
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIndium phosphasalen catalysts showing high isoselectivity and activity in racemic lactide and lactone ring opening polymerizations†
Nattawut
Yuntawattana
 a,
Thomas M.
McGuire
b,
Christopher B.
Durr
a,
Antoine
Buchard
*b and
Charlotte K.
Williams
*a
a,
Thomas M.
McGuire
b,
Christopher B.
Durr
a,
Antoine
Buchard
*b and
Charlotte K.
Williams
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
First published on 18th September 2020
Abstract
Stereoblock polylactide (PLA) shows higher melting temperatures and better mechanical properties than other PLA stereoisomers. More stereoselective and active catalysts are needed to polymerize racemic-lactide (LA) and produce stereoblock PLA. This work describes a series of phosphasalen indium catalysts (1–5) which result in very high isoselectivity, at room temperature, (Pi = 0.91, 25 °C) and high activity, at low catalyst loading (TOF = 100 h−1, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 catalyst:LA, [LA] = 1 M, THF, 25 °C). The catalyst structure–activity and structure–stereoselectivity relationships are investigated using various experimental methods and DFT calculations. The most isoselective catalyst features two different phosphasalen substituents, a tert-butyl and phenyl group, it forms an achiral, meso indium complex which operates by a chain end control mechanism. The work highlights the benefits of phosphasalen ligands and identifies new avenues for catalyst investigation by exploitation of asymmetrically substituted phosphorus atoms. The catalysts also show good activity and control for the ring-opening polymerizations of ε-caprolactone, β-butyrolactone, ε-decalactone and δ-hexalactone (γ-methyl-δ-valerolactone), demonstrating future potential for copolyester production.
:500 catalyst:LA, [LA] = 1 M, THF, 25 °C). The catalyst structure–activity and structure–stereoselectivity relationships are investigated using various experimental methods and DFT calculations. The most isoselective catalyst features two different phosphasalen substituents, a tert-butyl and phenyl group, it forms an achiral, meso indium complex which operates by a chain end control mechanism. The work highlights the benefits of phosphasalen ligands and identifies new avenues for catalyst investigation by exploitation of asymmetrically substituted phosphorus atoms. The catalysts also show good activity and control for the ring-opening polymerizations of ε-caprolactone, β-butyrolactone, ε-decalactone and δ-hexalactone (γ-methyl-δ-valerolactone), demonstrating future potential for copolyester production.
Introduction
Polymers are manufactured at >350 Mt/annum globally; almost all of these materials are currently sourced from virgin petrochemicals and designed without consideration for end-life options.1 There is an urgent need to improve sustainability throughout the polymer lifecycle, for example replacing petrochemicals with monomers derived from renewable resources or by designing polymer structures to facilitate recycling and enable eventual (bio)degradation.1 Polylactide (PLA) is one of the leading commercial, sustainable polymers: it is sourced from plants such as sugar-cane, shows similar properties to polystyrene and, because it is an aliphatic polyester, is susceptible to catalyzed hydrolyses or alcoholyses facilitating closed-loop recycling and, under appropriate conditions, enabling complete biodegradation. PLA is currently produced on a scale of ∼1–2 Mt/annum, but growing environmental concerns associated with pervasive plastic wastes and its significantly reduced greenhouse gas emissions, compared to alternative petrochemicals like polyethylene terephthalate or polystyrene, continue to drive growth. To expand the range of PLA applications both to its properties and production processes would benefit from improvements. Control over PLA tacticity is an attractive means to enhance its physical–chemical properties, for example stereoblock PLA, which comprises blocks of each enantiomer (PLLA-b-PDLA/P(S,S-LA)-b-P(R,R-LA)), shows a higher melting temperature (Tm = 170–200 °C) than isotactic PLLA (Tm = 130–160 °C).2 Blending stereoblock PLA into isotactic PLLA also increases crystallite nucleation rates and the resulting materials have a higher Young's modulus and tensile strength than PLLA.2e Blending it into mixtures of PLLA/PDLA accelerates stereocomplex crystallization and increases the plastic's temperature stability.3Stereoblock PLA can be prepared in one-step by the isoselective ring-opening polymerization (ROP) of racemic lactide (LA). The process stereoselectivity, rate and control are highly dependent upon the catalyst and by its mode of operation.4 Generally, there are two stereocontrol mechanisms: (1) enantiomorphic site control occurs when chiral catalysts react faster with one lactide enantiomer or (2) chain end control occurs using achiral catalysts and with stereocontrol being induced by interactions between the catalyst and the growing polymer chain end group.5 Currently, the most isoselective catalysts operate by enantiomorphic site control mechanisms and the stand-out exemplars are chiral salen aluminium catalysts (Al-salen).6 After optimization, chiral Al-salen catalysts may show probability of isotactic diad formation, Pi, values exceeding 0.95 (where Pi = 1 corresponds to a fully isotactic, stereoblock polymer).7 These high performance arise from very low rates of transesterification, and, hence, limited scrambling of stereo-sequences, but it comes with a significant down-side since rates of forward polymerization, which are also a form of intermolecular transesterification, are also slow.4a,5–8 Because of their low rates, most Al-salen complexes are applied at unacceptably high catalyst loadings (∼1 mol%) and show turn-over-frequencies (TOF) from 0.1 to 1 h−1 at 70 °C (1 mol% catalyst loading, [LA] = 1 M in toluene).4a,5–8 Enantiomorphic site control mechanisms inherently result in even slower polymerization of the ‘wrong’ enantiomer which further limits their application in stereoblock PLA production. Pioneering work by Nomura and co-workers established that achiral Al-salen catalysts also showed outstanding isoselectivity and operate by chain end control mechanisms (Pi = 0.92–0.98, TOF = 7 h−1, Al:LA 1:100, 70 °C).7,8 Although the stereoselectivity is impressive unfortunately activity values remain very low (TOF = 7 h−1).7
Catalysts combining high isoselectivity and rate are feasible using other elements. One difficulty for the field is a lack of mechanistic insight to underpin future catalyst design. It is also challenging to compare different catalysts, as conditions applied are variable and not all authors report common kinetic measures (rate laws). In this work, catalysts are only highlighted that show Pi > 0.8 at room temperature (20–30 °C). In fact to produce stereoblock PLA showing useful properties such as increased melting temperature, the PLA molar mass values should be high (Mn > 50–100 kg mol−1) and Pi values should exceed 0.9: very few catalysts meet both criteria. It is also essential that high stereocontrol is achieved under viable loading and, so, catalysts should operate successfully at low loading, be able to access high molar mass PLA and at accessible temperatures without requiring undue cooling. In order to compare catalyst activity data and to bench-mark it against the most stereoselective Al-salen catalysts, turn-over-frequencies (determined from conversion-time point kinetic data) are compared under conditions close to 1 mol% catalyst loading, [LA] = 1 M, T = 20–30 °C. It is worth emphasis that the three highest performing catalyst types, and those described herein, are all capable of operating under considerably lower, and more practically applicable, catalyst loadings than 1 mol% but these values are applied to allow for meaningful comparisons.9 At the upper end of these performance metrics are three different catalysts: 1) the benzimidazolyl aminophenolate zinc(II) alkoxide complexes (reported by Ma and co-workers, A);9a,d,10 2) the bis(phenolate)ether yttrium(III) alkoxide complexes (reported by Lu and co-workers, B);9b and 3) the phosphasalen indium(III) alkoxide complexes (reported by our group, C) (Fig. 1).9c,11 Ma and co-workers have also pioneered a series of high-performance chiral oxazolinyl aminophenolate zinc complexes.9d,10 The authors, however, point out that achiral ligands are more desirable and in 2019, an outstanding achiral benzimidazolyl zinc complex (A) was reported which shows Pi = 0.88 and TOF = 840 h−1 (cat.:LA, 1:200, [LA] = 1 M, 25 °C).9a The second class of high-performance catalysts, reported by Lu and co-workers, is represented by the bis(phenolate)ether yttrium alkoxide (B) which shows Pi = 0.84 and TOF = 2280 h−1 (cat.:LA, 1:200, [LA] = 1 M, 25 °C).9b The third class (phosphasalens) are represented by an indium(III) phosphasalen alkoxide complex (C), reported by our group, which shows Pi = 0.87 and TOF = 480 h−1 (cat.:LA 1:500, [LA] = 1 M, 25 °C).9c Other good catalysts, in some cases also showing excellent stereoselectivity, comprise complexes of Zn(II), Ca(II) or Fe(III) (Scheme 2).12 Several organocatalysts are also highly isoselective but their activities and the need for very high loadings mean they cannot compete against the best metal based catalysts (Scheme 2).13
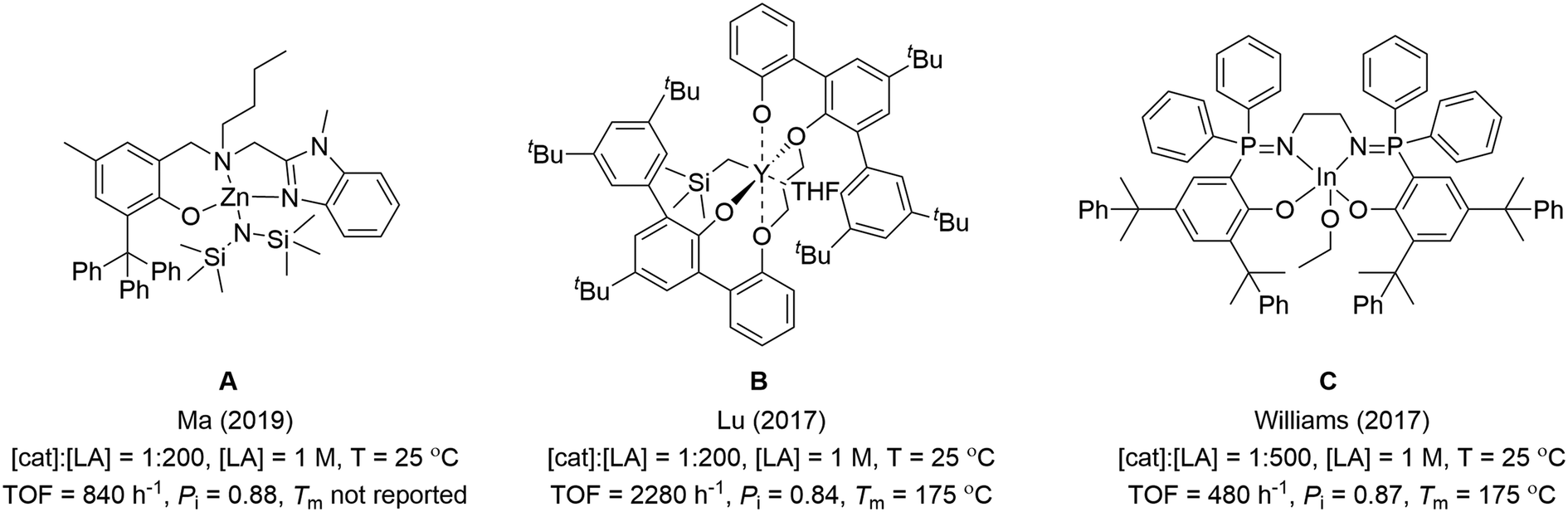 | ||
| Fig. 1 Previously reported metal catalysts (A–C) with high activity and isoselectivity.9a–c | ||
Our group has reported stereoselective phosphasalen metal catalysts, featuring yttrium(III), lutetium (III) and indium(III) (Fig. 1).9c,11,14 The phosphasalen ligand is important in conferring high rates and stereocontrol and generally significantly out-performs when compared with analogous salen ligands.9c,11,14,15 Phosphasalen ligands are both σ and π-donors and, so, are considerably more electron donating than salens. It is proposed that the enhanced electron donation increases the lability of the metal alkoxide propagating bond and accelerates monomer insertion reactions.15 In addition to increasing polymerization rates, phosphasalen ligands feature two phosphorus atoms and their substituents may enhance steric shielding and direct lactone coordination at the active site thereby enabling monomer stereoselectivity.9c,11,14,15d,e,16 The first isoselective catalyst of this class was a pentadentate phosphasalen yttrium complex, showing Pi = 0.71 and TOF = 531 h−1 (cat.:LA 1:500, [LA] = 1 M, 25 °C).11a Comparing a series of pentadentate phosphasalen lanthanide complexes revealed that the lutetium complex showed the highest isoselectivity with Pi = 0.82 and TOF = 52 h−1 (cat.:LA 1:500, [LA] = 1 M, 25 °C).11b It was also feasible to isolate a scandium-lactate complex which allowed for isolation of a possible catalytic intermediate.14 A tetradentate phosphasalen indium catalyst, C, showed even higher isoselectivity Pi = 0.87 and better rates TOF = 480 h−1 (cat.:LA 1:500, [LA] = 1 M, 25 °C).9c Notably, the same tetradentate ligand coordinated to yttrium resulted in a highly heteroselective catalyst, a finding whose explanation is not obvious and which underscores the need for detailed insight into structure–stereoselectivity mechanisms.15d,e Both isoselective yttrium and indium phosphasalen catalysts operate by chain end control mechanisms and the continued development of this type of catalyst is desirable since it obviates the expensive chiral ligands necessary in enantiomorphic site control mechanisms and allows for high rates throughout polymerization reactions. In this work, new phosphasalen indium catalysts are investigated, building upon the promising performance of catalyst C, and the influences of the amine linker group chemistry and of asymmetrically substituted phosphorus atoms on the catalysis are investigated.
Results and discussion
Catalyst synthesis
Previous research into phosphasalen yttrium catalysts utilized triamine and diamine donors, joined through various C2 or C3 linker groups.11 Initial scoping experiments using longer alkylene linkers, e.g. C4, resulted in difficulties isolating complexes likely due to formation of both coordination complexes and polymers. Henceforth, the linker group chemistry is constrained to ethylene and propylene diamines. Previously phosphasalen metal catalysts always applied phosphorus atoms substituted with two phenyl substituents and, furthermore, the fluxionality of these substituents appeared to be correlated with the high isoselectivity but exactly how these substituents influence tacticity control is unclear.11 To better understand their role, here asymmetric phosphorus substitution, i.e. where one substituent is phenyl and the second is another sterically hindered group, tert-butyl, is explored. To address both the influences of linker group chemistry and symmetric vs. asymmetric phosphorus substituents, five indium complexes (1–5) were prepared (Scheme 1).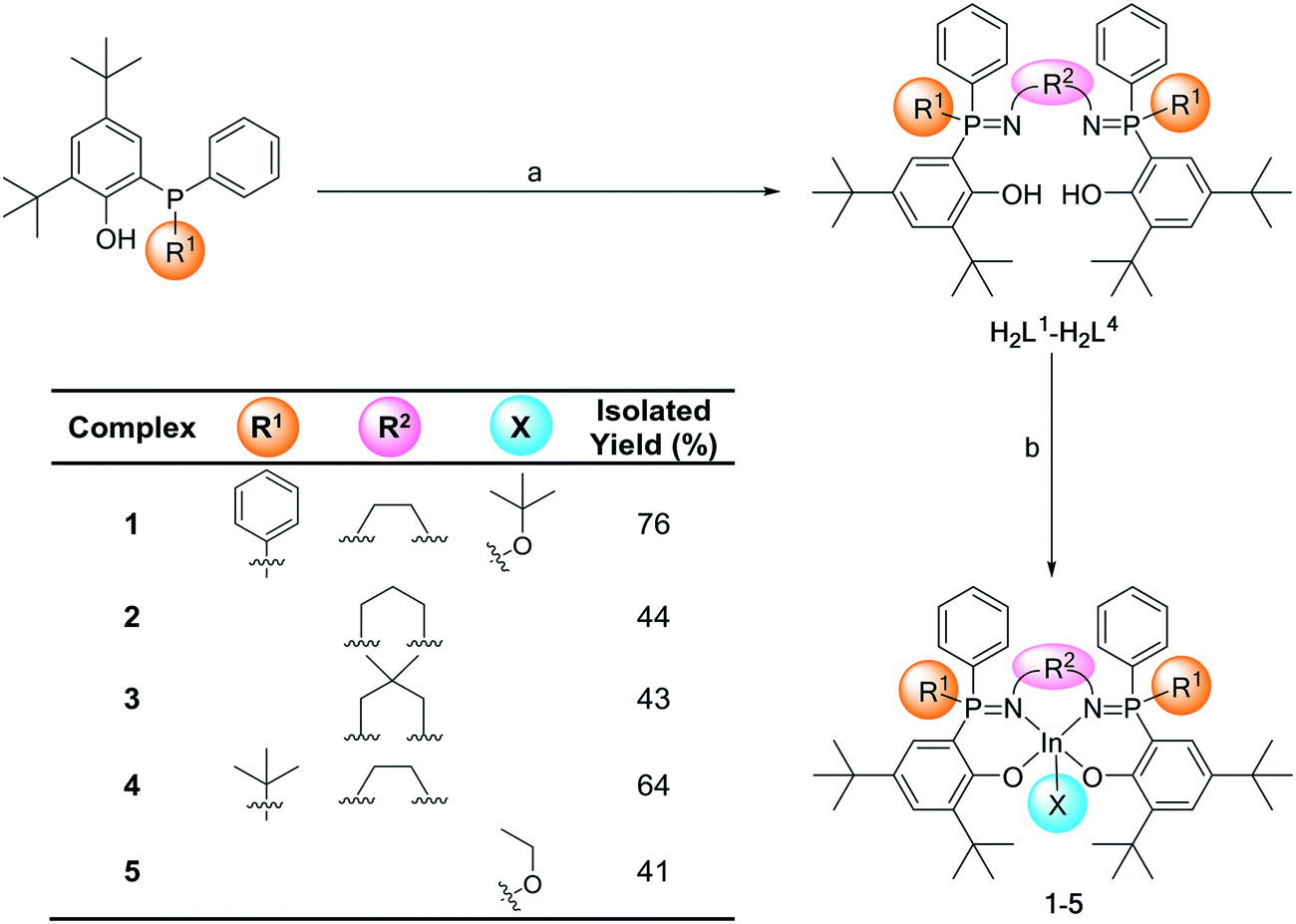 | ||
| Scheme 1 Illustrates the syntheses of indium complexes 1–5. Reagents and conditions: (a): i) Br2 (1 equiv.), DCM, −78 °C, 2 h; ii) nBu3N (0.5 equiv.), iii) diamine backbone (0.5 equiv.) DCM, −78 °C, 18 h and (b): iv) KN(SiMe3)2 (2 equiv.), THF, 25 °C, 2 h; v) InCl3 (1 equiv.), THF, 25 °C, 2 h; vi) KOtBu or KOEt (1 equiv.), THF, 25 °C, 16 h. | ||
To make the target complexes, a series of pro-ligands were prepared (H2L1–H2L4) from 2,4-di-tert-butyl-6-(diphenyl(phosphaenyl))phenol or 2,4-di-tert-butyl-6-(tert-butyl(phenyl)phosphaenyl)phenol (see the ESI† for the experimental details). The syntheses apply a modified Kirsanov reaction (bromination of the phosphine, followed by reaction with the diamine to form the iminophosphorane groups) and allow for isolation of the pure pro-ligands, after column chromatograph, in 10–22% yields. Ligands H2L1–H2L3 feature both phenyl substituents on the phosphorus atoms and differ in terms of the diamine linker groups (ethylene, propylene and 2,2-dimethyl propylene, respectively). Ligand H2L4 features two different substituents on the phosphorus atoms (phenyl and tert-butyl) and the highest performing ethylene diamine linker. Crystals of H2L4 were isolated by layering hexane onto a chloroform solution of the ligand and the molecular structure was determined by single crystal X-ray diffraction. The structure shows the ligand as a single diastereoisomeric pair (with R,S and S,R stereochemistry at each phosphorus, Fig. S45 and Tables S1 and S2†). As the crystals were isolated from a sample which had been purified by column chromatography and the isolated yields were low, it was important to establish whether the other set of diastereoisomers (R,R and S,S) were also formed. The crude reaction product (i.e. prior to any purification by column chromatography) showed a 1H NMR spectrum with identical signals to the purified product (after column chromatography) and identical signals to the isolated crystals (Fig. S18 and S19†). This data is consistent with the selective formation of only the R,S/S,R ligand diastereoisomeric pair.
The indium alkoxide catalysts (1–5) were isolated, after three sequential reactions, in 41–64% yield. The complexes were prepared from the appropriate pro-ligand by a deprotonation reaction, using KN(SiMe3)2 as the base, and the di-potassium complex was reacted in situ with InCl3. The indium chloride complex was reacted, again in situ, with an equivalent of KOtBu or KOEt to form the desired indium alkoxide catalyst.11a The series of complexation reactions were analyzed at each stage using 31P{1H} NMR spectroscopy and these analyses show that the overall conversions are very high, typically each reaction is >95% within the detection limits of NMR spectroscopy (Fig. 2). This high conversion means that the moderate isolated yields could likely be improved in the future through optimization of the salt washing and complex isolation procedures.
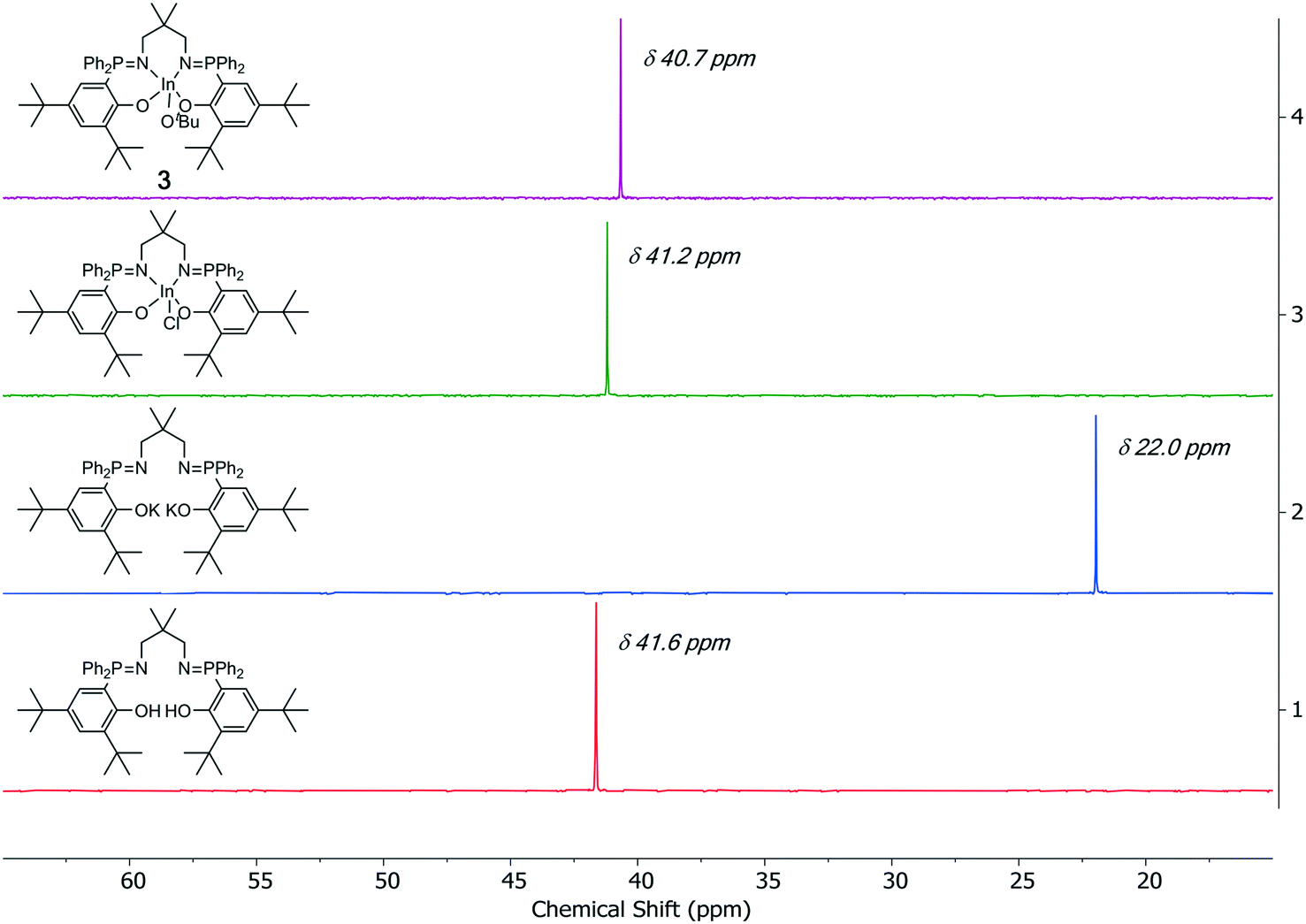 | ||
| Fig. 2 Selected regions of the 31P{1H} NMR spectra of the ligands, intermediates and complex 3. | ||
Solid state structures
The complexes' solid state structures were determined using single crystal X-ray diffraction techniques (Fig. S46–S49 and Tables S3–S9†). Complexes 2–5 are mononuclear complexes with indium atoms showing distorted square pyramidal geometries where the alkoxide ligand (initiating group) occupies the axial position (Fig. 3). The extent of distortion from the square pyramidal geometry is assessed by τ5 values of 0.36 for 2 and 0.33 for 3, both are closely comparable to complex 1 (τ5 = 0.33).9c Complexes 4 and 5 show much less distorted structures, with τ5 values of 0.06 (4) and 0.01 (5) (Fig. 3 and S49 and Tables S5 and S6†). Both 4 and 5 are meso isomers, with the ligands showing R,S stereochemistry at each phosphorus atom. In both structures, the two phosphorus tert-butyl substituents are both orientated in the opposite direction to the alkoxide co-ligand, i.e. they are mutually cis oriented but are trans to the alkoxide. It is important to emphasise that several different crystals were examined, from different synthetic batches, and only the R,S meso isomer was observed (Tables S5–S9†). The mutually cis-arrangement of the phosphorous substituents may be an important contributing factor in the improved stereoselectivity of catalysts 4 and 5 (vide infra).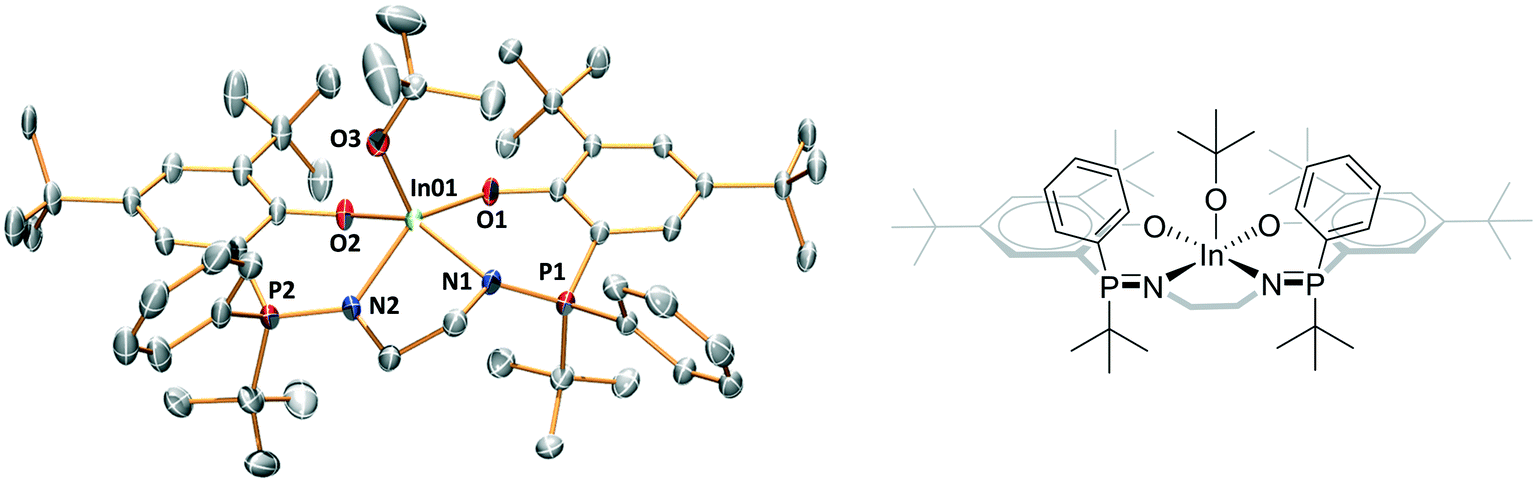 | ||
| Fig. 3 Representation of the molecular structure of complex 4. | ||
Characterization in solution
In THF solution, all the complexes (2–5) show a single resonance in the 31P{1H} NMR spectrum which is indicative of identical phosphorus environments on the timescale of the experiments (Fig. S28, S31, S35 and S38†). The 1H NMR spectra also show resonances and integrals consistent with symmetrical complex geometries. All spectra confirm the selective formation of indium alkoxide complexes and show new resonances at 1.00 ppm (OC(CH3)3) for 2–4, and at 3.71 (OCH2) and 0.82 (OCH2CH3) ppm for 5 (Fig. S26, S29, S32 and S36†). All complexes show a single diffusion coefficient in the DOSY NMR spectra, with diffusion rates correlating with mononuclear structures (Fig. S41–S44†).To gain further understanding of the catalyst solution structures, 1H rotating frame nuclear Overhauser effect spectroscopy (ROESY) was conducted using complex 4, in THF-d8 (Fig. 4). THF was chosen for the measurements because of it is expected to show similar Lewis basicity to the monomer lactide and hence improves understanding of speciation of the complex under conditions relevant to those used in catalysis. The ROESY NMR spectrum shows a correlation peak between the two phosphorus phenyl substituents and there is a correlation between the phosphorus phenyl and tert-butoxide groups. There is no correlation between the phosphorus tert-butyl substituents and the tert-butoxide groups but there is a correlation between the two phosphorus tert-butyl groups. Overall, the data is fully consistent with the solid state structure being retained in solution, i.e. the R,S meso isomer is present where the phenyl ligands are orientated cis to the alkoxide co-ligand.
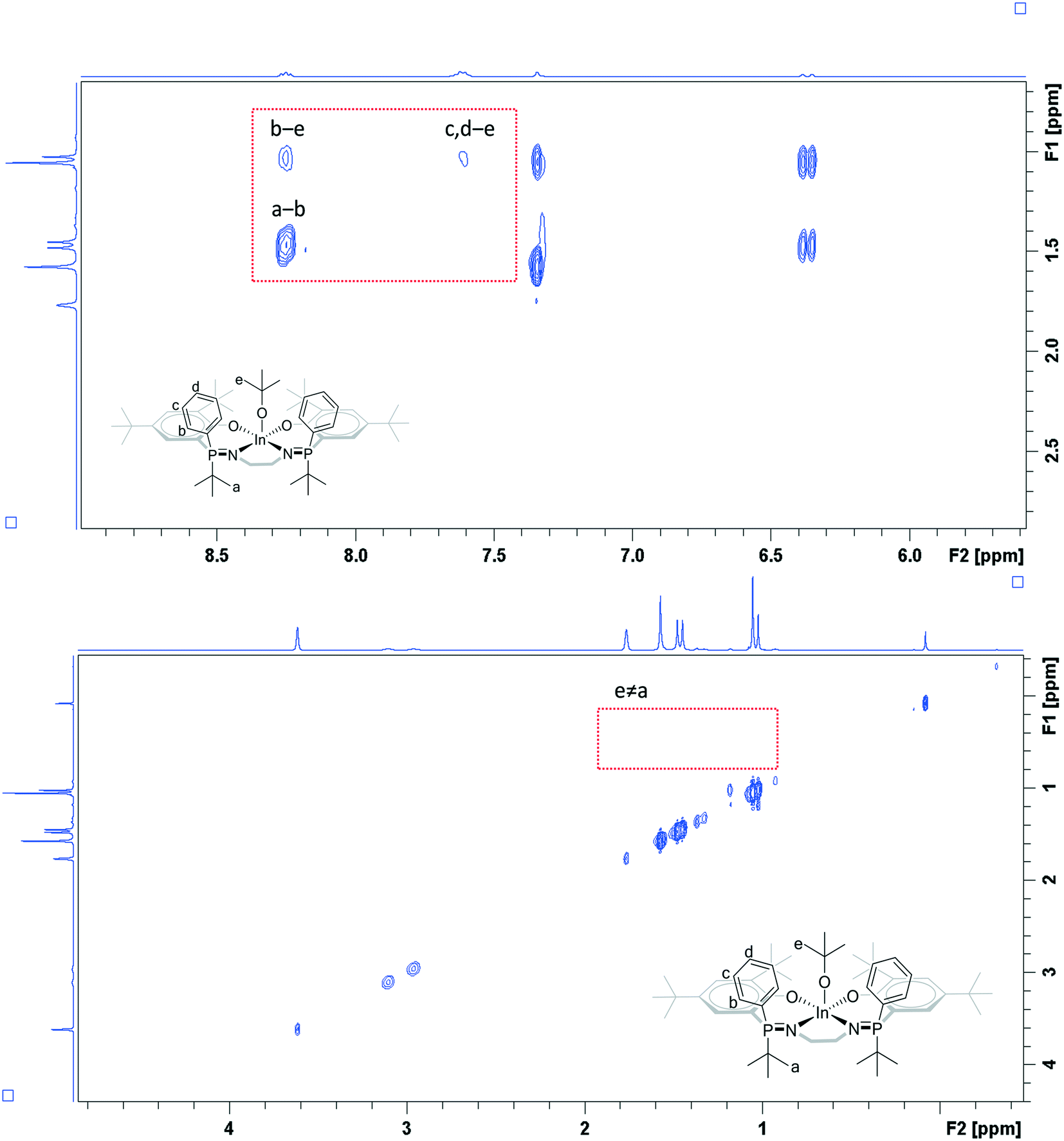 | ||
| Fig. 4 Selected regions of the 1H ROESY NMR spectrum of complex 4 (THF-d8). | ||
Polymerization catalysis
The series of indium complexes were tested as catalysts for LA ROP, using 1 M solution of LA, 1:500 molar ratio of indium complex:LA and with THF solvent, at 25 °C (consistent with the conditions previously used for 1).9c Complexes 2–5 are all active and isoselective catalysts, they also show good polymerization control, yielding PLA with monomodal molar mass distributions and narrow dispersity.
Catalytic activity
The catalysts can be compared either using point-kinetic measures, such as turn-over frequency, or from the rate coefficients, kobs (vide infra). Conversion and molar mass data were acquired by taking regular aliquots and using NMR spectroscopy and GPC analysis. Semi-logarithmic concentration–time plots show a linear fit to the data, consistent with a first order dependence of rate on LA concentration (Fig. S51–S54†). The pseudo first order rate coefficients (kobs) are the gradients of these plots and allow for more accurate understanding of catalyst performances. Comparing both the rate coefficients and TOF values reveals that catalyst 1 (ethylene linker) is faster than either 2 (propylene linker) or 3 (dimethyl propylene linker). In terms of the phosphorus substituents, complex 4 (tert-butyl and phenyl) is less active than complex 1 (phenyl) but is significantly more stereoselective and its overall activity is at the upper end of the series. Within the series of catalysts, the slowest is catalyst 3 and referencing rates against this complex shows that 2 is 16 times faster, 4 is 22 times faster, 5 is 25 times faster and 1 is 55 times faster.To better understand the kinetic data, the rate law was determined for catalyst 5. It shows a first order rate dependence in monomer concentration and the linear relationship between kobs and [In] is consistent with a first order dependence on the catalyst concentration (Fig. S55 and S56†). Therefore, an overall second order rate law is consistent with the data:
| ν = kp[LA][In] |
Isoselectivity
Complexes 1–3, featuring different diamine linkers, produced stereoblock PLA, with Pi values from 0.69–0.84 (Fig. S72 and S73†) and with the slowest catalyst, 3, being the most isoselective. Complex 2 (Pi = 0.69, 25 °C) shows a significantly lower isoselectivity perhaps due to the structure being fluxional on the polymerization timescale, as indicated by broadened resonances in its 1H NMR spectrum at room temperature. Complexes 4 and 5, featuring asymmetric phosphorus substituents, each show very high isoselectivity with Pi values exceeding 0.90 (Fig. S74–S77†). In addition, average isotactic block lengths were estimated by using Pi and Ps values, determined using Bernoullian statistics and by comparing integrals of carbonyl signals in the 13C{1H} NMR spectrum (hexad level) (δ 169.8 ppm for iso-rich hexad and δ 169.6–169.4 ppm for racemic-rich hexad) (Fig. S63–S65 and Table S10†).19,20 It should be noted that the Pi and Ps values used to determine the block lengths differ slightly to those reported in Table 1, where the values derive from the 1H{1H} NMR spectra (tetrad level). The calculated isotactic block length is 14 LA units for PLA produced using complex 5 at 25 °C (Table 1, entry 6). A slightly larger length of 19 LA units occurs for the higher molar mass sample (Table 1, entry 7). The largest average block length of 24 LA units is observed when the polymerization occurs at low temperature (Table 1, entry 5). Given such high stereoselectivity, the crystallinity and melting temperature of the resulting stereoblock PLA was investigated (Fig. S66–S71†). The DSC thermograms show melting temperatures (Tm) of 175 °C and 177 °C, for PLA, prepared from 4 and 5, respectively. Conducting the polymerization at 5 °C slightly increased the Pi value (4, Pi = 0.92) and increased the melting temperature of the resulting stereoblock PLA to 185 °C (Fig. S66, S69 and S70†). As part of a preliminary assessment of the feasibility to apply 4 under industry relevant manufacturing conditions, a polymerization was conducted at 130 °C (1:500, 4: LA and using neat LA). This led to the production of isotactic PLA (Pi = 0.78) (Fig. S78†) but the polymerization failed to increase beyond ∼20% conversion, suggesting possible thermal decomposition issues for catalyst 4 under these conditions. The influence of the ligand phosphorus substituents is appreciated when comparing the performance of 4 against 1, and manifests in a 20% increase in Pi value and 15 °C increase in PLA melting temperature. The data clearly show the benefit of substituting the phosphorus site with a tert-butyl substituent. Attempts were made to prepare the ligand featuring two tert-butyl phosphorous substituents but resulted in intractable product mixtures.
| Entry | Cat. | Time (h) | Conv.d (%) | TOFe (h−1) | M n,GPC (kg mol−1)(Đ) | M n,calc. (kg mol−1) | P i | k obs (×106 s−1) |
|---|---|---|---|---|---|---|---|---|
|
a Polymerization conditions: [In]:[LA] = 1:500, [LA] = 1 M, [In] = 2 mM, THF, 25 °C.
b The polymerization was conducted at 5 °C.
c This polymerization involved 3 sequential additions of rac-LA, each addition was made after >80% LA conversion, 3 × 500 equiv. of 1 M LA, THF, where the overall [In]:[LA] = 1:1500.
d Determined from the 1H NMR spectrum by comparison of the normalized integrals of the methine signals at δ 5.06–4.98 ppm (LA) and δ 5.22–5.08 ppm (PLA).
e TOF = (number of moles of LA consumed per mole of catalyst)/time (h).
f Determined by GPC analysis, calibrated using narrow MW polystyrene standards, in THF or CHCl3 (Fig. S57–S62†), and with a correction factor of 0.58.17
g Determined using the 1H{1H} NMR spectrum by integration of the methine tetrads (δ 5.22–5.11 ppm) and comparison of experimental integrals with the values predicted by Bernoullian statistics (Fig. S72–S77†).18
h
k
obs obtained from the gradient of semi-logarithmic plots of LA conversion and time (s). Errors are derived from the errors to the fit for each plot.
i Polymerization conditions: [In]:[LA] = 1:100, [LA] = 1 M, [In] = 10 mM.
|
||||||||
| 1 (ref. 9c) | 1 | 2.5 | 80 | 160 | 50.8 (1.19) | 57.7 | 0.75 ± 0.04 | 194 ± 4.00 |
| 2 | 2 | 8 | 81 | 51 | 36.1 (1.19) | 58.4 | 0.69 ± 0.02 | 56.5 ± 0.58 |
| 3 | 3 | 74 | 61 | 4 | 29.4 (1.18) | 44.0 | 0.84 ± 0.04 | 3.55 ± 0.08 |
| 4 | 4 | 8 | 88 | 55 | 90.4 (1.26) | 63.4 | 0.91 ± 0.05 | 77.8 ± 1.77 |
| 5b | 4 | 14.5 | 80 | 28 | 108.7 (1.15) | 57.7 | 0.92 ± 0.04 | NA |
| 6 | 5 | 4.5 | 90 | 100 | 57.0 (1.17) | 64.9 | 0.90 ± 0.05 | 134 ± 2.99 |
| 7c | 5 | 46 | 63 | — | 157.8 (1.25) | 136.2 | 0.92 ± 0.04 | NA |
| 8i | 5 | 40 min | 90 | 135 | 10.5 (1.11) | 13.3 | 0.87 ± 0.04 | 877 ± 19.7 |
Polymerization control
Complexes 2 and 3 produce PLA with slightly lower molar mass than predicted theoretically, whilst 4 shows the opposite result with a higher molar mass value than expected (Fig. S57–S59†). Since the complexes are analytically pure and the findings are reproducible using stock solutions of different batches of pure catalyst and lactide (i.e. systematic errors are unlikely to be responsible) the results are hard to explain. The higher than expected molar mass observed in PLA produced using 4 likely arises due to slow initiation from the sterically hindered tert-butoxide initiating group.11a Consistent with this notion, catalyst 5 features an ethoxide initiating group and shows molar mass values consistent with expected values and slightly higher overall rates of polymerization (Table 1 and Fig. S59 and S62†). All catalysts showed linear evolutions of molar mass with conversion and formed PLA with narrow dispersity (Fig. S57–S62†). These latter findings are consistent with good polymerization control.To understand the limits of catalyst 5, polymerizations were conducted at lower catalyst loading ([In]:[LA] = 1:1000) but reactions failed to progress beyond approximately 20% conversion. It appears that under more dilute conditions 5 is susceptible to decomposition processes, perhaps by reaction with residual water. Nonetheless, it is possible to prepare high molar mass PLA using 5 by exploiting the ability of controlled polymerizations to undergo re-initiation upon addition of more monomer. Thus, by reacting 5 with three successive LA batches whilst maintaining the overall catalyst loading at 0.2 mol%, it was possible to prepare PLA with molar mass of 160 kg mol−1 (i.e. consistent with 1:3 × 500 loading) (Table 1, entry 7). This latter finding is fully consistent with the high polymerization control afforded by these complexes and highlights their potential both to access high molar mass polymer and for block polymer formation.
Benchmarking catalyst performances against the literature
One issue for this field of catalysis is the overall lack of mechanistic insight into catalyst structure–performance and the ability to predictably design catalysts showing high stereoselectivity. Another difficulty is the comparison between different catalysts since different authors report isoselectivity and rate measurements under different conditions, and for point-kinetic measures such as turn-over-frequency (TOF), the precise conditions applied are not always the same, reproducibility is not always reported and thus comparisons are necessarily somewhat subjective. It is, of course, essential to properly contextualize and bench-mark new catalysts against the literature reports and, given the difficulties outlined, we have elected to use as points of comparison for this work high-performance group 13 catalysts as well as any catalyst, either metallic or organic, that shows a Pi > 0.8 at 25 °C. In terms of strengths and weaknesses of different methods used to report Pi values, the reader is referred to the useful analysis conducted by Mehrkhodavandi and co-workers.21 Here, all Pi values are reported as the average of values determined for each of the stereo-sequences (tetrads) and integral values are fit using the homonuclear decoupled NMR data using a global spectral deconvolution (GSD) algorithm (level 2), with 5 discrete fitting cycles. In all cases PLA melting temperatures are also determined and values are fully consistent with the high Pi values obtained by NMR spectroscopy. As mentioned in the introduction, to standardize the catalyst activity data, TOF values are compared, where possible, at catalyst: monomer loadings of 1:100 (or as close to this as was reported in the paper), [LA] = 1 M and 25 °C.
Catalyst 5 shows impressive performances when compared to other indium and aluminium catalyst systems (Fig. 5 and 6).4a,6a,c–9c,21–22 As outlined in the introduction, Al-salen catalysts show Pi values reaching 0.95, but they are very slow with typical TOF 0.1–10 h−1 (1:100, cat.:LA, [LA] = 1 M, 25 °C) (Scheme 2, F, G, H).4a,6a,c–8 Compared to this performance, catalyst 5 shows equivalent or better stereoselectivity. Indeed the melting point is higher for the resulting PLA than for polymers prepared from Al-salens purported to be more isotactic. Importantly, its rate is orders of magnitude faster, even accounting for the difficulties in making direct comparisons because Al-salen catalysts are applied at temperatures of 70 °C or higher. One useful strategy to increase rates has been to apply heavier group 13 element indium in place of Al(III), with the reduced Lewis acidity proposed to acclerate lactide insertion reactions. So far indium catalysts have shown isoselectivity values significantly lower than Al-counterparts.22a–c Catalyst 5 is notable since it combines the high rates of previous In(III) systems but significantly out-performs them in terms of isoselectivity and achieves tacticity control previously only possible for highly optimized Al-salens. For example, di-indium catalysts (D) show good rates but limited stereoselectivity.23 Chiral salen indium catalysts (E) show high rates and good isoselectivity (Pi = 0.77, 25 °C).21
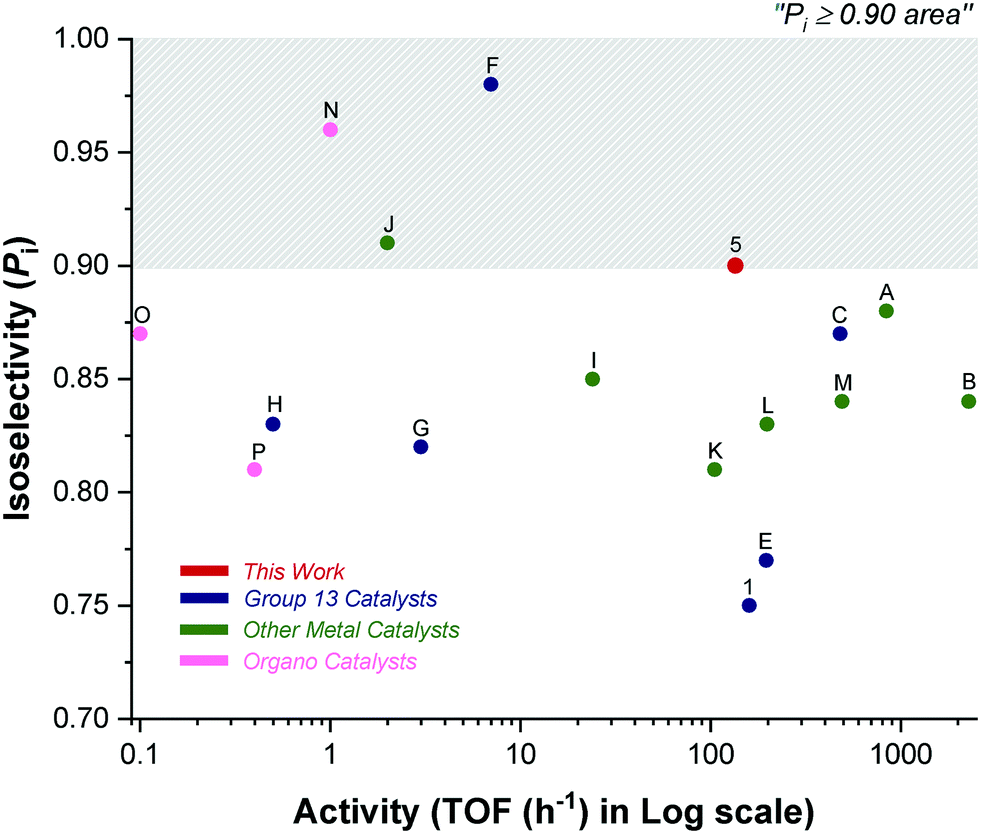 | ||
| Fig. 5 Plot showing isoselectivity values (Pi) against activity (TOF/h) for selected high-performance literature catalysts (Fig. 1 for structures A–C; Scheme 2 for structures D–P). | ||
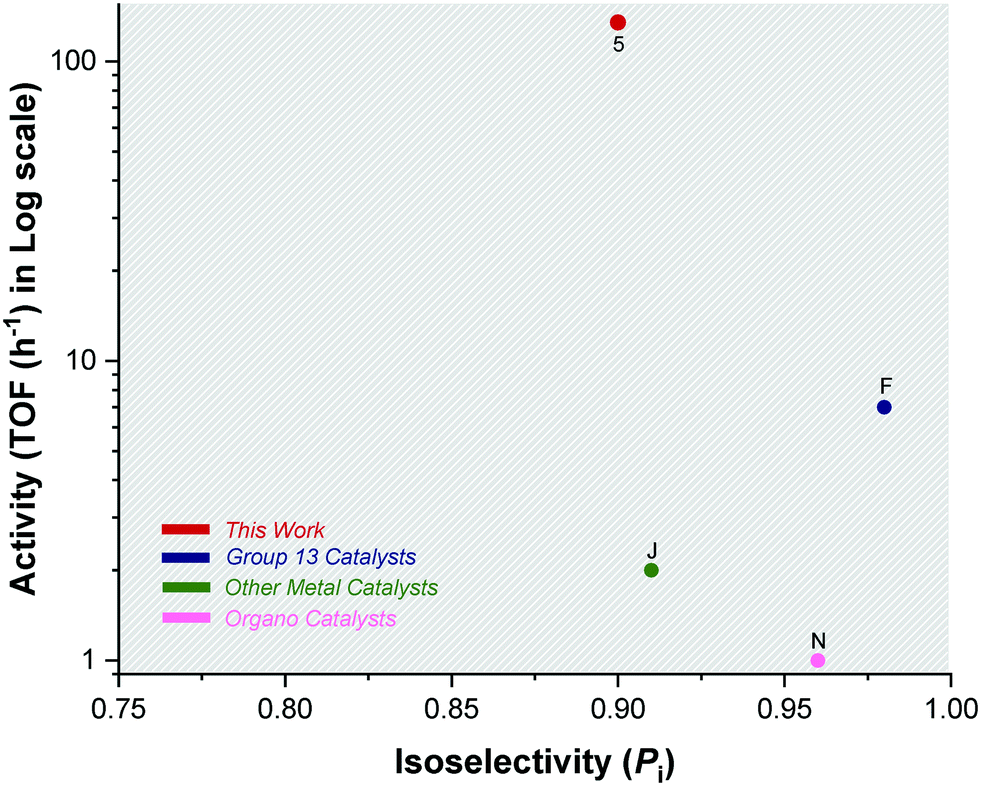 | ||
| Fig. 6 Plot showing the highest isoselectivity values (Pi > 0.9) against activity (TOF/h) for literature catalysts F, J, N and catalyst 5 (Scheme 2). | ||
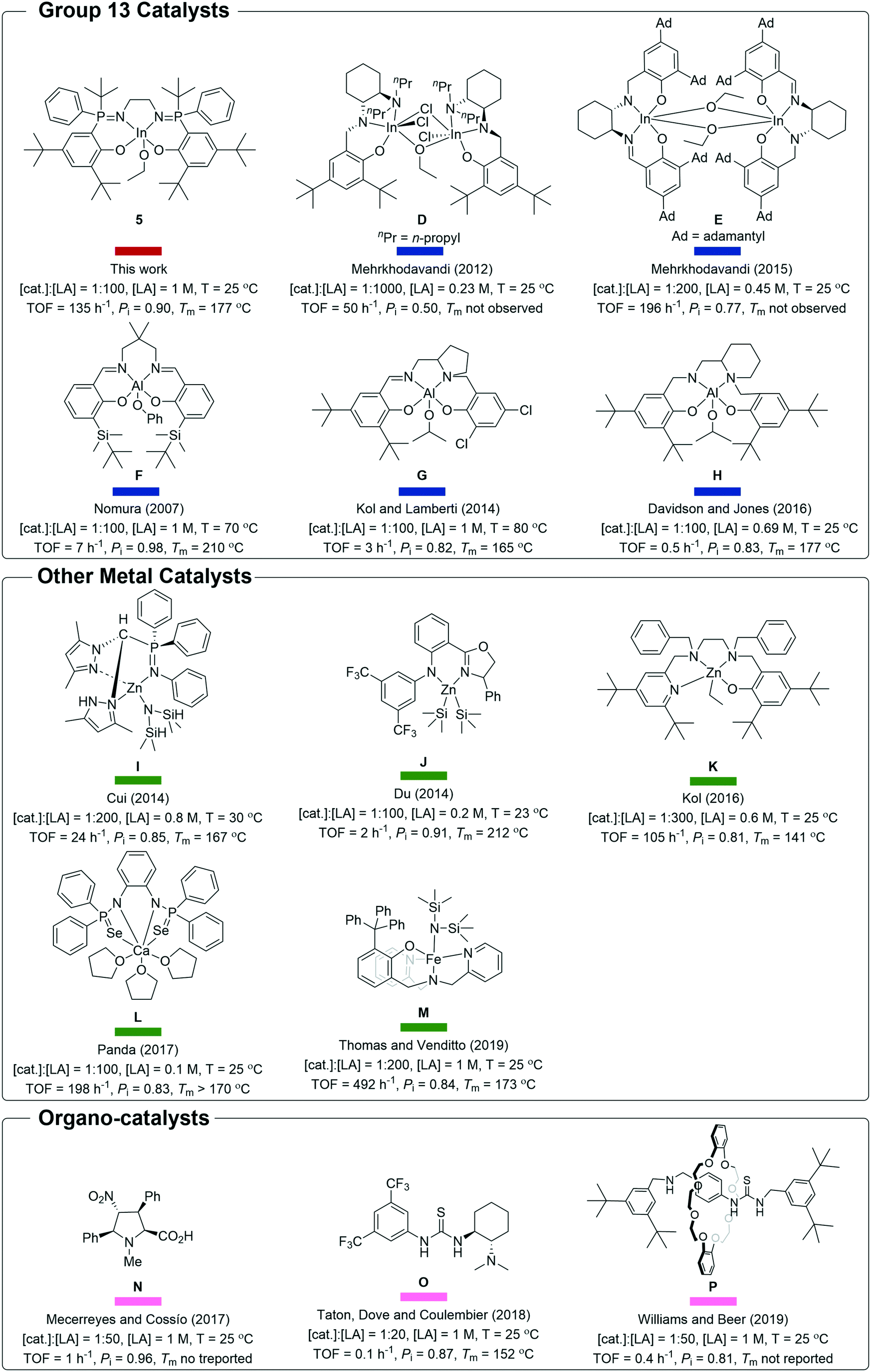 | ||
| Scheme 2 Structures of catalysts D–P and 5 (note catalysts A–C are illustrated in Fig. 1).7,8b,12a–c,e,f,13a,b,d,21,22d,24 | ||
Catalyst 5 is slower than zinc catalyst A (see Fig. 1) but it shows 5–10% higher isoselectivity.9a,10a Compared to the other high-performance yttrium catalyst B, it is slower but more isoselective.9b In comparison to indium phosphasalen catalyst 1, it increases isoselectivity by 20% and maintains a broadly similar rate. Considering the large number of other catalysts showing good rates and isoselectivity values, catalyst 5 performs very well. For example, it is both faster and more selective than heteroscorpionate zwitterionic zinc complexes which show Pi = 0.85 and TOF = 24 h−1 (cat.:LA 1:200, [LA] = 0.8 M, 30 °C).12a Catalyst 5 is around 70 times faster than the chiral amido oxazolinate zinc complexes and delivers equivalent isotacticity (Pi = 0.91, TOF = 2 h−1, cat.:LA 1:100, [LA] = 1 M, 23 °C).12b It gives around 10% improvement in isotacticity and similar rates to the phenolate triamine zinc catalysts (Pi = 0.81, TOF = 105 h−1, cat.:LA, 1:300, [LA] = 0.69 M, 25 °C).12c,d It is 10% more isoselective than bis(thio/seleno phosphinic amide) calcium complexes which were reported without an initiating group and presumably operate by an activated monomer mechanism (Pi = 0.83, TOF = 198 h−1, cat.:LA, 1:100, [LA] = 0.1 M, 25 °C).12f It is more isoselective but slower than the diamino-phenolate iron(II) complex recently reported by Thomas and co-workers (Pi = 0.84, TOF = 492 h−1, cat.:LA, 1:200, [LA] = 1 M, 25 °C).12e It matches the isoselectivity of the best organo-catalysts and shows significant improvements in rate, for example, it is 100–1000 times faster (at lower catalyst loading) than chiral prolines, chiral thioureas or mechanically interlocked [2]-rotaxanes.13a,b,d
Stereocontrol mechanism
Given the high degree of isoselectivity exhibited by 5, the mechanism of stereocontrol warrants investigation. Polymerization kinetics were determined, under identical conditions, using R,R and S,S-LA and show comparable rates, 132 ± 3.73 × 10−6 s−1 (R,R-LA) and 133 ± 2.26 × 10−6 s−1 (S,S-LA), which are both faster than rac-LA polymerization (kobs = 77.8 ± 1.76 × 10−6 s−1) (Fig. 7). The data are indicative of a chain end control mechanism since there is no particular discrimination for either lactide enantiomer and the slower rates for rac-LA arise from chain exchange processes.7,25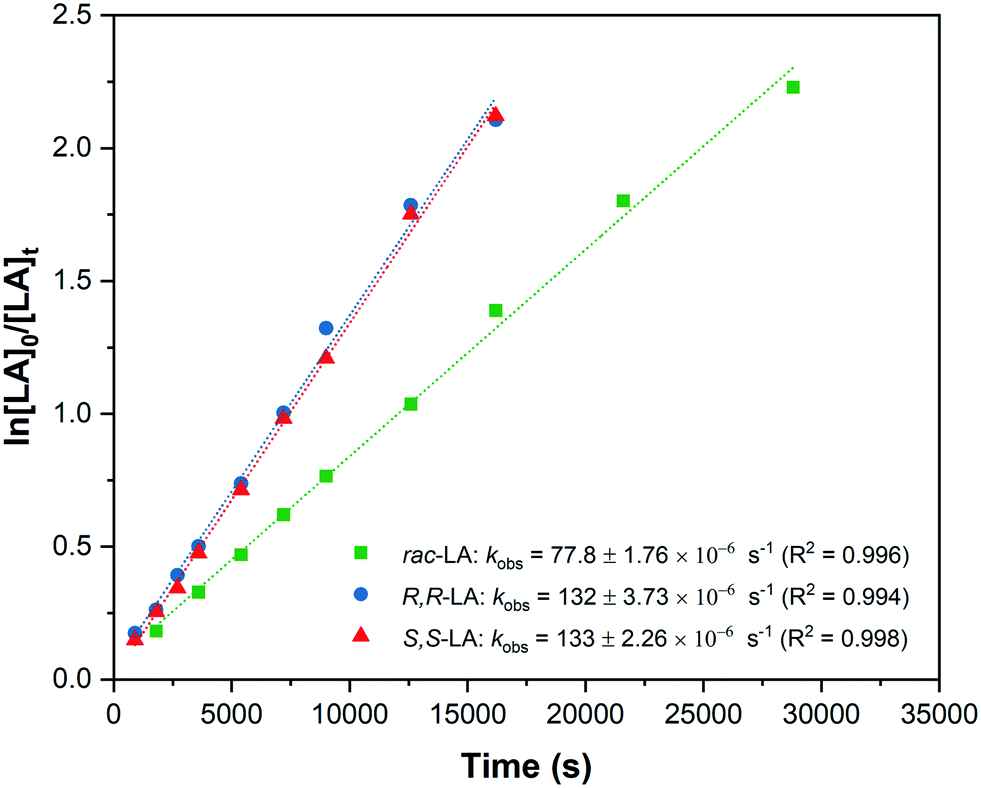 | ||
| Fig. 7 Semi-logarthimic plots of LA conversion vs. time for R,R-LA (D-LA); S,S-LA (L-LA) and rac-LA using catalyst 4. | ||
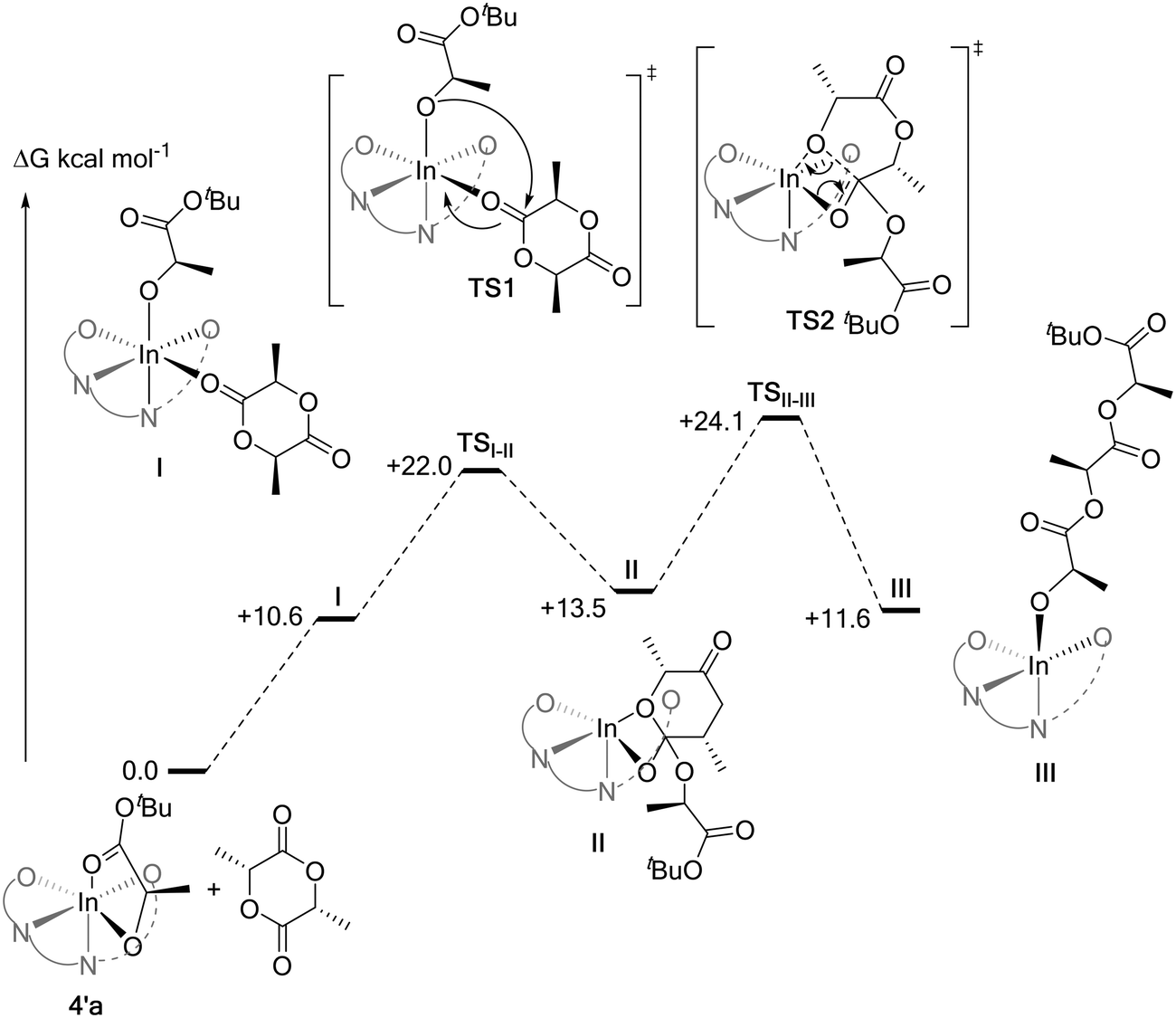
As part of attempts to understand the structure of the propagating catalytic intermediate, complex 4 was reacted with an equivalent of R,R-LA. Slow diffusion of hexane into the reaction solution in THF, allowed isolation of a crystal of a new complex, 4′a, and this species was analyzed using X-ray crystallography. The structure reveals an indium complex that is coordinated by both the phosphasalen ligand, binding in a cis β fashion, and by R-tert-butyl lactate. The lactate adopts a κ2 chelating mode occupying two cis coordination sites around the octahedral indium center (Fig. 8 and Tables S7 and S9†). The precise mechanism by which this complex forms is not clear and it should also be emphasized it is not quantitatively formed. Nonetheless, its formation is consistent with facile polymeryl chain exchange, as implicated from the kinetic studies, and likely forms by a process involving the tert-butyl alkoxide ligand of complex 4 attacking and ring-opening R,R-lactide and forming a ring-opened intermediate (i.e.4-LA-LA). The intermediate may undergo bimolecular transesterification to form 4′a (4-LA) and a trimeric propagating alkoxide (4-LA-LA-LA) (Scheme S3†). It is proposed that 4′a crystallizes more readily than other intermediates and hence is isolable. Crystals of 4′a were dissolved in THF-d8 and its 31P{1H} NMR spectrum shows two equal intensity singlets, at 54.3 and 54.0 ppm, as might be expected from the solid state structure since the two phosphorus environments are inequivalent (Fig. S39†). The structure of 4′a is significant since it provides a model structure for the propagating alkoxide species during polymerization.
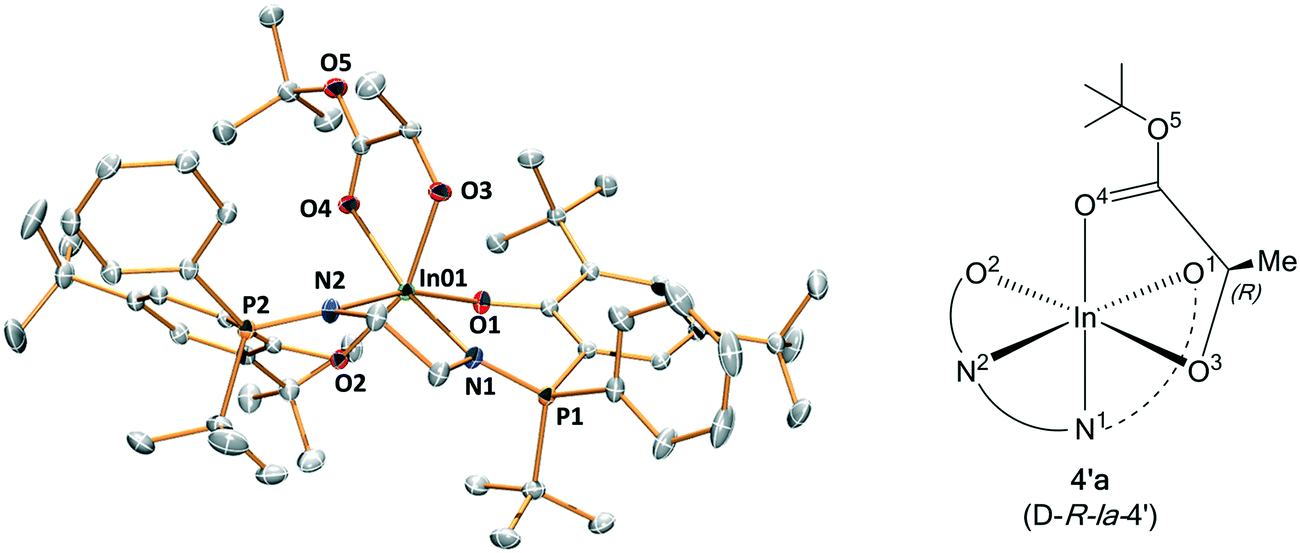 | ||
| Fig. 8 Representation of the molecular structure of key intermediate 4′a. | ||
DFT studies
Computational modelling was undertaken using DFT calculations to shed light on the increased isoselectivity of 4 compared with 1. The structure of the lactate complex 4′a provided an excellent starting point for modelling rac-LA propagation. However, 4′a is only one of the four stereoisomers possible for such model lactate indium complexes. Based on the molecular structure of 4′a revealed by X-ray crystallography, the tetradentate phosphasalen coordination mode (Λ vs. Δ) and the lactate stereochemistry (R vs. S) give rise to two pairs of diastereoisomers, namely Λ-R-la-4′(4′a) and Δ-R-la-4′(4′b), as well as Λ-S-la-4′ and Δ-S-la-4′ (Fig. 9). By assuming that enantiomers should show similar potential energy surfaces, only a single diastereotopic pair, 4′a and 4′b, is considered, consistent with the experimental data.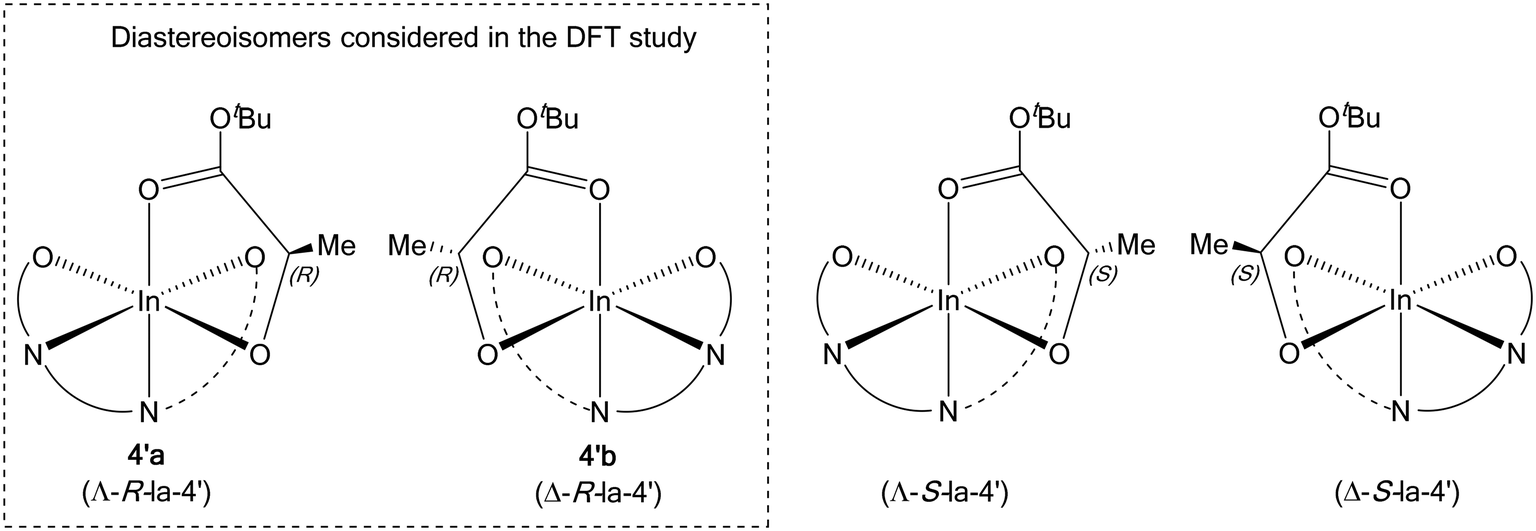 | ||
| Fig. 9 Four possible stereoisomers of 4′. | ||
For each starting lactate complex and a given LA enantiomer, assuming the phosphasalen ligand coordinates in a cis β fashion, four feasible pathways were determined per monomer ring-opening reaction, depending on the initial coordination site of the lactide molecule. The LA carbonyl can be cis or trans to the phenoxide moiety of the phosphasalen ligand, with either the re or si faces of the lactide facing the alkoxide ligand (which models the site occupied by the growing polymer chain) (Fig. 10; see Fig. S84† for the full sixteen possibilities arising from the coordination of D or L-LA to 4′a or 4′b).
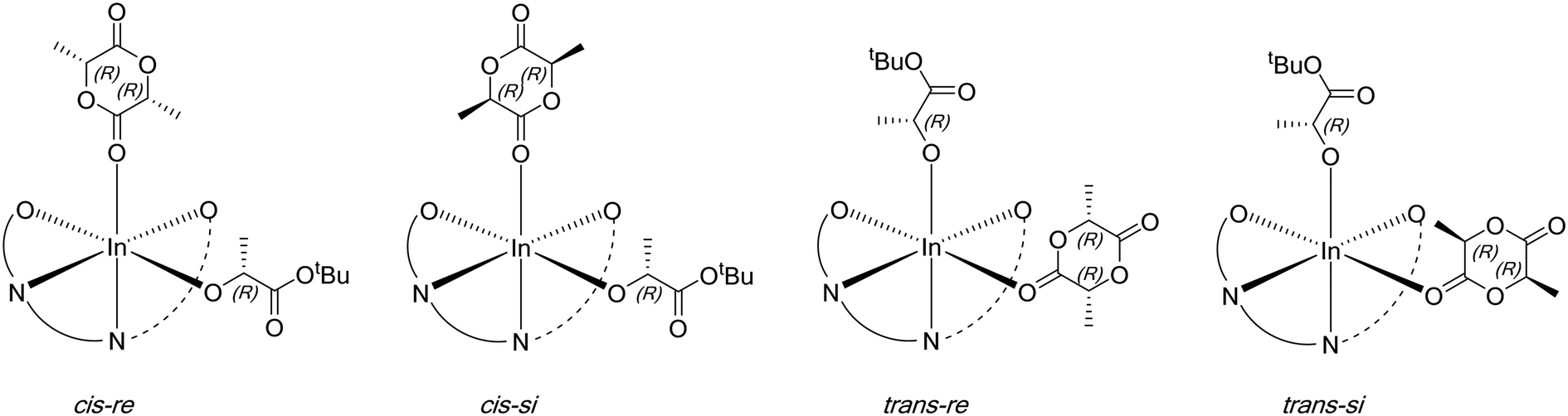 | ||
| Fig. 10 Four possible coordination modes of D-LA to 4′a, resulting in four alternative pathways for ring-opening. | ||
All possible pathways for the ring-opening of rac-LA by 4′a (eight in total) were calculated using the M06-L functional (Fig. S84, S85 and Table S13†). In agreement with experiments, an isotactic preference was apparent (ΔΔG‡isotactic − ΔΔG‡heterotactic = −3.5 kcal mol−1; i.e. the difference between the lowest activation barriers for the isotactic or heterotactic ring-opening pathways, respectively). Isotactic ring-opening via the trans re pathway was the most favourable (Fig. 11). 4′b was also found to favor isoselectivity (ΔΔG‡isotactic − ΔΔG‡heterotactic = −6.5 kcal mol−1), although this time the lowest energy pathway was the trans si one. Both these results contrast with calculations for the diphenyl complexes 1′a and 1′b, used to model propagation using 1, which did not show any isotactic preference (ΔΔG‡isotactic − ΔΔG‡heterotactic for 1′a = +0.6 kcal mol−1; for 1′b = +5.1 kcal mol−1) (Fig. S84 and S85 and Table S13†). The greater isoselectivity of 4 compared to 1 was further corroborated with benchmarking studies performed using the PBE0 and ωb97XD functionals. While 1′a shows some isoselectivity (ΔΔG‡isotactic − ΔΔG‡heterotactic = −2.6 and −1.1 kcal mol−1 for PBE0 and ωb97XD, respectively), it was much lower than for 4′a (ΔΔG‡isotactic − ΔΔG‡heterotactic = −10.4 and −9.8 kcal mol−1 for PBE0 and ωb97XD, respectively) (Schemes S4–S6 and Table S18†). Similarly, 1′b shows no isoselectivity (ΔΔG‡isotactic − ΔΔG‡heterotactic = +1.8 and +4.2 kcal mol−1 for PBE0 and ωb97XD, respectively) contrasting with 4′b which exhibits a strong isoselective preference (ΔΔG‡isotactic − ΔΔG‡heterotactic = −4.2 and −6.6 kcal mol−1 for PBE0 and ωb97XD, respectively).
 | ||
| Fig. 11 Lowest energy pathway computed for the isotactic ring opening of D-lactide by 4′a. Calculations were performed using the M06L functional (with empirical dispersion correction factor, GD3, applied), basis set 6-31+G(d,p) for N, P and O atoms, 6-31G(d,p) for C and H atoms, and basis set and pseudo potential lanl2dz for In atom. Solvent effects were accounted for using the SMD continuum model for THF at 298.15 K. | ||
Further comparison between the lowest computed energy pathways reveal that across the range of functionals, the nucleophilic attack of the indium alkoxide on the lactide carbonyl (TSI-II) is the rate determining transition state for the complexes bearing phosphorus diphenyl substituents. However, for these complexes the differences between the associated TSI-II barriers for isotactic or heterotactic pathways are small (Table S19 and Fig. S86†). For the complex featuring both phenyl and tert-butyl substituents on the phosphorus atoms, the barriers for TSI-II diverge: increasing for the heterotactic pathway and decreasing for the isotactic pathway. Consistent with the correct identification of the rate limiting step, there was no such correlation between the improved isoselectivity, of 4′a–b over 1′a–b, and the activation barriers for TSII-III. Therefore, the catalytic pocket around the indium centre in TSI-II was further analyzed using steric maps, generated from the SambVca 2 web tool developed by Cavallo and coworkers (Fig. S87†).26 No specific interaction could be identified as causing the changes in TSI-II barriers for 4′a–b, however, the TSI-II structures computed from 4′a and 4′b for the heterotactic pathway showed higher buried volumes than any others (Fig. S88 and Table S20†), suggesting that the improved isoselectivity of 4 over that of 1 might be of steric origin.
Monomer scope
The catalysts were also evaluated for various other lactone ring opening polymerizations, including using ε-caprolactone (CL), β-butyrolactone (BL), ε-decalactone (DL) and δ-hexalactone (HL) (Table 2).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Monomer | Complex | Time (h) | Lactone conv.d (%) | M n,GPC (kg mol−1) | Đ |
| a Conditions: [monomer] = 1 M, [In] = 2 mM, THF, [monomer]/[In] = 500. b Conditions: [monomer] = 1 M, [In] = 5 mM, THF, [monomer]/[In] = 200. c Conditions: [δ-HL] = 5 M, [In] = 10 mM, THF, [δ-HL]/[In] = 500. d Determined by integration of the methine or methylene region of the 1H NMR spectrum (CL: δ 4.17–4.12 ppm, PCL: δ 4.02–3.92 ppm, BL: δ 4.59–4.48 ppm, PBL: δ 5.18–5.01 ppm, DL: δ 4.18–4.03 ppm, PDL: δ 4.80–4.63 ppm, HL: δ 4.43–4.27 ppm, PHL: δ 4.91–4.68 ppm). e Determined by GPC analysis, against polystyrene standards, in THF and for PCL a correction factor of 0.56 was applied.17 | ||||||
| 1 | CLa | 1 | 6.5 min | 82 | 105.3 | 1.43 |
| 2 | 2 | 4.8 min | 99 | 117.9 | 1.19 | |
| 3 | 3 | 55 min | 95 | 63.8 | 1.09 | |
| 4 | 5 | 5 min | 90 | 60.8 | 1.53 | |
| 5 | BLb | 1 | 7 | 81 | 25.9 | 1.16 |
| 6 | 2 | 4 | 99 | 27.0 | 1.21 | |
| 7 | 3 | 48 | Trace | NA | NA | |
| 8 | 5 | 33 | 82 | 11.9 | 1.13 | |
| 9 | DLb | 1 | 24 | 97 | 34.4 | 1.14 |
| 10 | 2 | 120 | 99 | 28.2 | 1.13 | |
| 11 | 3 | 120 | 6 | NA | NA | |
| 12 | 5 | 80 | 70 | 18.0 | 1.17 | |
| 13 | HLc | 1 | 30 | 87 | 86.0 | 1.48 |
| 14 | 2 | 120 | 30 | 25.8 | 1.29 | |
| 15 | 3 | 120 | Trace | NA | NA | |
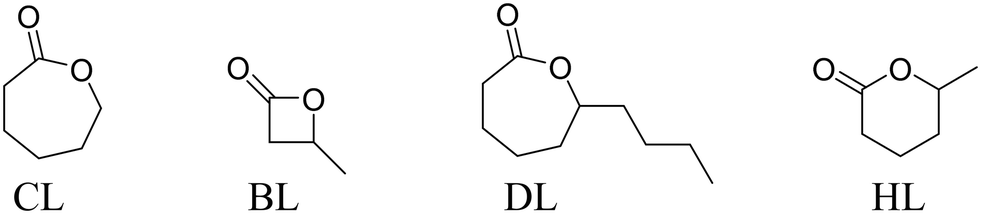
For CL ROP, catalysts 1 and 2 achieved almost full conversion of 500 equivalents in only a few minutes but 3 was much slower and required ∼1 h to reach complete conversion. Complexes 1 and 2 produce PCL with molar mass values higher than expected (Mn,calc. = 60 kg mol−1 at 100% conversion), and consistent with slower initiation than propagation.27 Also consistent with this notion was the finding that complex 3, which is qualitatively slower, shows good molar mass control and close agreement between theoretical and experimental Mn values. Catalyst 5 shows very fast rates but produces PCL with a molar mass close to the expected value (consistent with initiation problems being overcome by using the ethoxide co-ligand). The molar mass distribution is somewhat broadened which may indicate transesterification side-reactions using this catalyst. For BL ROP, a higher catalyst loading (0.5 mol%) was applied since it is generally found to polymerize slower than LA or CL.28 Under these conditions, the order of activity is 2 > 1 > 5 ≫ 3 and in all cases the PBL produced is atactic (Fig. S79†). Both findings are quite different to LA ROP using these catalysts and signal that caution must be applied in generalizing any findings even between apparently similar monomers. The PBL shows Mn values (uncorrected by GPC) which are in reasonable agreement with theoretical values (Mn,calc. = 18 kg mol−1 at 100% conversion). Furthermore, BL ROP occurs mostly through acyl bond cleavage and there was no evidence for any crotonyl or alkyl chain end-groups by NMR spectroscopy (Fig. S80†). For DL ROP once again higher catalyst loadings were applied (0.5 mol%) as polymerizations are typically slower,29 and the order of activity values are 1 > 2 ∼ 5 > 3. These catalysts all operate at room temperature which is in contrast to most other high activity DL ROP catalysts which require high temperatures.29b,30 Polymerization control is good and the PDL shows molar mass values (GPC, uncorrected) in reasonable agreement with calculated values (Mn,calc. = 34 kg mol−1 at 100% conversion) and with monomodal, narrow dispersity distributions. HL ROP was investigated using 1 (0.2 mol%) but only reached 20% conversion (over 20 h). The slow conversion may arises from the high equilibrium monomer conversion, due to a reduced polymerization enthalpy for HL. Indeed, using the polymerization enthalpy and entropy values, reported by Hillmyer and coworkers, for [HL] = 1 M, at 25 °C, an equilibrium monomer concentration = 0.72 M is determined which is in good agreement with the experimental value measured here.31 Conducting HL ROP at higher monomer concentration (1:HL 1:500, [HL] = 5 M) allowed polymerizations to reach >85% conversion (30 h). HL ROP, under these higher conversion conditions, using 2 or 3 were extremely slow. PHL, isolated from ROP using 1, shows molar mass values (GPC, uncorrected) in reasonable agreement with theoretical values (Mn,calc. = 50 kg mol−1 at 85% conversion), although dispersity values were broad.
Conclusions
The series of five new indium phosphasalen complexes show promising performances in racemic-lactide ring-opening polymerizations, combining high activity, high isoselectivity and good polymerization control. The most isoselective catalysts apply asymmetrical phosphorus substituents, with a phenyl and tert-butyl group attached to each phosphorus, but form achiral meso complexes. Using these ligands allows for Pi values >0.90, at 25 °C, and with turn-over-frequencies of 100 h−1 (0.2 mol% catalyst loading). Kinetic investigations, an isolated catalytic intermediate and DFT calculations implicate a chain end control mechanism. The complexes also show good activity in other lactone ring-opening polymerizations and are effective catalysts to produce a range of other aliphatic polyesters. This work highlights the potential for achiral ligands with asymmetrical substitution at phosphorus as a future design strategy to optimize both rates and stereoselectivity in lactide polymerizations. The catalysts also warrant further exploration for the rapid and controllable preparation of partially crystalline block polyesters.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
N. Y. acknowledges funding from the Development and Promotion of Science Talents Project under the Royal Thai Government. Research funding from the EPSRC (EP/S018603/1; EP/K014668/1, EP/R027129/1, EP/N509589/1), the Royal Society (UF/160021 fellowship to AB) and from SCG Chemical is also acknowledged.References
- (a) Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 Search PubMed; (b) X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 Search PubMed.
- (a) J. C. Worch, H. Prydderch, S. Jimaja, P. Bexis, M. L. Becker and A. P. Dove, Nat. Rev. Chem., 2019, 3, 514–535 Search PubMed; (b) Z. B. Li, B. H. Tan, T. T. Lin and C. B. He, Prog. Polym. Sci., 2016, 62, 22–72 Search PubMed; (c) K. Fukushima and Y. Kimura, Polym. Int., 2006, 55, 626–642 Search PubMed; (d) H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 Search PubMed; (e) S. C. Schmidt and M. A. Hillmyer, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 300–313 Search PubMed.
- (a) K. Fukushima, Y. H. Chang and Y. Kimura, Macromol. Biosci., 2007, 7, 829–835 Search PubMed; (b) M. Hirata and Y. Kimura, Polymer, 2008, 49, 2656–2661 Search PubMed.
- (a) P. J. Dijkstra, H. Z. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 Search PubMed; (b) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 Search PubMed; (c) S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem. – Eur. J., 2015, 21, 7988–8003 Search PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 Search PubMed.
- (a) Z. Y. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 Search PubMed; (b) P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343–15348 Search PubMed; (c) K. Majerska and A. Duda, J. Am. Chem. Soc., 2004, 126, 1026–1027 Search PubMed; (d) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 Search PubMed.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 Search PubMed.
- (a) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 Search PubMed; (b) A. Pilone, K. Press, I. Goldberg, M. Kol, M. Mazzeo and M. Lamberti, J. Am. Chem. Soc., 2014, 136, 2940–2943 Search PubMed.
- (a) Y. M. Gong and H. Y. Ma, Chem. Commun., 2019, 55, 10112–10115 Search PubMed; (b) T. Q. Xu, G. W. Yang, C. Liu and X. B. Lu, Macromolecules, 2017, 50, 515–522 Search PubMed; (c) D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 Search PubMed; (d) C. Kan, J. W. Hu, Y. Huang, H. B. Wang and H. Y. Ma, Macromolecules, 2017, 50, 7911–7919 Search PubMed.
- (a) J. W. Hu, C. Kan, H. B. Wang and H. Y. Ma, Macromolecules, 2018, 51, 5304–5312 Search PubMed; (b) H. B. Wang and H. Y. Ma, Chem. Commun., 2013, 49, 8686–8688 Search PubMed; (c) H. B. Wang, Y. Yang and H. Y. Ma, Inorg. Chem., 2016, 55, 7356–7372 Search PubMed.
- (a) C. Bakewell, T. P. A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 Search PubMed; (b) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 Search PubMed.
- (a) Z. H. Mou, B. Liu, M. Y. Wang, H. Y. Xie, P. Li, L. Li, S. H. Li and D. M. Cui, Chem. Commun., 2014, 50, 11411–11414 Search PubMed; (b) S. Abbina and G. D. Du, ACS Macro Lett., 2014, 3, 689–692 Search PubMed; (c) T. Rosen, Y. Popowski, I. Goldberg and M. Kol, Chem. – Eur. J., 2016, 22, 11533–11536 Search PubMed; (d) D. E. Stasiw, A. M. Luke, T. Rosen, A. B. League, M. Mandal, B. D. Neisen, C. J. Cramer, M. Kol and W. B. Tolman, Inorg. Chem., 2017, 56, 14366–14372 Search PubMed; (e) P. Marin, M. J.-L. Tschan, F. Isnard, C. Robert, P. Haquette, X. Trivelli, L.-M. Chamoreau, V. Guérineau, I. del Rosal, L. Maron, V. Venditto and C. M. Thomas, Angew. Chem., Int. Ed., 2019, 58, 12585–12589 Search PubMed; (f) J. Bhattacharjee, A. Harinath, H. P. Nayek, A. Sarkar and T. K. Panda, Chem. – Eur. J., 2017, 23, 9319–9331 Search PubMed.
- (a) B. Orhan, M. J. L. Tschan, A.-L. Wirotius, A. P. Dove, O. Coulembier and D. Taton, ACS Macro Lett., 2018, 7, 1413–1419 Search PubMed; (b) J. Y. C. Lim, N. Yuntawattana, P. D. Beer and C. K. Williams, Angew. Chem., Int. Ed., 2019, 58, 6007–6011 Search PubMed; (c) S. Liu, H. Li, N. Zhao and Z. Li, ACS Macro Lett., 2018, 7, 624–628 Search PubMed; (d) A. Sanchez-Sanchez, I. Rivilla, M. Agirre, A. Basterretxea, A. Etxeberria, A. Veloso, H. Sardon, D. Mecerreyes and F. P. Cossio, J. Am. Chem. Soc., 2017, 139, 4805–4814 Search PubMed.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2015, 54, 2204–2212 Search PubMed.
- (a) I. M. Marín and A. Auffrant, Eur. J. Inorg. Chem., 2018, 15, 1634–1644 Search PubMed; (b) T. P. A. Cao, G. Nocton, L. Ricard, X. F. Le Goff and A. Auffrant, Angew. Chem., Int. Ed., 2014, 53, 1368–1372 Search PubMed; (c) T. P. A. Cao, S. Labouille, A. Auffrant, Y. Jean, X. F. Le Goff and P. Le Floch, Dalton Trans., 2011, 40, 10029–10037 Search PubMed; (d) T. P. A. Cao, A. Buchard, X. F. Le Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157–2169 Search PubMed; (e) C. Bakewell, T. P. A. Cao, X. F. Le Goff, N. J. Long, A. Auffrant and C. K. Williams, Organometallics, 2013, 32, 1475–1483 Search PubMed; (f) T. Cheisson, T. P. A. Cao, X. F. Le Goff and A. Auffrant, Organometallics, 2014, 33, 6193–6199 Search PubMed; (g) I. M. Marin, T. Cheisson, R. Singh-Chauhan, C. Herrero, M. Cordier, C. Clavaguera, G. Nocton and A. Auffrant, Chem. – Eur. J., 2017, 23, 17940–17953 Search PubMed.
- A. Thevenon, A. Cyriac, D. Myers, A. J. P. White, C. B. Durr and C. K. Williams, J. Am. Chem. Soc., 2018, 140, 6893–6903 Search PubMed.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 Search PubMed.
- J. Coudane, C. Ustariz-Peyret, G. Schwach and M. Vert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1651–1658 Search PubMed.
- M. H. Rahaman and H. Tsuji, Polym. Degrad. Stab., 2013, 98, 709–719 Search PubMed.
- (a) F. Chabot, M. Vert, S. Chapelle and P. Granger, Polymer, 1983, 24, 53–59 Search PubMed; (b) N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 Search PubMed.
- D. C. Aluthge, J. M. Ahn and P. Mehrkhodavandi, Chem. Sci., 2015, 6, 5284–5292 Search PubMed.
- (a) A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 Search PubMed; (b) K. M. Osten and P. Mehrkhodavandi, Acc. Chem. Res., 2017, 50, 2861–2869 Search PubMed; (c) S. Dagorne, M. Normand, E. Kirillov and J. F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 Search PubMed; (d) P. McKeown, M. G. Davidson, G. Kociok-Kohn and M. D. Jones, Chem. Commun., 2016, 52, 10431–10434 Search PubMed.
- (a) A. B. Kremer, K. M. Osten, I. Yu, T. Ebrahimi, D. C. Aluthge and P. Mehrkhodavandi, Inorg. Chem., 2016, 55, 5365–5374 Search PubMed; (b) K. M. Osten, I. Yu, I. R. Duffy, P. O. Lagaditis, J. C. C. Yu, C. J. Wallis and P. Mehrkhodavandi, Dalton Trans., 2012, 41, 8123–8134 Search PubMed.
- I. Yu, A. Acosta-Ramirez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 Search PubMed.
- A. Pilone, N. De Maio, K. Press, V. Venditto, D. Pappalardo, M. Mazzeo, C. Pellecchia, M. Kol and M. Lamberti, Dalton Trans., 2015, 44, 2157–2165 Search PubMed.
- (a) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 Search PubMed; (b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 Search PubMed.
- S. M. Quan and P. L. Diaconescu, Chem. Commun., 2015, 51, 9643–9646 Search PubMed.
- (a) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 Search PubMed; (b) H.-L. Chen, H.-J. Chuang, B.-H. Huang and C.-C. Lin, Inorg. Chem. Commun., 2013, 35, 247–251 Search PubMed; (c) J. S. Klitzke, T. Roisnel, E. Kirillov, O. D. L. Casagrande and J.-F. Carpentier, Organometallics, 2014, 33, 309–321 Search PubMed.
- (a) D. Bandelli, C. Weber and U. S. Schubert, Macromol. Rapid Commun., 2019, 40, 1900306 Search PubMed; (b) M. P. F. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 Search PubMed.
- (a) P. Olsén, T. Borke, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2013, 14, 2883–2890 Search PubMed; (b) Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 Search PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2018635–2018640. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy01484b |
| This journal is © The Royal Society of Chemistry 2020 |