Pantaleo and A. Martorana, J. Synchrotron Radiat., 2005, 12, 499–505 CrossRef CAS
- S. B. Rasmussen, R. López-Medina, R. Portela, E. Mikolajska, M. Daturi, P. Ávila and M. A. Bañares, Catal. Sci. Technol., 2015, 5, 4942–4945 RSC
- N. S. Marinkovic, Q. Wang and A. I. Frenkel, J. Synchrotron Radiat., 2011, 18, 447–455 CrossRef CAS
- K. A. Beyer, H. Zhao, O. J. Borkiewicz, M. A. Newton, P. J. Chupas and K. W. Chapman, J. Appl. Crystallogr., 2014, 47, 95–101 CrossRef CAS
- S. Yao, K. Mudiyanselage, W. Xu, A. C. Johnston-Peck, J. C. Hanson, T. Wu, D. Stacchiola, J. A. Rodriguez, H. Zhao, K. A. Beyer, K. W. Chapman, P. J. Chupas, A. Martínez-Arias, R. Si, T. B. Bolin, W. Liu and S. D. Senanayake, ACS Catal., 2014, 4, 1650–1661 CrossRef CAS
- G. Agostini, D. Meira, M. Monte, H. Vitoux, A. Iglesias-Juez, M. Fernandez-Garcia, O. Mathon, F. Meunier, G. Berruyer, F. Perrin, S. Pasternak, T. Mairs, S. Pascarelli and B. Gorges, J. Synchrotron Radiat., 2018, 25, 1745–1752 CrossRef CAS
- G. L. Chiarello, M. Nachtegaal, V. Marchionni, L. Quaroni and D. Ferri, Rev. Sci. Instrum., 2014, 85, 074102 CrossRef
- E. K. Dann, E. K. Gibson, C. R. A. Catlow, V. Celorrio, P. Collier, T. Eralp, M. Amboage, C. Hardacre, C. Stere, A. Kroner, A. Raj, S. Rogers, A. Goguet and P. P. Wells, J. Catal., 2019, 373, 201–208 CrossRef CAS
- M. A. Newton, Top. Catal., 2009, 52, 1410–1424 CrossRef CAS
- J. Yue, J. C. Schouten and T. A. Nijhuis, Ind. Eng. Chem. Res., 2012, 51, 14583–14609 CrossRef CAS
- E. Cao, S. Firth, P. F. McMillan and A. Gavriilidis, Catal. Today, 2007, 126, 119–126 CrossRef CAS
- E. Cao, M. Sankar, S. Firth, K. F. Lam, D. Bethell, D. K. Knight, G. J. Hutchings, P. F. McMillan and A. Gavriilidis, Chem. Eng. J., 2011, 167, 734–743 CrossRef CAS
- G. Sankar, E. Cao and A. Gavriilidis, Catal. Today, 2007, 125, 24–28 CrossRef CAS
- B. A. Rizkin, F. G. Popovic and R. L. Hartman, J. Vac. Sci. Technol., 2019, 37, 050801 CrossRef
- S. Zhao, Y. Li, E. Stavitski, R. Tappero, S. Crowley, M. J. Castaldi, D. N. Zakharov, R. G. Nuzzo, A. I. Frenkel and E. A. Stach, ChemCatChem, 2015, 7, 3683–3691 CrossRef CAS
- S. J. A. Figueroa, D. Gibson, T. Mairs, S. Pasternak, M. A. Newton, M. Di Michiel, J. Andrieux, K. C. Christoforidis, A. Iglesias-Juez, M. Fernandez-Garcia and C. Prestipino, J. Appl. Crystallogr., 2013, 46, 1523–1527 CrossRef CAS
- D. E. Doronkin, S. Baier, T. Sheppard, F. Benzi and J. D. Grunwaldt, J. Phys.: Conf. Ser., 2016, 712, 012030 CrossRef
- A. Urakawa, N. Maeda and A. Baiker, Angew. Chem., Int. Ed., 2008, 47, 9256–9259 CrossRef CAS
- A. Urakawa, F. Trachsel, P. R. von Rohr and A. Baiker, Analyst, 2008, 133, 1352–1354 RSC
- P. Beato, R. Kraehnert, S. Engelschalt, T. Frank and R. Schlögl, Chem. Eng. J., 2008, 135, S247–S253 CrossRef CAS
- C. K. C. Tan, W. N. Delgass and C. D. Baertsch, Appl. Catal., B, 2009, 93, 66–74 CrossRef CAS
- C. Daniel, M. O. Clarté, S. P. Teh, O. Thinon, H. Provendier, A. C. Van Veen, B. J. Beccard, Y. Schuurman and C. Mirodatos, J. Catal., 2010, 272, 55–64 CrossRef CAS
- I. P. Silverwood, N. Al-Rifai, E. Cao, D. J. Nelson, A. Chutia, P. P. Wells, S. P. Nolan, M. D. Frogley, G. Cinque, A. Gavriilidis and C. R. A. Catlow, Rev. Sci. Instrum., 2016, 87, 024101 CrossRef CAS
- E. Gross, X.-Z. Shu, S. Alayoglu, H. A. Bechtel, M. C. Martin, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 3624–3629 CrossRef CAS
- J.-D. Grunwaldt and B. S. Clausen, Top. Catal., 2002, 18, 37–43 CrossRef CAS
- S. Baier, A. Rochet, G. Hofmann, M. Kraut and J.-D. Grunwaldt, Rev. Sci. Instrum., 2015, 86, 065101 CrossRef CAS
- J.-D. Grunwaldt, B. Kimmerle, S. Hannemann, A. Baiker, P. Boye and C. G. Schroer, J. Mater. Chem., 2007, 17, 2603–2606 RSC
- N. Richards, J. H. Carter, E. Nowicka, L. A. Parker, S. Pattisson, Q. He, N. F. Dummer, S. Golunski and G. J. Hutchings, Appl. Catal., B, 2020, 264, 118501 CrossRef
- A. J. Dent, G. Cibin, S. Ramos, S. A. Parry, D. Gianolio, A. D. Smith, S. M. Scott, L. Varandas, S. Patel, M. R. Pearson, L. Hudson, N. A. Krumpa, A. S. Marsch and P. E. Robbins, J. Phys.: Conf. Ser., 2013, 430, 012023 CrossRef
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS
- R. Burch, F. J. Urbano and P. K. Loader, Appl. Catal., A, 1995, 123, 173–184 CrossRef CAS
- P. Briot and M. Primet, Appl. Catal., 1991, 68, 301–314 CrossRef CAS
- R. Burch and P. K. Loader, Appl. Catal., B, 1994, 5, 149–164 CrossRef CAS
- N. Mouaddib, C. Feumi-Jantou, E. Garbowski and M. Primet, Appl. Catal., A, 1992, 87, 129–144 CrossRef CAS
- S. K. Matam, G. L. Chiarello, Y. Lu, A. Weidenkaff and D. Ferri, Top. Catal., 2013, 56, 239–242 CrossRef CAS
- J. C. van Giezen, F. R. van den Berg, J. L. Kleinen, A. J. van Dillen and J. W. Geus, Catal. Today, 1999, 47, 287–293 CrossRef CAS
- F. H. Ribeiro, M. Chow and R. A. Dallabetta, J. Catal., 1994, 146, 537–544 CrossRef CAS
- W. R. Schwartz, D. Ciuparu and L. D. Pfefferle, J. Phys. Chem., 2012, 116, 8587–8593 CAS
- M. Schmal, M. M. V. M. Souza, V. V. Alegre, M. A. P. da Silva, D. V. César and C. A. C. Perez, Catal. Today, 2006, 118, 392–401 CrossRef CAS
- H. Na, Z. Liu and T. Zhu, React. Kinet., Mech. Catal., 2014, 111, 137–148 CrossRef CAS
- O. Demoulin, M. Navez and P. Ruiz, Appl. Catal., A, 2005, 295, 59–70 CrossRef CAS
- C. Brieger, J. Melke, N. van der Bosch, U. Reinholz, H. Riesemeier, A. Guilherme Buzanich, M. K. Kayarkatte, I. Derr, A. Schökel and C. Roth, J. Catal., 2016, 339, 57–67 CrossRef CAS
- S. K. Matam, E. V. Kondratenko, M. H. Aguirre, P. Hug, D. Rentsch, A. Winkler, A. Weidenkaff and D. Ferri, Appl. Catal., B, 2013, 129, 214–224 CrossRef CAS
- M. Bowker, Chem. Soc. Rev., 2008, 37, 2204–2211 RSC
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS
- R. H. Nibbelke, M. A. J. Campman, J. H. B. J. Hoebink and G. B. Marin, J. Catal., 1997, 171, 358–373 CrossRef CAS
-
M. Campman, PhD thesis, University of Eindhoven, 1996 Search PubMed
- N. W. Cant, P. C. Hicks and B. S. Lennon, J. Catal., 1978, 54, 372–383 CrossRef CAS
- C. Daniel, M.-O. Clarté, H. Provendier, A. C. Van Veen, Y. Schuurman, B. J. Beccard and C. Mirodatos, C. R. Chim., 2009, 12, 647–653 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01608j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |