
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdenum and tungsten carbides can shine too†
M.
Führer
,
T.
van Haasterecht
and
J. H.
Bitter
 *
*
Department of Agrotechnology and Food Sciences, Wageningen University and Research, PO Box 17, 6700 AA Wageningen, The Netherlands. E-mail: harry.bitter@wur.nl
First published on 7th September 2020
Abstract
In this perspective, we argue that carbides of transition metals such as molybdenum and tungsten hold great potential for the catalytic conversions of future feedstocks due to their ability to remain active in the presence of impurities in the feedstock. The presence of N and S impurities, found in increasing amounts in fossil-based feedstocks and also in new renewable feedstocks (such as biomass) may cause the carbides to convert to their respective nitrides or sulphides. These phases are catalytically active for similar reactions to the carbides and so these impurities would not lead to complete catalyst deactivation as they do for noble metal catalysts. Establishing the full potential of transition metal carbides as catalysts requires studies that use real feedstocks to look into the role of heteroatoms during the processing of fossil and novel feedstocks.
1. Introduction
Catalyzing chemical conversions to produce chemicals and fuels is becoming more demanding since the used feedstocks are changing. Fossil-based feedstocks are becoming heavier and now contain more impurities with N, S and P compounds1,2 and new renewable feedstocks such as biomass are emerging. An important class of catalysts used to convert the current feedstocks is based on metals (e.g. Pt, Ni, Co, Fe). Particularly noble metal catalysts are highly active and have been widely studied, but their limited availability is leading to an increasing demand for alternative catalysts and it also makes them expensive. In addition, noble metal catalysts have difficulty dealing with the impurities present in the newer feedstocks; these impurities interact strongly with the metal and cause catalyst deactivation.Transition metal carbides, such as those of molybdenum and tungsten, are considered viable alternatives to noble metal catalysts.3,4 This is based on the groundbreaking work by Levy and Boudart in 1973 who showed that W carbide and Pt have similarities in electronic structure and catalytic behaviour in the formation of water from H2 and O2 at room temperature.5 Subsequently, these carbides have been shown applicable for a wide variety of catalytic reactions, such as hydrogenation (HYD), hydrodeoxygenation (HDO), hydrodesulphurisation (HDS), hydrodenitrogenation (HDN) and isomerisation.1,6,7
In this perspective, we discuss the potential of tungsten and molybdenum carbides as an alternative for noble metal catalysts for use in the conversion of traditional fossil feedstocks, heavier fossil feedstocks and renewable biomass feedstocks. We point out the tolerance of transition metal carbides with respect to N and S impurities and the ability to (partially) convert the carbides to their respective nitrides or sulphides. The availability and catalytic activity of these carbide catalysts have both been mentioned before, but their relative stability in the presence of such impurities barely has. Although carbides can (partially) convert to nitrides and sulphides under the relevant reaction conditions, that does not necessarily lead to a complete loss of catalyst performance, as is the case for noble metals.4 Thus, taking the compositions of some fossil as well as most new feedstocks into account, Mo and W carbides may become the preferred choice over noble metals.
1.1 Need for non-noble metal catalysts
Noble metals (group 8 in the periodic table) are often used as catalytically active metals because of their good performance (activity/selectivity) and excellent stability.8 These metals are less prone to oxidation and therefore also less prone to leaching (dissolution) than non-noble metals, which is especially relevant for liquid-phase reactions. However, a major drawback of noble metals is their limited availability. Even though spent catalysts can and should be recycled, the expected growth of applications such as in electrolysers and fuel cells will require a vast amount of catalyst material and thus of noble metals.9–11Fig. 1 shows the abundance in the earth's crust of some relevant metals used in catalysis.12,13 As expected, each metal's abundance is inversely proportional to its price and its CO2 footprint, which makes non-noble metals an obvious choice as replacements for noble metals.14,15 Well known is the use of nickel for reactions for which Pt used to be the preferred catalyst.16–18 For example, Ni-based catalysts have been applied in the production of renewable hydrogen via aqueous-phase reforming.18–20 However, the stability of Ni for its use in the aqueous phase is still an issue.18 Cobalt and copper have also been considered as a replacement for noble metal catalysts. For example, cobalt and copper are highly active in the (steam) reforming of methane, methanol and ethanol.21
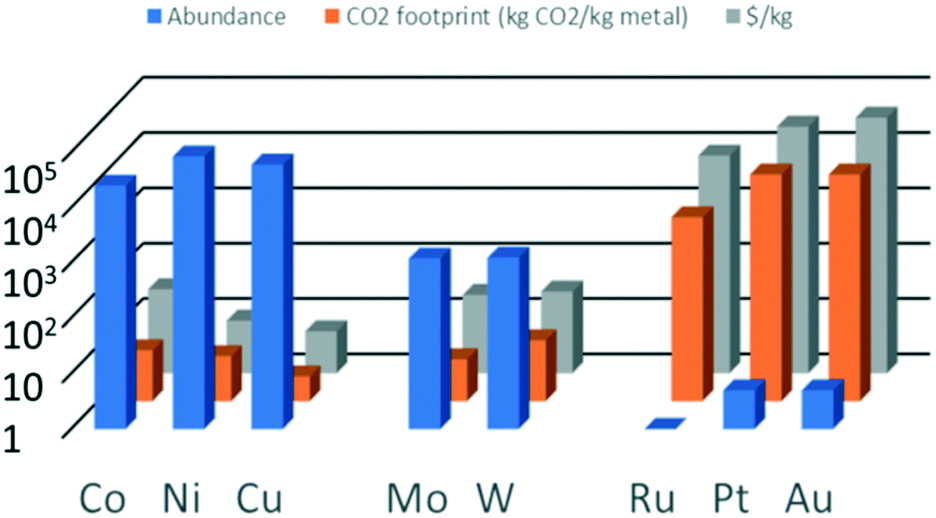 | ||
| Fig. 1 Abundance, CO2 footprint and price of some metals relevant for catalysis. | ||
Based on the abundance of the non-noble metals, iron (not shown in Fig. 1) is always the preferred choice as it is the most abundant of all metals (5% by weight). However, not all catalytic conversions can be performed with that single element. Therefore, a plethora of catalysts based on different metals is both needed and currently available. Next to an element's abundance on earth, the efforts associated with recovery, processing as well as accessibility are points of attention. For example, the European Union has published a list of critical raw materials.22 Their supply is not necessarily limited in terms of (future) abundance, but can also be restricted by their (currently) limited accessibility due to, for example, geopolitical circumstances. Therefore, adopting a general strategy of diversification, i.e. having alternatives for a given metal, is highly recommended. We argue that Mo and W, each having an elemental abundance that lies between that of the non-noble and the noble metals and a price comparable to that of non-noble metals, should be further explored as alternatives for noble metals.
2. State of the art of tungsten and molybdenum carbides
2.1 Mo and W carbides versus noble metal catalysts
Since the 1970s, tungsten and molybdenum carbides have emerged as alternatives for noble metal catalysts.23,24 From a characterisation point of view25–27 and from a performance5,28 point of view, these carbides resemble noble metals (especially Pt). For instance, in 1992, Oyama made a comparison between transition metal carbides and nitrides and noble metals.3 The crystallographic structure of these nitrides and carbides is determined by geometric and electronic factors. The geometric factor as identified by Hägg29 determines that simple structures (i.e. fcc, hcp and hex) are formed if the ratio between the atomic radii of the non-metal and the metal is less than 0.59; this is the case for carbides and nitrides of transition metals. The electronic factor finds its basis in the Engel–Brewer theory,30 which states that nitrogen and carbon atoms combine their valence s–p electrons with those of the interstitial sites of the host metal (s–d–p bands of Mo and W). This explains why the catalytic activity of carbides and nitrides resembles that of noble metals.3,4,31 Mo and W nitrides and carbides have excellent catalytic activities in reactions such as HYN, HDO, isomerisation and methanation. Particularly in HDO reactions, transition metal carbides reach yields close to those of noble metal catalysts. However, the selectivity of these newer catalysts differs from those of the noble metal catalysts.3 Some carbide catalysts enable unique reaction pathways that result in valuable products (e.g. enhanced HDO and isomerisation selectivity); this leads to the suggestion that carbides and nitrides catalysts can even be more desirable than noble metals.4 For instance, for the n-hexane reaction (614–630 K, excess H2), WC was twice as active as W2C and 0.5 wt% Pt/Al2O3 and showed enhanced selectivity for isomerisation products.3 Further, Stellwagen and Bitter32 studied the activity and selectivity of W and Mo carbide supported on carbon nanofibers in stearic acid HYD (batch reactor, 350 °C, 30 bar of H2 pressure). Interestingly, supported W carbides catalysts were selective (>50%) towards highly valued alkenes at the intermediate conversion level, while supported Mo carbides were selective for oxygenates (octadecanol and octadecanal). Both intermediate products (the alkenes and the oxygenates) are platform chemicals for synthesising a wide range of value-added products32 and are not normally observed with noble metal catalysts. For example, the conversion of stearic acid over Pd and Pt catalysts yields primarily heptadecane via the decarboxylation pathway.332.2 Challenges and opportunities for Mo and W carbides
Mo and W carbides are versatile and diverse catalysts, yet even after fifty years of research, captured in many reviews on the use of metal carbides in catalysis,4,6,7,34–42 it is still often unclear what the exact nature of the active site in these catalysts is. For example, Sullivan et al.7 reviewed the role of different synthesis techniques (temperature-programmed reduction and ultra-high vacuum) in the performance and characteristics of metal carbide catalysts, especially supported and unsupported W and Mo carbides. They emphasised that metal carbides undergo changes in their morphology and surface composition during synthesis and/or when used for a reaction under oxidative conditions. The authors stressed the need for in situ studies7 to enable preventing the influence of O2 on the material.Recently, a number of review papers have described the use of metal carbide-based catalysts to upgrade biobased feedstocks.6,7,41–43 For example, Chan-Thaw and Villa6 reviewed both the influence of the synthesis techniques (temperature-programmes reduction, ball milling and carbothermal hydrogen reduction) for unsupported and supported metal carbides and their use in the transformation of biomass to biofuels and fine chemicals. Examples of HYD and HDO of first-generation (vegetable oils) and second-generation (cellulose, hemicellulose and lignin) biomass with molybdenum and tungsten carbide catalysts were given. The carbides exhibit similar catalytic performances as Pt-group metals for HDO, HYD and isomerisation of biomass feedstocks. Although carbides are more resistant towards sulphur and nitride poisoning (see section 3), the carbide catalysts become deactivated due to coke deposition, leaching and over-reduction. The use of a support, e.g. a porous support (carbon nanotubes or mesoporous carbon), increases the stability of the carbides and leads to better control of the particle size.6,36
Pang et al.40 also reviewed the use of metal carbides (supported and unsupported W & Mo) for biomass conversion and indicated their great potential in HDO of cellulose, lignin and other platform chemical conversions. They emphasised that the use of carbides in biomass conversions needs to be explored more thoroughly and suggested to explore traditional carbides (Re, Ti, V or Zr carbides) further to gain a fundamental understanding regarding structure–performance relationships of Mo and W carbides for biomass conversions.40 In addition, Pang et al. looked at issues encountered during carbide synthesis.40 Obtaining highly dispersed and phase-pure (WC vs. W2C) metal carbides is still challenging. Carbon decomposition during the reaction and the existence of mixed carbide phases (WC or W2C) hamper the comprehension of the structure–performance correlation of these materials, which again stresses the need for in situ investigations.40,44
To gain more insight into the property–performance relationships of carbides, also quantum mechanical calculations have been applied. For example, Alaba et al.38 reviewed different density functional theory (DFT) studies on Mo carbide nanoparticles (as a catalyst for hydrogenation and hydrogen production) to identify and categorise the different existing Mo carbide phases. Depending on the preparation method and the carburisation agent, five different crystal structures were characterised: α-MoC1−x, α-Mo2C, β-Mo2C, γ-MoC and η-MoC. These structure differences do not only affect the stability of the catalyst but also influence the electrochemical activity. Among the different Mo carbide phases, α-MoC1−x and β-Mo2C are the most stable and they display remarkable catalytic behaviours in electrochemical catalysis due to their large ionic contribution.38
The aforementioned studies and reviews have demonstrated the applicability of transition metal carbides in a wide range of reactions and their potential as a replacement for noble metal catalysts for fossil and biomass feedstocks. However, almost all of these studies also highlighted the need for further (in situ) investigations to obtain information on structure–performance relationships.
2.3 Carbides, nitrides and sulphides in reactions involving H2 activation/transfer
Carbides are efficient catalysts for reactions involving H2 activation/transfer, i.e., carbides are able to split hydrogen and transfer the hydrogen atoms to different reactant molecules in a reversible manner. That makes them suitable catalysts for reactions that involve hydrogen activation, such as ammonia synthesis and decomposition, hydrogenation, hydrogenolysis, hydro-isomerisation, methanation and hydroprocessing.45 The ESI† contains a summary of those thermocatalytic reactions.It is noteworthy that aside from the transition metal carbides, also nitrides and sulphides have emerged as catalysts with activity for H2 activation and transfer reactions.31,46 For example, Monnier et al. tested γ-Al2O3-supported molybdenum, tungsten and vanadium nitrides for the HDO of oleic acid and canola oil.47 The molybdenum nitride showed a superior oleic acid HDO performance in comparison with the other nitrides; after 20 h of steady state conditions, the Mo nitride yielded a three times higher selectivity for n-alkane than was obtained by V and W nitride. Furthermore, the Mo nitride was operated for 450 h in the continuous operation of canola oil hydrotreatment with only minimal deactivation. Grilc et al.48 studied the hydrodeoxygenation of liquefied lignocellulosic biomass over unsupported MoS2, MoO2, Mo2C and WS2. MoS2 showed the highest hydrogenolysis selectivity as nearly 85% hydroxyl group conversion was reached within 30 min at 300 °C (∼30% for MoO2, ∼40% for Mo2C, ∼60% for WS).48 This shows that Mo and W nitrides and sulphides can be active for the same type of reactions as their respective carbides.
3. Potential of Mo and W carbide catalysts for use with S- and N-containing feedstock
Clearly, Mo and W carbides (as well as nitrides and sulphides) are versatile catalysts and potential replacements for noble metal catalysts even though the exact working mechanism and the nature of the active site of the carbide catalyst are not always known. However, with respect to stability, more research efforts are required.Often, four pathways are defined for catalyst deactivation i.e., 1) blocking of the active site (e.g. coke deposition), 2) crystallite growth, 3) leaching and 4) oxidation. These pathways have also been reviewed for metal carbides in liquid phase reactions by our group.36 For Mo and W carbides, minor changes in the atmosphere of the catalyst can already result in different catalytic behaviours. A prime example is the exposure to air which can change the surface of the carbide through the formation of oxy-carbides and oxides.39,49,50
3.1 S and N impurities in novel feedstocks
Sulphur and nitrogen compounds are present in fossil feedstocks such as crude oil and coal. The exact amounts of sulphur and nitrogen in crude oil and coal depend on origin and type. For example, Furimsky et al.51 stated that conventional crude oil contains 1.8 wt% sulphur and 0.1 wt% nitrogen, whereas the sulphur content of coal samples lies between 0.1 and 10 wt% depending on the source of the coal, while the nitrogen content ranging from 0.5 to 1.5 wt% (see ESI†).52–54 Traditional catalysts based on noble metals cannot cope well with these heteroatoms. Catalysts based on metals like Fe, Pt, Ru, Ni and Co as used in (de)hydrogenation, (steam) reforming and ammonia and Fischer–Tropsch (FT) synthesis are poisoned by H2S and NH3.55 The removal of such harmful impurities from the feedstock is therefore essential to maintain catalyst activity and selectivity. In current refineries, (reduced) metal catalysts are protected by upstream hydrotreating steps, i.e. hydrodesulphurisation (HDS) and hydrodenitrogenation (HDN).56–58Significant amounts of sulphur/sulphide (S) and ammonia (N) can also be present in biomass-derived feedstocks before and after processing (gasification, pyrolysis, digestion). According to Robinson et al., the sulphur content ranges from ∼14 to 2200 ppm depending on the biomass source and the season.59,60 Even pure vegetable oils still contain ∼10 ppmw of sulphur.61
Converting biomass or a fossil resource to syngas (H2/CO) followed by conversion of the syngas to the desired products (alcohols, alkenes, alkanes) via FT synthesis is gaining interest. When using syngas obtained from biomass, additional gas cleaning steps should be implemented to protect the FT catalyst (based on Fe or Co) against poisoning with S. This issue is exemplified by the increased interest in the fermentation of syngas62 as an alternative to metal-based conversions.
Mo and W carbide catalysts could be employed as alternatives for reduced metal catalysts in the upgrading of many of the biomass-derived sources (either syngas or other more complex molecules). The use of Mo2C in gas-to-liquid (GTL) processes has already been established for the synthesis of MeOH,63 higher alcohols64 and FT of fuel/diesel/hydrocarbons65,66 from pure syngas sources. The use of Mo and W carbides in the decarboxylation and hydrodeoxygenation of vegetable oils is another example.67 Both the hydrogenation of pyrolysis oil68 and reforming of methane69–73 with Mo2C and W2C have been demonstrated. To evaluate the true potential of Mo and W carbides, the effect of the S and N content in these feed sources needs to be taken into account.
3.2 Stability and activity of carbides, nitrides and sulphides
For reduced metal catalysts, the formation of a strong metal–S bond inevitably results in surface sulphidation and consequently in deactivation.74 Especially noble metals, such as platinum and palladium, suffer from deactivation in the presence of sulphur compounds.75 During simultaneous HDS, HDO and HYD (450 °C, 200 ppm O2, 5 wt% cumene, 95% tetradecane), a Pt catalyst supported on alumina deactivated immediately upon addition of sulphur.56Also the effect of nitrogen compounds on noble metal catalysts has been studied. Augusto et al. showed that Pt or Pd supported on zeolite Y catalysts used for tetralin HYD was deactivated by quinoline (499 ppm) as well as by dibenzothiophene (100 ppm).76 Similarly, the catalytic activity of Rh-based catalysts is strongly inhibited by sulphur compounds during methane oxidation or steam reforming.77 Already after addition of 1 ppmv of SO2 or H2S, the catalytic activity of Rh/γ-Al2O3 during the methane/steam reforming can become significantly decreased; after adding 10 ppmw, the catalytic activity decreases to zero.77 Transition metals such as Co and Ni are also prone to deactivation by sulphur poisoning. As an example, a drop in methanation activity of more than 3 orders of magnitude can occur for Co, Ni and Ru in the presence of only 15 ppb of H2S.78
For Mo and W carbides, their response to S and N impurities also needs to be considered. The presence of S or N in the feedstock can result in the transformation of carbide to a nitride and sulphide under relevant reaction conditions. Fig. 2 shows that the transition of MoS2 to Mo2C occurs in a temperature range of 400 to 700 °C in the presence of 0.01 to 10 ppm H2S, while the stability of WS2 already is affected at 600 °C at only 0.01 ppm. In general, equilibrium calculations using the HSC Chemistry package show that, thermodynamically, W2C is unstable in the presence of both S (as H2S) as well as N (as NH3) and strongly favours the formation of W sulphides and nitrides. Mo2C is only resistant to sulphur at very high temperatures and low sulphur concentrations (e.g. above 650 °C at 1 ppm H2S). Thus, the conversion of carbides to sulphides or nitrides is thermodynamically possible at conditions relevant for crude oil processing and biomass upgrading (with the exception of high-temperature steam reforming). It should be noted that the thermodynamic calculations are valid for the bulk phase and that the results will be different for more reactive (supported) nanoparticles. The rate at which a favourable transformation occurs also depends on other factors besides bulk versus supported catalyst, e.g., support type and particle size. Furthermore, partial transformations might occur, e.g., surface sulphidation might result in a passivation layer, serving as a diffusion barrier that prevents or slows down the sulphidation of the whole particle.79
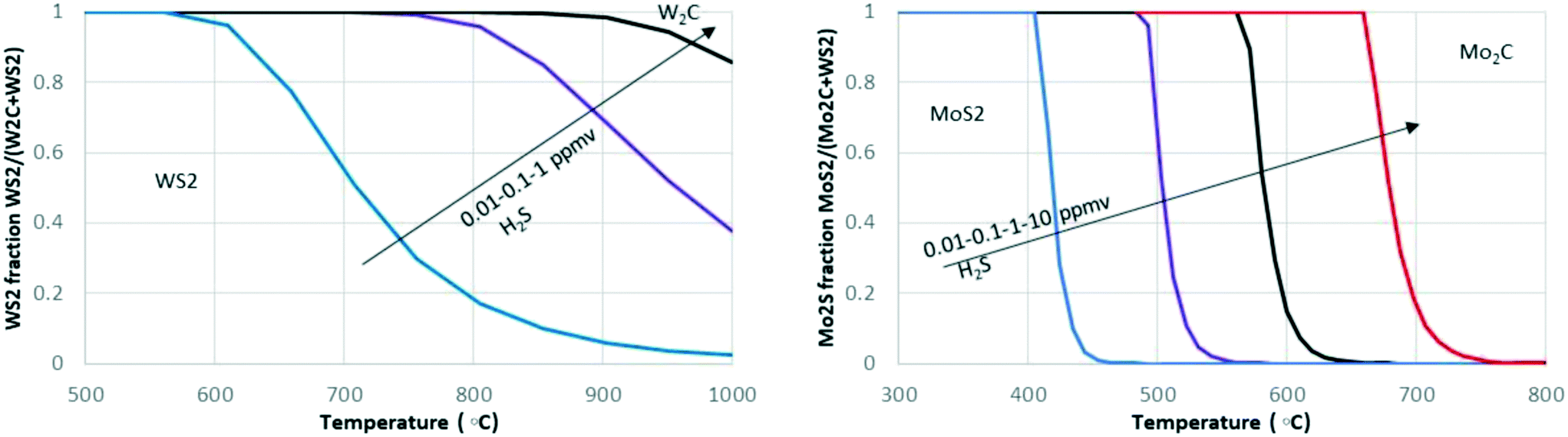 | ||
| Fig. 2 The stability regions for Mo and W sulphides and carbides as the fraction of sulphide versus temperature for WS2–W2C (left) and MoS2–Mo2C (right) in the presence of 0.01–10 ppm of H2S in H2 calculated with the HSC Chemistry software (metallic species omitted). | ||
Transformation from carbide to sulphide or nitride might, therefore, occur during processing of heavy oil-based feedstocks (which contain more sulphur)80–82 or biomass (which contains N and S in the feed). Interestingly, the (partial) conversion of Mo or W carbides to sulphides or nitrides should not necessarily result in deactivation because also the nitrides and sulphides are active for reactions involving hydrogen transfer (hydrogenation/dehydrogenation), as we already mentioned.56,83–86
This is further illustrated by the fact that prime examples of Mo catalysts are hydrotreating catalysts (CoMo- and NiMo-based), which are active in the sulphide state. That the relevant reactions are hydrogenation reactions shows that Mo sulphides could also play a role in the catalysis of other (de)hydrogenation reactions. For instance, the Mo sulphides traditionally used for hydrotreating reactions (NiMoS2, CoMoS2) are also active for HDO reactions of fatty acids and bio oils.87 In addition, there are examples of the use of W2C (ref. 58) and especially Mo2C (ref. 88–92) in HDS (see Table S1 in the ESI†). In addition, Mo and W nitrides are active for HDO, HDN and HDS reactions and in some cases (CoMo and NiMo) are even superior to the sulphide catalysts.93 Although Mo2C carbides display greater activity in HDS reactions than MoSx, the difference in activity can either be explained by the change in the number of active sites on the catalyst surface or is due to the difference in intrinsic chemical activity.79 However, this clearly shows that for Mo- and W-based catalysts, sulphur is not a poison, although it is for noble metal catalysts. For both the Mo carbide and the Mo nitride catalysts, their activity is retained when the surface becomes sulphated during HDS of thiophene at 400 °C.94
Considering the above, one might expect that Mo and W carbide and/or sulphide catalysts are a good choice for other hydrogen transfer reactions in which S or N impurities are expected. However, this potential advantage appears to have been overlooked so far since not many studies have focused on the effect of impurities. Therefore, for reactions other than HDS, only limited experimental information is available on the actual performance of Mo and W carbides in the presence of real feedstocks containing S and N. Already in 2002, Furimsky et al. mentioned that experimental information on hydrogen adsorption in the presence of H2S or by partially sulphided carbides and nitrites was lacking.1
A few studies have been conducted with bulk Mo2C and the addition of S compounds to the feed; Table 1 summarises them. Two of these studies (steam reforming at 1000 °C (ref. 71) and dry methane reforming at 1050 °C (ref. 73 and 95)) used conditions in which, according to Fig. 2, sulphide formation is not expected. Indeed, the authors reported no MoS2 formation for increased temperatures and the activity of the catalyst remained intact at high sulphur concentrations. On the other hand, the introduction of sulphur during steam reforming of MeOH resulted in an immediate but limited decrease in activity (of about 30%) and the formation of surface sulphur was detected with XPS.63 Also in the liquid phase HYD of furfural, trace amounts of S were found with XPS while the catalytic activity remained. In the hydrogenation of cumene, the formation of a surface carbosulphide phase was identified with XPS, while XRD showed that the bulk Mo2C was unaffected,56 revealing that only surface modification had occurred.
| Reaction | T/p | S-Source | Performance/remarks | Ref. |
|---|---|---|---|---|
| Steam reforming, oxidative-stream reforming of tri-methyl pentane | 1000 °C | ≤1000 ppm thiophene | - Activity remains up to 100 ppm | 95 |
| 1 bar | - Higher concentrations of S result in surface oxidation and coke formation | |||
| CO2/CH4 reforming | 1050 °C | 250–500 ppm dimethyl sulphide | - Mo2C deactivates at 800 °C due to reversible CS2 chemisorption | 73 |
| 1 bar | - Rh is S-tolerant under these conditions | |||
| Steam reforming of methanol | 185–240 °C | 5 ppm H2S | - S decreases the activity but the catalyst does not become fully deactivate | 63 |
| 1 bar | - Deactivation is reversible | |||
| Hydrogenation of cumene | 250 °C | 30–100 ppm S (thiophene, (di-)benzo-thiophene) | - With 30–60 ppm S, Mo2C is superior to noble metals | 56 |
| 51 bar | - At 100 ppm, Mo2C deactivates | |||
| - S present as surface sulphide | ||||
| Aqueous phase hydrogenation of furfural | 120–150 °C | 380 ppm thiophene | - Similar activity in presence and absence of S | 80 |
| 120 bar | ||||
| Steam reforming of hexadecane | 965 °C | 125–500 ppm benzo-thiophene | - Deactivation dependent on sulphur concentration | 101 |
| 1 bar | ||||
| - Deactivation minimal at S concentration below 100 ppm | ||||
| Water gas shift reaction | 200–240 °C | Without and with 5 ppm H2S | - MoS2 sites active in the presence of sulphur | 60 |
| 1 bar | ||||
| - Mo2C catalyst quickly poisoned by sulphur, but partly regenerated | ||||
| Partial oxidation of methane to syngas | 750 °C | 0.1% of H2S | - High S concentration leads to carbon deposition and sulphidation of the catalyst (catalyst deactivation) | 102 |
| 8 bar | ||||
| - At lower S concentration (<0.1%), no change in catalyst phase, but carbon acceleration on the reactor wall | ||||
| Tetralin hydrogenation | 300 °C | 200 ppm H2S (dimethyl-sulphide) | - Decease in tetralin conversion upon adding H2S | 96 |
| 40 bar | ||||
| - Minimal deactivation of supported Mo2C and WC | ||||
| - Near-complete deactivation of Pt/Al2O3 |
Thompson et al. investigated the effect of sulphur on Mo2C and Pt/Mo2C during the water–gas-shift reaction.60 Both catalysts became deactivated by exposing them to H2S but the Mo2C became only partially deactivated due to the formation of a still active MoS2 phase, while the Pt/Mo2C catalyst showed irreversible deactivation of the platinum. We found only one study that included Mo and W carbides as supported catalysts.96 In this work, da Costa et al. compared the effect of H2S on Mo2C and WC supported on Al2O3. The carbide activity was tested and compared in the presence and absence of 200 ppm H2S during tetralin HYD (300 °C, 4 MPa). In the absence of sulphur, the supported Mo2C and WC reached a yield of 6.5 mol% and 8 mol%, respectively. In the presence of sulphur, the conversion was slightly lower (4.5 mol% for Mo2C/Al2O3; 6 mol% for WC/Al2O3), but there was no complete deactivation of the carbide catalysts.96
While these studies concern very different reactions and conditions, they show that the carbides are able to tolerate sulphur in the feedstock. For the further development of stable carbide-based catalysts, it is essential to explore the mode of interaction of the N/S compound with the catalyst first. The partial transformation of a carbide to a sulphide or nitride might have a limited influence on catalytic performance as argued before; however, blocking of the active sites by a nitrogen/sulphur compound may still lead to deactivation. Therefore, a fundamental understanding of the interaction between the carbides surfaces and the sulphur/nitrogen-containing molecules is needed to understand the catalytic performance of these materials.4,97
A few articles have been published regarding the behaviour of Mo carbide towards sulphide adsorption/desorption.79 For instance, Rodriguez et al. used photoemission and XANES to establish the chemistry of SO2, H2S, and CH3SH on Mo carbide (and metallic Mo). The adsorption of SO2 on Mo2C at around −123 to 26 °C first led to the formation of SO3 and SO4 and, with increasing temperatures, to the formation of sulphided and oxidised carbides. The interaction between H2S and the Mo carbides was strong and led to substantial sulphidation, even at low temperatures. For the CH2S, the methyl group dissociated with increasing temperature while CHyS species coexisted on the carbide surface. However, the carbide was not modified by the sulphide. This shows that molybdenum carbides are either tolerant towards sulphur-containing molecules or form a still active MoSx phase, depending on the molecule in question and the temperature. Thus, the adsorption/desorption behaviour of carbide material depends on the sulphur compound and conditions (e.g. temperature).
Limited research has been conducted towards the effect of sulphur on bulk tungsten carbide.98–100 In 1981, Ko et al. published a study on the effect of oxygen and surface on the bonding and reactivity of carbon monoxide, hydrogen, formaldehyde, and methanol on tungsten and tungsten carbide surfaces. They found that the W(100)–(5 × 1)C surface rapidly adsorbed H2S which led to site competition with CO. The surface reactivity for formaldehyde and methanol decreased significantly after the sulphur treatment; however, it was still more active than metallic tungsten.98 Schulz-Ekloff and co-workers (1975) showed that in the presence of 25 μg m−2 H2S, the ammonia yield was reduced by 50%. After adding 50 μg m−2 H2S, the complete inhibition of the tungsten carbide occurred.99
In summary, based on thermodynamic arguments carbide catalysts might change the nature of their active site when used in the presence of S and N impurities during the processing of crude oil or renewable feedstock. However, they can remain catalytically active since the sulfide and nitride phases also possess activity for the same type of reactions. This distinguishes them from noble metal catalysts which quickly deactivate in the presence of sulfur or nitrogen containing compounds. The limited direct evidence available from experiments appears to support this view. Under which conditions the different types of carbides remain (partially) active and how the impurities interact with the carbides needs further investigation to define an operational window of the carbides.
4. Outlook
Future feedstock for chemicals and fuels, whether fossil or renewable in nature, will put new demands on the robustness of catalysts used in their processing. Tungsten and molybdenum carbides hold great potential for catalytic applications as replacements for noble metals. Both W and Mo have higher abundance and as a result lower cost compared to their noble metal counterparts. However, it has previously been stressed in a number of reviews that more detailed investigations into the nature of the active sites of these materials is still required.7,40,43It is our view that W and Mo carbides possess advantages beyond their availability and lower cost which should be the focus of further research to advance the field of W and Mo carbide catalysis and move towards industrial applications. We, therefore, stress that there is the need to study and gain more insight into the role of heteroatoms in the performance of carbide catalysts. We have argued that Mo and W carbide and/or sulphide catalysts can become the preferred choice for a wide range of hydrogen transfer reactions relevant to the upgrading of both novel crude oil and renewable feedstock in which S or N impurities are present. This is because Mo and W carbides are either less susceptible to poisoning by these S-containing compound compared to noble metals or can be converted into their still active sulfide/nitride phases. Although not many studies have focused on the effect of impurities, the limited experimental evidence available for mainly bulk molybdenum carbide in the presence of thiols/thioesthers supports this view.
Therefore, we propose that more studies are needed that focus on realistic feedstocks to show the true potential of these catalysts under real conditions, especially considering the effect that impurities like N and S in real feedstocks may have on the nature of the active site and the stability of these catalysts.
Secondly, more fundamental studies are still needed to understand the interaction of different S and N compounds, e.g. sulphide versus sulphate species, with the various types of Mo and W carbide catalysts. For this also the use of in situ surface-sensitive spectroscopic techniques would be required, understandably involving controlled conditions and the use of model compounds and feedstocks. In addition, theoretical calculations can provide us with a broader insight into adsorption of sulphur and sulfidation of Mo and W carbide catalysts in relation to their structural and electronic properties.
We propose these as the next avenues to explore. In any case, the potential for Mo and W carbides as supported catalysts is evident. It is not only noble metals that shine.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Dutch Research Council (NWO) for its financial support.References
- E. Furimsky, Appl. Catal., A, 2003, 240, 1–28 CrossRef CAS.
- M. Breysse, G. Djega-Mariadassou, S. Pessayre, C. Geantet, M. Vrinat, G. Pérot and M. Lemaire, Catal. Today, 2003, 84, 129–138 CrossRef CAS.
- S. Oyama, Catal. Today, 1992, 15, 179–200 CrossRef CAS.
- J. G. Chen, Chem. Rev., 1996, 96, 1477–1498 CrossRef CAS.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS.
- C. E. Chan-Thaw and A. Villa, Appl. Sci., 2018, 8, 259 CrossRef.
- M. M. Sullivan, C. J. Chen and A. Bhan, Catal. Sci. Technol., 2016, 6, 602–616 RSC.
- G. Ertl, H. Knözinger and J. Weitkamp, Handbook of heterogeneous catalysis, 1997 Search PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- R. Guil-Lopez, M. V. Martinez-Huerta, O. Guillen-Villafuerte, M. A. Pena, J. L. G. Fierro and E. Pastor, Int. J. Hydrogen Energy, 2010, 35, 7881–7888 CrossRef CAS.
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef.
- D. R. Lide, CRC handbook of chemistry and physics, CRC press, 2004 Search PubMed.
- P. G. Harrison, N. C. Lloyd and W. Azelee, Stud. Surf. Sci. Catal., 1995, 96, 487–496 CrossRef CAS.
- L. Shi-Yao and L. Bei-Lu, React. Kinet. Catal. Lett., 1996, 57, 183–190 CrossRef.
- J. Shabaker, G. Huber and J. Dumesic, J. Catal., 2004, 222, 180–191 CrossRef CAS.
- T. van Haasterecht, M. Swart, K. P. de Jong and J. H. Bitter, J. Energy Chem., 2016, 25, 289–296 CrossRef.
- T. Van Haasterecht, C. Ludding, K. De Jong and J. Bitter, J. Catal., 2014, 319, 27–35 CrossRef CAS.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180–191 CrossRef CAS.
- J. W. Shabaker, D. A. Simonetti, R. D. Cortright and J. A. Dumesic, J. Catal., 2005, 231, 67–76 CrossRef CAS.
- T. van Haasterecht, C. C. I. Ludding, K. P. de Jong and J. H. Bitter, J. Energy Chem., 2013, 22, 257–269 CrossRef CAS.
- B. G. S. EU Commission, Deloitte Sustainability, Bureau de Recherches Géologiques et Minières, Netherlands Organisation for Applied Scientific Research, 2017.
- L. Leclercq, K. Imura, S. Yoshida, T. Barbee and M. Boudart, Synthesis of new catalytic materials: Metal carbides of the group VI B elements, in Studies in Surface Science and Catalysis, Elsevier, 1979, vol. 3, pp. 627–639 Search PubMed.
- L. Volpe and M. Boudart, J. Solid State Chem., 1985, 59, 348–356 CrossRef CAS.
- L. H. Bennett, J. R. Cuthill, A. J. Mcalister, N. E. Erickson and R. E. Watson, Science, 1974, 184, 563–565 CrossRef CAS.
- W. Wu, Z. Wu, C. Liang, X. Chen, P. Ying and C. Li, J. Phys. Chem. B, 2003, 107, 7088–7094 CrossRef CAS.
- B. Ingham, C. D. A. Brady, G. T. Burnstein and M. P. Ryan, J. Phys. Chem., 2009, 113, 17407–17410 CAS.
- M. D. Scanlon, X. Bian, H. Vrubel, V. Amstutz, K. Schenk, X. Hu, B. Liu and H. H. Girault, Phys. Chem. Chem. Phys., 2013, 15, 2847–2857 RSC.
- G. Hägg, Z. Phys. Chem., Abt. B, 1931, 12, 33 Search PubMed.
- W. Hume-Rothery, Prog. Mater. Sci., 1968, 13, 229–265 CrossRef.
- J. Hargreaves, Coord. Chem. Rev., 2013, 257, 2015–2031 CrossRef CAS.
- D. R. Stellwagen and J. H. Bitter, Green Chem., 2015, 17, 582–593 RSC.
- M. Snåre, I. Kubičkova, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- A. L. Stottlemyer, T. G. Kelly, Q. H. Meng and J. G. G. Chen, Surf. Sci. Rep., 2012, 67, 201–232 CrossRef CAS.
- E. Lam and J. H. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS.
- L. S. Macedo, D. R. Stellwagen, V. T. da Silva and J. H. Bitter, ChemCatChem, 2015, 7, 2816–2823 CrossRef.
- G. A. Urzhuntsev, E. G. Kodenev and G. V. Echevskii, Catalysis in Industry, 2016, 8, 224–230 CrossRef.
- P. A. Alaba, A. Abbas, J. Huang and W. M. A. W. Daud, Renewable Sustainable Energy Rev., 2018, 91, 287–300 CrossRef CAS.
- E. Iglesia, F. H. Ribeiro, M. Boudart and J. E. Baumgartner, Catal. Today, 1992, 15, 307–337 CrossRef CAS.
- J. Pang, J. Sun, M. Zheng, H. Li, Y. Wang and T. Zhang, Appl. Catal., B, 2019, 254, 510–522 CrossRef CAS.
- Z. Lin, W. Wan, S. Yao and J. G. Chen, Appl. Catal., B, 2018, 233, 160–166 CrossRef CAS.
- A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS.
- K. J. Smith, Curr. Opin. Green Sustain. Chem., 2020, 22, 47–53 CrossRef.
- H. Fang, A. Roldan, C. Tian, Y. Zheng, X. Duan, K. Chen, L. Ye, S. Leoni and Y. Yuan, J. Catal., 2019, 369, 283–295 CrossRef CAS.
- S. T. Oyama, J. Solid State Chem., 1992, 96, 442–445 CrossRef CAS.
- A. B. Dongil, Nanomaterials, 2019, 9(8), 1111 CrossRef CAS.
- J. Monnier, H. Sulimma, A. Dalai and G. Caravaggio, Appl. Catal., A, 2010, 382, 176–180 CrossRef CAS.
- M. Grilc, G. Veryasov, B. Likozar, A. Jesih and J. Levec, Appl. Catal., B, 2015, 163, 467–477 CrossRef CAS.
- A. Kumar and A. Bhan, Chem. Eng. Sci., 2019, 197, 371–378 CrossRef CAS.
- M. M. Sullivan, J. T. Held and A. Bhan, J. Catal., 2015, 326, 82–91 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- T. Grzybek, R. Pietrzak and H. Wachowska, Fuel Process. Technol., 2002, 77, 1–7 CrossRef.
- J. Hou, Y. Ma, S. Li, J. Shi, L. He and J. Li, Fuel, 2018, 231, 134–144 CrossRef CAS.
- Z. Wang, Q. Li, Z. Lin, R. Whiddon, K. Qiu, M. Kuang and K. Cen, Fuel, 2016, 164, 254–261 CrossRef CAS.
- M. Argyle and C. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- B. Dhandapani, T. S. Clair and S. Oyama, Appl. Catal., A, 1998, 168, 219–228 CrossRef CAS.
- Y.-J. Zhang, Q. Xin, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Appl. Catal., A, 1999, 180, 237–245 CrossRef CAS.
- B. Pawelec, R. Mariscal, J. L. G. Fierro, A. Greenwood and P. T. Vasudevan, Appl. Catal., A, 2001, 206, 295–307 CrossRef CAS.
- J. M. Robinson, S. R. Barrett, K. Nhoy, R. K. Pandey, J. Phillips, O. M. Ramirez and R. I. Rodriguez, Energy Fuels, 2009, 23, 2235–2241 CrossRef CAS.
- J. A. Schaidle, A. C. Lausche and L. T. Thompson, J. Catal., 2010, 272, 235–245 CrossRef CAS.
- B. He, J. Van Gerpen and J. Thompson, Appl. Eng. Agric., 2009, 25, 223–226 Search PubMed.
- J. Daniell, M. Köpke and S. Simpson, Energies, 2012, 5, 5372–5417 CrossRef CAS.
- A. C. Lausche, J. A. Schaidle and L. T. Thompson, Appl. Catal., A, 2011, 401, 29–36 CrossRef CAS.
- Q. Wu, J. M. Christensen, G. L. Chiarello, L. D. L. Duchstein, J. B. Wagner, B. Temel, J. D. Grunwaldt and A. D. Jensen, Catal. Today, 2013, 215, 162–168 CrossRef CAS.
- D. V. N. Vo and A. A. Adesina, Fuel Process. Technol., 2011, 92, 1249–1260 CrossRef CAS.
- J. A. Schaidle and L. T. Thompson, J. Catal., 2015, 329, 325–334 CrossRef CAS.
- R. W. Gosselink, S. A. W. Hollak, S. W. Chang, J. Van Haveren, K. P. De Jong, J. H. Bitter and D. S. Van Es, ChemSusChem, 2013, 6, 1576–1594 CrossRef CAS.
- W. Li, Z. Zhao, P. Ren and G. Wang, RSC Adv., 2015, 5, 100865–100872 RSC.
- A. J. Brungs, A. P. E. York, J. B. Claridge, C. Márquez-Alvarez and M. L. H. Green, Catal. Lett., 2000, 70, 117–122 CrossRef CAS.
- J. Huang, T. Huang, L. Liu, W. Huang and R. Ma, Energy Sources, Part A, 2011, 33, 2249–2256 CrossRef CAS.
- H. Gao, Z. Yao, Y. Shi, R. Jia, F. Liang, Y. Sun, W. Mao and H. Wang, Inorg. Chem. Front., 2018, 5, 90–99 RSC.
- A. P. E. York, J. B. Claridge, C. Marquez-Alvarez, A. J. Brungs, S. C. Tsang and M. L. H. Green, Synthesis of early transition metal carbides and their application for the reforming of methane to synthesis gas, in Studies in Surface Science and Catalysis, Elsevier, 1997, vol. 110, pp. 711–720 Search PubMed.
- M. L. Pritchard, R. L. McCauley, B. N. Gallaher and W. J. Thomson, Appl. Catal., A, 2004, 275, 213–220 CrossRef CAS.
- J. Dunleavy, Platinum Met. Rev., 2006, 50, 110 CrossRef CAS.
- C. N. Satterfield, Heterogeneous catalysis in industrial practice, 1991 Search PubMed.
- C. C. C. Augusto, J. L. Zotin and A. da Costa Faro, Catal. Lett., 2001, 75, 37–43 CrossRef CAS.
- G. Mancino, S. Cimino and L. Lisi, Catal. Today, 2016, 277, 126–132 CrossRef CAS.
- F. H. Ribeiro, A. E. Schach von Wittenau, C. H. Bartholomew and G. A. Somorjai, Catal. Rev.: Sci. Eng., 1997, 39, 49–76 CrossRef CAS.
- J. A. Rodriguez, J. Dvorak and T. Jirsak, J. Phys. Chem. B, 2000, 104, 11515–11521 CrossRef CAS.
- Z. Li, J. S. Choi, H. Wang, A. W. Lepore, R. M. Connatser, S. A. Lewis, H. M. Meyer, D. M. Santosa and A. H. Zacher, Energy Fuels, 2017, 31, 9585–9594 CrossRef CAS.
- H. Wang and Y. Wang, Top. Catal., 2016, 59, 65–72 CrossRef CAS.
- X. Ma, L. Sun and C. Song, Catal. Today, 2002, 77, 107–116 CrossRef CAS.
- S. Boullosa-Eiras, R. Lødeng, H. Bergem, M. Stöcker, L. Hannevold and E. A. Blekkan, Catal. Today, 2014, 223, 44–53 CrossRef CAS.
- J. S. Lee, M. H. Yeom, K. Y. Park, I. S. Nam, J. S. Chung, Y. G. Kim and S. H. Moon, J. Catal., 1991, 128, 126–136 CrossRef CAS.
- P. Da Costa, C. Potvin, J. M. Manoli, M. Breeysse and G. Djega-Mariadassou, Catal. Lett., 2003, 86, 133–138 CrossRef CAS.
- F. E. Massoth and C. L. Kibby, J. Catal., 1977, 47, 300–315 CrossRef CAS.
- J. M. Christensen, L. D. L. Duchstein, J. B. Wagner, P. A. Jensen, B. Temel and A. D. Jensen, Ind. Eng. Chem. Res., 2012, 51, 4161–4172 CrossRef CAS.
- G. Jin, J. Zhu, X. Fan, G. Sun and J. Gao, Chin. J. Catal., 2006, 27, 899–903 CrossRef CAS.
- J. S. Lee and M. Boudart, Appl. Catal., 1985, 19, 207–210 CrossRef CAS.
- K. R. McCrea, J. W. Logan, T. L. Tarbuck, J. L. Heiser and M. E. Bussell, J. Catal., 1997, 171, 255–267 CrossRef CAS.
- H. K. Park, D. S. Kim and K. L. Kim, Korean J. Chem. Eng., 1998, 15, 625–630 CrossRef CAS.
- E. Puello-Polo, A. Gutierrez-Alejandre, G. Gonzalez and J. L. Brito, Catal. Lett., 2010, 135, 212–218 CrossRef CAS.
- G. Dolce, P. Savage and L. Thompson, Energy Fuels, 1997, 11, 668–675 CrossRef CAS.
- P. A. Aegerter, W. W. C. Quigley, G. J. Simpson, D. D. Ziegler, J. W. Logan, K. R. McCrea, S. Glazier and M. E. Bussell, J. Catal., 1996, 164, 109–121 CrossRef CAS.
- P. K. Cheekatamarla and W. J. Thomson, Appl. Catal., A, 2005, 287, 176–182 CrossRef CAS.
- P. Da Costa, J.-L. Lemberton, C. Potvin, J.-M. Manoli, G. Perot, M. Breysse and G. Djega-Mariadassou, Catal. Today, 2001, 65, 195–200 CrossRef CAS.
- P. Liu, J. A. Rodriguez and J. T. Muckerman, J. Mol. Catal. A: Chem., 2005, 239, 116–124 CrossRef CAS.
- E. Ko and R. Madix, J. Phys. Chem., 1981, 85, 4019–4025 CrossRef CAS.
- G. N. Schulz-Ekloff, D. Baresel and W. Sarholz, J. Catal., 1976, 43, 353–355 CrossRef CAS.
- M. Lewandowski, A. Szymanska-Kolasa, C. Sayag, P. Beaunier and G. Djega-Mariadassou, Appl. Catal., B, 2014, 144, 750–759 CrossRef CAS.
- P. K. Cheekatamarla and W. J. Thomson, J. Power Sources, 2006, 158, 477–484 CrossRef CAS.
- T. C. Xiao, H. T. Wang, A. P. E. York and M. L. H. Green, Catal. Lett., 2002, 83, 241–246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01420f |
| This journal is © The Royal Society of Chemistry 2020 |