
Low-temperature hydrogen production from methanol over a ruthenium catalyst in water†
Mahendra K.
Awasthi
a,
Rohit K.
Rai
 b,
Silke
Behrens
c and
Sanjay K.
Singh
*a
b,
Silke
Behrens
c and
Sanjay K.
Singh
*a
aCatalysis Group, Discipline of Chemistry, Indian Institute of Technology Indore, Simrol, Indore 453552, M.P., India. E-mail: sksingh@iiti.ac.in
bKAUST Catalysis Center and Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Kingdom of Saudi Arabia
cInstitut für Katalyseforschung und – Technologie (IKFT), Karlsruher Institut für Technologie (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 26th October 2020
Abstract
Traditionally, methanol reforming at a very high temperature (>200 °C) has been explored for hydrogen production. Here, we show that in situ generated ruthenium nanoparticles (ca. 1.5 nm) from an organometallic precursor promote hydrogen production from methanol in water at low temperature (90–130 °C), which leads to a practical and efficient approach for low-temperature hydrogen production from methanol in water. The reactivity of ruthenium nanoparticles is tuned to achieve a high rate of hydrogen gas production from methanol. Notably, the use of a pyridine-2-ol ligand significantly accelerated the hydrogen production rate by 80% to 49 mol H2 per mol Ru per hour at 130 °C. Moreover, the studied ruthenium catalyst exhibits appreciably long-term stability to achieve a turnover number of 762 mol H2 per mol Ru generating 186 L of H2 per gram of Ru.
Introduction
Hydrogen is a potential clean energy carrier, and when used in fuel cells, it only produces water as a by-product. Unfortunately, the presence of hydrogen gas in the earth's atmosphere is extremely low (≈1 ppm by volume). Therefore, one of the major hurdles in exploring hydrogen economy with full potential is the safe production and storage of hydrogen gas. Notably, carrying big and heavy hydrogen cylinders with high pressure has critical safety and economical challenges. On the other hand, using liquid hydrogen storage materials (such as HCHO, CH3OH, and HCOOH) in the fuel tank of existing vehicles (using petroleum products) to generate hydrogen on-board to supply to fuel cells is not only a viable concept but also very economical.1–12 In this context, methanol, which contains an appreciably high gravimetric content of hydrogen (12.5 wt%), is a promising candidate for on-board and off-board (stationary) large-scale production of hydrogen gas.13–20 Notably, methanol offers several advantages, such as being an inexpensive liquid, low carbon content (C1 alcohol), easy to store, and being produced on a large scale from biomass resources and hydrogen and carbon monoxide, or from industrial by-products.13–20 Traditionally, hydrogen gas is being produced from a methanol steam reforming process at a high-temperature range (200–350 °C), while catalyst assisted hydrogen production from methanol in water is more energy efficient as it operates at low temperatures (<190 °C).In principle, hydrogen production from methanol is mildly endothermic; therefore, a suitable catalyst may activate methanol to produce hydrogen gas.14–24 Experimental evidence revealed that in the presence of a catalyst, the dehydrogenation of methanol may follow three consecutive pathways: initially, the dehydrogenation of methanol to formaldehyde and hydrogen (eqn (1)), followed by the dehydrogenation of formaldehyde in the presence of water (gem-diol) to hydrogen and formic acid (eqn (2)), and finally, the dehydrogenation of formic acid to hydrogen and CO2 (eqn (3)).
| CH3OH → H2CO + H2 (ΔH = 129.8 kJ mol−1) | (1) |
| H2CO + H2O → HCOOH + H2 (ΔH = −30.7 kJ mol−1) | (2) |
| HCOOH → CO2 + H2 (ΔH = 31.6 kJ mol−1) | (3) |
A wide range of catalysts has been explored to utilize methanol, as a potential liquid hydrogen storage material, for the production of hydrogen gas at low temperature with regulated emission of unwanted CO and methane.21,25 Recently, homogeneous catalysts based on Ru, Ir, Fe, and Mn have been explored to dehydrogenate methanol in the presence of water and produce H2 without or with very low ppm CO at temperatures below 100 °C.14,21,25–30 In particular, ruthenium-pincer based molecular catalysts exhibited higher activity for producing hydrogen from methanol under basic conditions.25,26 In contrast to the above, heterogeneously catalyzed reforming of methanol to produce hydrogen and carbon dioxide has been continuously studied and developed, using different metal-based catalysts such as CuO/ZnO/Al2O3,31 Pd/CeO2–ZrO2,32 Pt3Ni,33 and Ni–Fe–Mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 alloys, but most of these catalysts require higher temperature over 200 °C and pressure. On the other hand, industrially viable heterogeneous catalysts for low-temperature hydrogen production are rarely explored, until recently when the Pt/MoC catalyst was explored for hydrogen production from methanol, but this catalyst only worked effectively at higher temperature (150–190 °C) and uses an expensive Pt catalyst.35
34 alloys, but most of these catalysts require higher temperature over 200 °C and pressure. On the other hand, industrially viable heterogeneous catalysts for low-temperature hydrogen production are rarely explored, until recently when the Pt/MoC catalyst was explored for hydrogen production from methanol, but this catalyst only worked effectively at higher temperature (150–190 °C) and uses an expensive Pt catalyst.35
Notably, the development of catalysts for the selective transformation of methanol into hydrogen gas (eqn (1)) with the generation of formic acid is of critical importance, as it eliminates the energy-intensive process of CO2 scrubbing from a H2 and CO2 mixture obtained from complete transformation of methanol (eqn (1) and (2)). Furthermore, the by-product formic acid is also a valuable product and a potential hydrogen storage material.9 Herein, we synthesized ruthenium nanoparticles in situ from an organometallic ruthenium complex and a ligand and we utilized it to achieve efficient catalytic activity for producing hydrogen gas and formate/formic acid from methanol in water at lower temperatures (90–130 °C). The structure of the ruthenium catalyst was established by TEM, XPS, PXRD, ICP, and TGA and several reaction parameters were evaluated to achieve high catalytic activity for the low-temperature conversion of methanol to hydrogen gas and formate over the ruthenium catalyst in water.
Results and discussion
Initially, we employed [{(η6-benzene)RuCl2}2] ([Ru]-1) as a pre-catalyst for the hydrogen production from methanol (2:1 molar ratio of CH3OH:H2O) at 110 °C in the presence of 1.2 equiv. KOH, where we observed the release of 73 mol H2 per mol Ru (initial TOF: 9 h−1) (Table 1, entry 1). The evolved gas was identified as hydrogen by GC-TCD. Notably, the initial dark brown color of the reaction solution turned into a black suspension, which was identified as Ru nanoparticles (ca. 18.7 nm) by TEM (Fig. S1†). Further, recent studies revealed that pyridine-based ligands can act as an internal base and play a crucial role in the C–H activation reaction.36,37 We, therefore, investigated the role of 2-hydroxypyridine (L1) as a promoter in the ruthenium-catalyzed hydrogen production from methanol. For the Ru/L1 catalyst (pre-catalyst [Ru]-1 in the presence of L1), we observed a significant enhancement of ca. 82% in the initial TOF with the release of 0.66 mol H2 per mol methanol (a TON of 106 mol H2 per mol Ru) (Table 1, entry 2 and Fig. 1a). When the reaction was performed using a CH3OH:H2O molar ratio of 1:1, the intrinsic activity increased further yielding a TON of 134 mol H2 per mol Ru (0.83 mol H2 per mol methanol) with an improved initial TOF of 20 h−1 (Table 1, entry 5 and Fig. 1b). Notably, further increasing the water content (1:2 molar ratio of CH3OH:H2O) resulted in a lower catalytic activity for the H2 production from methanol over the Ru/L1 catalyst (Table 1, entry 6, and Fig. 1b), as compared to the reaction performed with a high methanol content (CH3OH:H2O molar ratio of 1:1, 2:1 or 4:1) (Table 1 and Fig. 1a). The literature reports on aqueous methanol dehydrogenation outlined the beneficial role of water in the methanol dehydrogenation reaction.11,14 For instance, Grützmacher et al. achieved dehydrogenation of methanol using a CH3OH/H2O 1:1 molar ratio, and mentioned that in the presence of water, a high gravimetric content of hydrogen can be achieved from methanol.11 Beller and other researchers also highlighted the role of water-promoted dehydrogenation of formaldehyde to formic acid and H2 during the methanol dehydrogenation reaction.25,38 Notably, Lin et al. also reported efficient hydrogen production from methanol over Pt/α-MoC with a higher water content (CH3OH/H2O molar ratio of 1:3 and 1:1).35
| Entry | Cat. | n(alc.)/n(H2O) | T (°C) | KOH (equiv.) | n(H2)/n(alc.) | n(H2)/n(cat.) | TOFh (h−1) |
|---|---|---|---|---|---|---|---|
|
a Reaction conditions: alcohol (16.08 mmol), Ru catalyst (0.625 mol%, n([Ru]-1)/n(L1) 1:2), KOH (1.2 equiv.), 10 h, argon.
b In the absence of ligand L1.
c Reaction with NaOMe in place of CH3OH.
d With [Ru]-2 and L1 (n([Ru]-2)/n(L1) 1:2).
e With the pre-synthesised Ru nanoparticles and L1 (1 equiv.).
f Using ethanol as a reactant.
g Using n-propanol as a reactant.
h Average turnover frequency (TOF) at 1 h.
|
|||||||
| 1b | Ru | 2:1 |
110 | 1.2 | 0.45 | 73 | 9 |
| 2 | Ru/L1 | 2:1 |
110 | 1.2 | 0.66 | 106 | 18 |
| 3 | Ru/L1 | 4:1 |
110 | 1.2 | 0.68 | 109 | 13 |
| 4 | Ru/L1 | Neat | 110 | 1.2 | 0.63 | 102 | 14 |
| 5 | Ru/L1 | 1:1 |
110 | 1.2 | 0.83 | 134 | 20 |
| 6 | Ru/L1 | 1:2 |
110 | 1.2 | 0.25 | 40 | 14 |
| 7c | Ru/L1 | 1:1 |
110 | — | 0.67 | 107 | 19 |
| 8 | Ru/L1 | 1:1 |
130 | 1.2 | 1.42 | 229 | 49 |
| 9 | Ru/L1 | 1:1 |
90 | 1.2 | 0.24 | 38 | 5 |
| 10d | Ru/L1 | 1:1 |
110 | 1.2 | 0.50 | 81 | 11 |
| 11e | RuNP/L1 | 1:1 |
110 | 1.2 | 0.13 | 21 | 12 |
| 12f | Ru/L1 | 1:1 |
110 | 1.2 | 0.61 | 98 | 20 |
| 13g | Ru/L1 | 1:1 |
110 | 1.2 | 0.69 | 111 | 16 |
 | ||
| Fig. 1 Effect of (a) ligand L1 and (b) methanol to the water molar ratio on the Ru/L1 catalyzed hydrogen production from methanol at 110 °C. (c) Comparative catalytic activity of various ruthenium catalysts with/without ligands for producing hydrogen from methanol at 110 °C (reaction conditions: methanol (16.08 mmol), Ru catalyst (0.625 mol%, n([Ru]-1)/n(L1) 1:2), KOH (1.2 equiv.), methanol to water molar ratio (1:1), argon; the amethanol to water molar ratio is 2:1, and the b[Ru]-2 precursor is used). | ||
Notably, we found that the base played a crucial role in generating the active Ru nanoparticles in the initial hour of the reaction. Moreover, lower activity was observed when using a less content of KOH (0.42 equiv.) (Table S1†). Results inferred that compared to KOH, the reaction with other bases such as NaOH, KtOBu, and K2CO3 exhibited either lower activity or no reaction (Table S2†). It is worth noting that an almost similar amount of H2 was released when NaOMe was used as a substrate instead of methanol, in the absence of a base, suggesting that presumably the content of the base is crucial for the deprotonation of methanol (Table 1, entry 7, and Fig. S2†). Moreover, kinetic isotope effect (KIE) studies indicated that CD3OD is more influential than D2O in tuning the reaction rate of the Ru/L1 catalyzed hydrogen production from methanol (Fig. S3 and Table S3†). These results inferred that the activation of methanol C–H bonds is presumably the rate-determining step and not the proton assisted release of hydrogen gas from methanol. Further, 13C NMR of the reaction aliquot after the completion of the catalytic reaction inferred the presence of formate with traces of carbonate (Fig. S4a†), suggesting that the decomposition of formate does not take place. Notably, performing the reaction with the spent Ru/L1 catalyst resulted in no formate decomposition and no traces of carbonate was detected in 13C NMR (Fig. S4b and S5, and Table S4†). Furthermore, the amount of formate formed as a co-product was also quantified to nearly half of the mol of H2 gas generated during the reaction (Fig. S5†). GC-TCD analysis of the evolved gas is in good agreement with the observation for purified hydrogen gas only (Fig. S6†). When the reaction temperature was increased to 130 °C, the initial TOF increased by over ten-folds to 49 mol H2 per mol Ru per hour as compared to the reaction performed at 90 °C (a TOF of 5 mol H2 per mol Ru per hour) (Table 1, entries 5, 8, and 9, and Fig. S7†). Notably, the Ru/L1 catalyst exhibited the generation of 1.42 mol H2 per mol methanol (∼71% conv.) (at 130 °C), which is several-folds higher than that reported for the 0.2%Pt/α-MoC catalyst (0.037 mol H2 per mol methanol at 190 °C).35 The apparent activation energy for the conversion of methanol to H2 over the Ru/L1 catalyst was estimated as 18.3 kcal mol−1 (Fig. S7†). Therefore, it is evident from these results that Ru/L1 is active even at lower reaction temperatures of 90–130 °C.
Further, to detect various species that might form during the dehydrogenation of methanol, the reaction mixture at various time intervals (0, 15 and 60 min) was analyzed by 1H and 13C NMR, where no traces of formaldehyde, methanediol, paraformaldehyde and trioxane were detected (Fig. S5†). Consistent with the literature reports, this can be attributed to the faster transformation of formaldehyde into formic acid and hydrogen in water as compared to methanol dehydrogenation to formaldehyde (Fig. S5†).11,18,38 Furthermore, we performed the catalytic dehydrogenation of aqueous formaldehyde (37 wt%) under analogous reaction conditions to methanol dehydrogenation and analyzed the evolved gas by GC-TCD and the reaction aliquots by NMR. Results inferred that indeed in the presence of a base, only pure hydrogen and formate was produced from formaldehyde dehydrogenation as confirmed by GC-TCD and NMR, respectively. Therefore, these results evidenced that during the methanol dehydrogenation under the optimized reaction conditions, the possible intermediates such as formaldehyde or methanediol formed during the reaction may also undergo faster transformation into formic acid and H2 gas, and therefore formaldehyde was not detected in the reaction solution. However, we have detected and isolated the formic acid generated during the methanol dehydrogenation reaction under the studied reaction conditions (Fig. S5†).
Further, to obtain insights into the role of the pyridine ligand, catalytic conversion of methanol to H2 was also examined using 2-methoxypyridine (L2) where a significant decline in the initial TOF (15 mol H2 per mol Ru per hour) was observed with the Ru/L2 catalyst as compared to the TOF of 20 mol H2 per mol Ru per hour obtained with the Ru/L1 catalyst (Table S5 and Fig. S8†). Further, the reaction performed using Ru/pyridine (L3) and Ru/phenol (L4) showed even lower activity (Fig. 1c, Table S5 and Fig. S8†). These observations suggested the possible involvement of the 2-hydroxypyridine ligand (L1) in promoting the facile activation of methanol over Ru nanoparticles to produce hydrogen gas with higher activity. Notably, performing the catalytic reaction with the larger content of the ligand L1 (Ru/L1 1:4) resulted in a lower activity yielding only 46 mol H2 per mol Ru in 8 h as compared to 62 mol H2 per mol Ru for the Ru/L1 ratio of 1:2, presumably because excess ligands may poison the catalyst (Fig. S9†). Moreover, using the [{(η6-p-cymene)RuCl2}2] ([Ru]-2) precursor instead of [Ru]-1 could not improve the catalytic activity (Table 1, entry 10, Fig. 1c and S10†). Further, performing the catalytic reaction in the presence of the pre-synthesized Ru nanoparticles with L1 resulted in lower activity, suggesting that in situ generation of Ru nanoparticles in the presence of L1 is the more suitable condition to generate active Ru catalysts (Table 1, entry 11, and Fig. 1c). Moreover, to know the nature of the real catalyst for methanol dehydrogenation, we performed a series of poisoning experiments using Whitesides' mercury test.25 Results inferred that the catalytic reaction quenched in the presence of Hg(0) (>300 equiv.) due to the poisoning of the Ru/L1 catalyst by amalgam formation. In contrast to the control reaction, the addition of Hg(0) (>300 equiv.) at the beginning of the reaction resulted in the complete quenching of the catalytic methanol dehydrogenation, suggesting the heterogeneous nature of the Ru/L1 catalyst (Fig. S11a†). Further, in another experiment, the spent Ru/L1 catalyst was also stirred with an excess of added Hg(0) (>300 equiv.), before employing it as a catalyst for the methanol dehydrogenation reaction under the optimized reaction conditions, but no gas release was observed (Fig. S11b†). It is worth noting here that the spent Ru/L1 catalyst was found to be highly active (in the absence of Hg(0)), supporting the heterogeneous nature of the studied catalysts (Fig. S11b†). Moreover, the Ru/L1 catalyst also exhibited appreciable long-term stability, where a total turnover number (TON) of 762 mol H2 per mol Ru was achieved in a 7-cycle recyclability experiment generating 186 L of H2 gas per gram of Ru (Fig. 2). On the other hand, continuing the reaction with the reaction solution (supernatant), obtained after the removal of the Ru/L1 catalyst, resulted in no release of gas even after extending the catalytic reaction for a longer duration (10 h) (Fig. S12†). The literature revealed that the reaction performed under reducing conditions may result in the transformation of the organometallic Ru precursor into Ru(0) nanoparticles.39–43 Chaudret et al. established the mechanism and the process for the transformation of organometallic ruthenium complexes into Ru nanoparticles.42,43 Similarly, some other groups have also established the transformation of organometallic ruthenium complexes into Ru nanoparticles.40 Therefore, the absence of the induction period during the in situ transformation of [Ru]-1 into Ru nanoparticles can be attributed to the highly reducing reaction conditions due to the presence of KOH and H2 gas at 110–130 °C. Further, ICP-OES analysis of the recovered nanoparticles inferred the presence of ∼54 wt% Ru, whereas no traces of metals were detected (a detection limit of 0.01 ppm) in the supernatant, indicating that the absence of ruthenium species in the reaction mixture also supports the complete transformation of the [Ru]-1 precursor into Ru nanoparticles. Moreover, the liquid portion (supernatant) was also analyzed by ESI-MS, where any isotopic pattern corresponding to ruthenium was not observed, further suggesting that the ruthenium complex is converted to ruthenium nanoparticles during the reaction and no residual ruthenium species is present in the solution (Fig. S13†). Therefore, the above findings evidenced the heterogeneous nature of the Ru/L1 catalyst for methanol dehydrogenation. Furthermore, ethanol and n-propanol were also investigated under catalytic reaction conditions and the results showed that the studied Ru/L1 catalysts can also efficiently generate hydrogen gas along with value-added products such as acetic acid (Table 1, entries 12 and 13, and Fig. S14†). The activity of the catalyst slightly decreased with subsequent catalytic runs, attributed to the surface oxide coating of the Ru catalyst, as also confirmed by XPS analysis (Fig. S15†). Although the turnover number was relatively low for the studied Ru/L1 catalyst, it is evident that the Ru/L1 catalysts exhibited the generation of higher equivalents of hydrogen gas per mole methanol (n(H2)/n(MeOH): 1.42) as compared to the earlier reported catalysts (Table S6†). Moreover, in the studied catalytic system, formate/formic acid is obtained as a valuable by-product. Advantageously, this resulted in the generation of hydrogen gas in high purity from methanol. Therefore, the studied Ru/L1 catalytic system could be a promising candidate for low-temperature bulk hydrogen production from methanol.
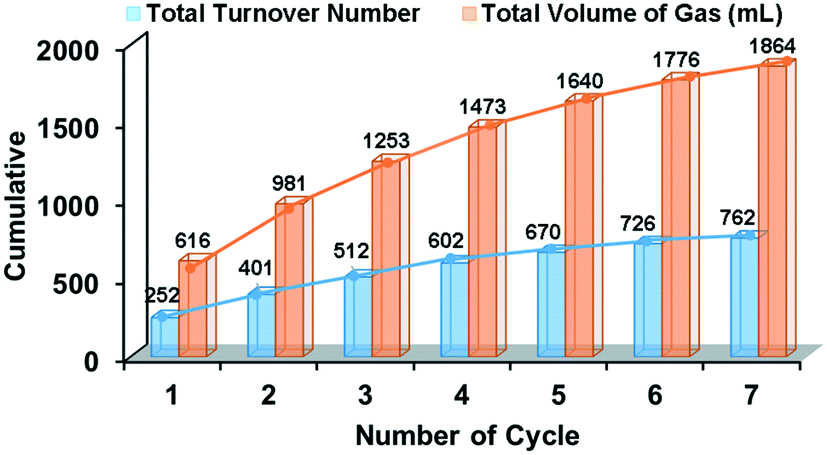 | ||
| Fig. 2 Long-term stability and recyclability experiment results for the Ru/L1 catalyzed production of H2 from methanol at 130 °C. | ||
To obtain insights into the structural and chemical natures of the in situ generated Ru/L1 catalyst, we employed several characterization methods. Powder X-ray diffraction of the Ru nanoparticles obtained after the catalytic reaction showed a broad peak at 25–45°, corresponding to the highly dispersed small Ru nanoparticles over the carbon support (Fig. S16†).44 Transmission electron microscopy (TEM) images confirm the existence of homogenously dispersed Ru nanoparticles with a particle size of ca. 1.5 nm on the carbon support (Fig. 3a and S17†). Furthermore, the dispersion of the in situ generated ruthenium nanoparticles in the presence of the L1 ligand was also estimated as 86%, as calculated from the generalized equations using the mean particle size (dTEM) as obtained by TEM by considering ∼110 particles (Table S7†).45,46 In sharp contrast, without the ligand, the Ru nanoparticles have an average particle size of ca. 18.7 nm with poor dispersion (Fig. S1†). High angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) images and energy-dispersive X-ray spectroscopy (EDS) analysis inferred the presence of the Ru element (Fig. 3b–d). Moreover, the EDS line scan also confirmed the even distribution of the Ru nanoparticles over the carbon support (Fig. 3d).
 | ||
| Fig. 3 (a) TEM image (inset is the particle size distribution), (b) EDS point analysis spectrum, (c) HAADF image and the corresponding (d) EDS line scan analyses for the Ru/L1 catalyst. | ||
Thermogravimetric analysis of the in situ generated Ru/L1 catalyst inferred the presence of a large organic content (∼30% more) as compared to the Ru nanoparticles obtained in the absence of ligand L1 (Fig. S18†). Further, the presence of 0.74% nitrogen content observed for Ru/L1 in the elemental analysis further suggests the presence of ligand L1 in the catalyst. Such a behavior is consistent with the stabilization of smaller Ru nanoparticles with the 2-hydroxypyridine ligand (L1). This phenomenon was also revealed earlier where Ru nanoparticles were stabilized by small ligands.47,48 In addition, the binding energy of Ru/L1 as obtained from X-ray photoelectron spectroscopy (XPS) experiment for both Ru(3d) and Ru(3p) core levels is assigned to the oxidation state of Ru in the catalyst using CuO as a standard (Cu2+3p3/2 = 933.5 eV in CuO). In the XPS spectra of the Ru/L1 catalyst, the peak maxima observed at the binding energy values of 461.7 eV (Ru 3p3/2) and 279.8 eV (Ru 3d5/2) are assigned to the metallic ruthenium (Fig. 4 and S16†). Moreover, the low-intensity XPS peak at 399.07 eV, corresponding to N 1s for the XPS of the Ru/L1 catalyst, indicated the presence of the ligand over the ruthenium nanoparticles (Fig. 4).
 | ||
| Fig. 4 XPS spectra corresponding to the (a) Ru 3p3/2 (b) Ru 3d5/2 and (c) N 1s core levels of the Ru/L1 catalyst. | ||
Notably, XPS of the Ru nanoparticles obtained in the absence of ligand L1 inferred the higher content of ruthenium oxide (Fig. S15†), further suggesting that the presence of ligand L1 over the Ru nanoparticles prevented the facile oxidation of the surface in the Ru/L1 catalyst. Hence, the observed enhanced catalytic performance of the Ru/L1 catalyst can be attributed to the smaller particle sized ruthenium nanoparticles and ligand L1.
Experimental
Catalytic hydrogen production from methanol
Typically, an appropriate amount of [{(η6-benzene)RuCl2}2] [Ru]-1 (0.05 mmol) (or other precursors) and a suitable ligand (0.1–0.2 mmol) were added in a methanol–water solution (n(CH3OH)/n(H2O) = 1:0 to 1:2), then it was placed in a 5 mL test tube reaction vessel and an appropriate base (1.1 equiv. with respect to methanol) was added. Then, the reaction vessel, equipped with a condenser (−10 °C) and water displacement setup, was de-aerated and flushed with Ar. Further, the reaction mixture was stirred at a suitable temperature in an oil bath (Fig. S19†). The amount of gas generated per unit time was quantified by the water displacement method, and the composition of the released gas was confirmed by GC-TCD. The turnover number (TON) was calculated by the formula [n(H2)/n(catalyst)]. The turnover frequency (TOF) was calculated as TON per time. After the catalytic reaction, the supported ruthenium nanoparticles were collected by centrifugation and dried in a vacuum oven and the weight was ∼14 mg of catalysts (as obtained from 25 mg of [Ru]-1 used in the catalytic reaction), which can be used for the further catalytic cycles.
Catalytic hydrogen production from ethanol/n-propanol
Catalytic hydrogen production from ethanol/n-propanol was performed by following the procedure used for methanol, by using ethanol/n-propanol (16.08 mmol), the Ru/L1 catalyst (0.625 mol%, n([Ru]-1)/n(L1) = 1:2) and potassium hydroxide (1.2 equiv.) in water (1 equiv.). Then, the 5 mL test tube reaction vessel, equipped with a condenser (−10 °C) and water displacement setup, was de-aerated and flushed with Ar. Further, the reaction mixture was stirred at 110 °C in an oil bath. The amount of gas generated per unit time was quantified by the water displacement method, and the composition of the released gas was confirmed by GC-TCD. The turnover number (TON) was calculated by the formula [n(H2)/n(catalyst)]. The turnover frequency (TOF) was calculated as TON per time.
Synthesis of ruthenium nanoparticles
Ru nanoparticles were synthesized by adding dropwise an aqueous solution of NaBH4 (0.025 g, in 5 mL of water) in an aqueous solution of RuCl3·3H2O (0.026 g, 0.1 mmol, in 5 mL of water) and PVP (0.05 g). The content of the flask was sonicated for 10 min to obtain a black suspension of Ru nanoparticles, which were collected by centrifugation and were washed with distilled water (10 mL × 02).Long-term stability and recyclability experiments
Initially, the Ru/L1 catalyst (0.625 mol%, n([Ru]-1)/n(L1) = 1:2) in methanol (16.08 mmol) and water (1 equiv.) was placed in a 5 mL test tube reaction vessel, and KOH (1.2 equiv.) was added. Then, the reaction vessel, equipped with a condenser (−10 °C) and water displacement setup, was de-aerated and flushed with Ar. Further, the reaction mixture was stirred at 130 °C in an oil bath. The amount of gas generated per unit time was quantified by the water displacement method. For the subsequent catalytic run, the reaction mixture was centrifuged to separate the catalyst. Further, the catalyst was transferred to the reaction vessel, and methanol (16.08 mmol), water (1 equiv.) and KOH (1.2 equiv.) were added to the reaction vessel, and the reaction mixture was stirred at 130 °C in an oil bath in an argon atmosphere. The release of gas was monitored by the water displacement process, and the composition of the released gas was confirmed by GC-TCD.
Conclusion
We developed an efficient new Ru/L1 catalyst that comprises ligand capped ruthenium nanoparticles homogeneously dispersed over the carbon support, in situ generated from the ruthenium arene precursor and 2-hydroxypyridine (L1) ligand. The Ru/L1 catalytic system displayed excellent catalytic activity for CO2 free hydrogen production from methanol (1.43 mol H2 per mol methanol) at low temperature (110–130 °C). This process also generated formic acid as a by-product. The studied catalyst exhibited outstanding long-term stability and generated 186 L of H2 gas per gram of Ru. This catalytic system may encourage the search for an efficient industrially viable process for the low-temperature transformation of methanol into hydrogen gas and formic acid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the SERB, the CSIR and the IIT Indore for financial support. The instrumentation facility at the SIC (IIT Indore), the MRC (MNIT Jaipur), the ACMS (IIT Kanpur) and the IKFT (KIT) are gratefully acknowledged. M. K. A. thanks the DST, New Delhi for his INSPIRE senior research fellowship.References
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schuth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- S. K. Singh and Q. Xu, Catal. Sci. Technol., 2013, 3, 1889–1900 RSC.
- S. K. Singh, A. K. Singh, K. Aranishi and Q. Xu, J. Am. Chem. Soc., 2011, 133, 19638–19641 CrossRef CAS.
- S. K. Singh and Q. Xu, J. Am. Chem. Soc., 2009, 131, 18032–18033 CrossRef CAS.
- S. K. Singh, X. B. Zhang and Q. Xu, J. Am. Chem. Soc., 2009, 131, 9894–9895 CrossRef CAS.
- S. Patra and S. K. Singh, Inorg. Chem., 2020, 59, 4234–4243 CrossRef CAS.
- L. E. Heim, N. E. Schlorer, J.-H. Choi and M. H. G. Prechtl, Nat. Commun., 2014, 5, 3621–3625 CrossRef.
- M. Trincado, V. Sinha, R. E. Rodriguez-Lugo, B. Pribanic, B. D. Bruin and H. Grützmacher, Nat. Commun., 2017, 8, 14990 CrossRef CAS.
- M. Trincado, H. Grützmacher and M. H. G. Prechtl, Phys. Sci. Rev., 2018, 3, 20170013 Search PubMed.
- L. E. Heim, D. Thiel, C. Gedig, J. Deska and M. H. G. Prechtl, Angew. Chem., Int. Ed., 2015, 54, 10308–10312 CrossRef CAS.
- F. F. van de Watering, M. Lutz, W. I. Dzik, B. de Bruin and J. N. H. Reek, ChemCatChem, 2016, 8, 2752–2756 CrossRef CAS.
- H. H. Kung and W.-H. Cheng, in Methanol production and use, ed. H. H Kung and W.-H. Cheng, Marcel Dekker, New York, 1994 Search PubMed.
- I. U. Dina, M. S. Shaharunb, M. A. Alotaibia, A. I. Alharthia and A. Naeem, J. CO2 Util., 2019, 34, 20–33 CrossRef.
- S. G. Jadhava, P. D. Vaidyaa, B. M. Bhanageb and J. B. Joshi, Chem. Eng. Res. Des., 2014, 92, 2557–2567 CrossRef.
- E. Alberico and M. Nielsen, Chem. Commun., 2015, 51, 6714–6725 RSC.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS.
- R. E. Rodriguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grutzmacher, Nat. Chem., 2013, 5, 342–347 CrossRef CAS.
- S. Sá, H. Silva, L. Brandão, J. M. Sousa and A. Mendes, Appl. Catal., B, 2010, 99, 43–57 CrossRef.
- W. Setthapun, S. K. Bej and L. T. Thompson, Top. Catal., 2008, 49, 73–80 CrossRef CAS.
- D. R. Palo, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS.
- P. Hu, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2014, 4, 2649–2652 CrossRef CAS.
- E. A. Bielinski, M. Forster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. C. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- C. Prichatz, E. Alberico, W. Baumann, H. Junge and M. Beller, ChemCatChem, 2017, 9, 1891–1896 CrossRef CAS.
- J. Campos, L. S. Sharninghausen, M. G. Manas and R. H. Crabtree, Inorg. Chem., 2015, 54, 5079–5084 CrossRef CAS.
- M. Anderez-Fernandez, L. K. Vogt, S. Fischer, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 559–562 CrossRef CAS.
- J. K. Lee, J. B. Ko and D. H. Kim, Appl. Catal., A, 2004, 278, 25–35 CrossRef CAS.
- M. Zhao, H. Zhang, X. Li and Y. Chen, J. Energy Chem., 2014, 23, 755–760 CrossRef.
- P. Du, P. Wu and C. Cai, J. Phys. Chem. C, 2017, 121, 9348–9360 CrossRef CAS.
- H. Mitani, Y. Xu, T. Hirano, M. Demura and R. Tamura, Catal. Today, 2017, 281, 669–676 CrossRef CAS.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS.
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489–494 CrossRef CAS.
- C. Binnani, R. K. Rai, D. Tyagi, S. M. Mobin and S. K. Singh, Eur. J. Inorg. Chem., 2018, 1435–1445 CrossRef CAS.
- M. Wakizaka, T. Matsumoto, R. Tanaka and H.-C. Chang, Nat. Commun., 2016, 7, 12333 CrossRef CAS.
- A. D. Dwivedi, R. K. Rai, K. Gupta and S. K. Singh, ChemCatChem, 2017, 9, 1930–1938 CrossRef CAS.
- J. A. Widegren, M. A. Bennett and R. G. Finke, J. Am. Chem. Soc., 2003, 125, 10301–10310 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS.
- K. Philippot, P. Lignier and B. Chaudret, Top. Organomet. Chem., 2014, 48, 319–370 CrossRef.
- L. M. Martínez-Prieto and B. Chaudret, Acc. Chem. Res., 2018, 51, 376–384 CrossRef.
- J. Ohyama, D. Kumada and A. Satsuma, J. Mater. Chem. A, 2016, 4, 15980–15985 RSC.
- G. Bergeret and P. Gallezot, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2008, pp. 738–765 Search PubMed.
- A. Borodzinski and M. Bonarowska, Langmuir, 1997, 13, 5613–5620 CrossRef CAS.
- J. Creus, S. Drouet, S. Suriñach, P. Lecante, V. Collière, R. Poteau, K. Philippot, J. García-Antón and X. Sala, ACS Catal., 2018, 8, 11094–11102 CrossRef CAS.
- J. García-Antón, M. R. Axet, S. Jansat, K. Philippot, B. Chaudret, T. Pery, G. Buntkowsky and H. H. Limbach, Angew. Chem., Int. Ed., 2008, 47, 2074–2078 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01470b |
| This journal is © The Royal Society of Chemistry 2021 |