
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural dynamics in Ni–Fe catalysts during CO2 methanation – role of iron oxide clusters†
Marc-André
Serrer
ab,
Abhijeet
Gaur
 ab,
Jelena
Jelic
b,
Sebastian
Weber
ab,
Charlotte
Fritsch
ab,
Adam H.
Clark
c,
Erisa
Saraçi
ab,
Felix
Studt
ab and
Jan-Dierk
Grunwaldt
*ab
ab,
Jelena
Jelic
b,
Sebastian
Weber
ab,
Charlotte
Fritsch
ab,
Adam H.
Clark
c,
Erisa
Saraçi
ab,
Felix
Studt
ab and
Jan-Dierk
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
cSuperXAS beamline, Paul Scherrer Institut (PSI), 5232 Villigen, Switzerland
First published on 30th September 2020
Abstract
Bimetallic Ni–Fe catalysts show great potential for CO2 methanation concerning activity, selectivity and long-term stability even under transient reaction conditions as required for Power-to-X applications. Various contrary suggestions on the role of iron in this system on CO2 activation have been proposed, hence, its actual task remained still unclear. In this study, we used X-ray absorption spectroscopy (XAS) combined with X-ray diffraction (XRD), XAS in combination with modulation excitation spectroscopy (MES) and density functional theory (DFT) to shed detailed light on the role of iron in a bimetallic Ni–Fe based CO2 methanation catalyst. During catalyst activation we observed a synergistic effect between nickel and iron that led to higher fractions of reduced nickel compared to a monometallic Ni-based catalyst. By XAS–XRD combined with DFT, we found formation of FeOx clusters on top of the metal particles. Modulation excitation coupled XAS data complemented with DFT calculations provided evidence of a Fe0 ⇌ Fe2+ ⇌ Fe3+ redox mechanism at the interface of these FeOx clusters. This may promote CO2 dissociation. This is the first time that this highly dynamic role of iron has been experimentally confirmed in bimetallic Ni–Fe based catalysts with respect to CO2 activation during the methanation reaction and may also be at the origin of better performance of other CO2-hydrogenation catalysts. The insight into the structural surface changes reported in this study show the dynamics of the Fe–Ni system and allow the development of realistic surface models as basis for CO2 activation and possible intermediates in these bimetallic systems.
Introduction
In future, the amount of renewable energy generated from wind and solar power will grow significantly.1 In order to maintain grid stability on the one hand and not to waste excess electricity on the other, there is the demand for efficient energy storage technologies.2 The “Power-to-X” concept describes an ideal case for short and long-term storage by sector coupling of electricity, heat, gas, chemistry and transport.3,4 Ideally, the (chemical energy) carriers suggested in this approach can be produced using CO2 as feedstock in order to close the carbon cycle. One part of this concept is “power-to-gas” in which methane is catalytically synthesized from CO2 and renewable hydrogen via the Sabatier reaction (eqn (1)):5,6| CO2 + 4H2 ⇌ CH4 + 2H2O ΔH298K = −165 kJ mol−1 | (1) |
Methane can be stored and transported cost efficiently to the end consumer via the already existing gas grid without the need of a new infrastructure. Furthermore, CH4 provides a three times higher volumetric energy density than H2 and can, in contrast to H2, be directly used in conventional gas power plants for reconversion into electrical power.7 However, the weather related generation of power and the thereto related generation of renewable H2 might lead to challenging operation conditions of the respective applied catalyst systems. This is critical, as catalysts are highly dynamic systems.8 For the industrial methanation of CO2 cheap and yet efficient Ni-based catalysts are widely used.9–15 Mutz et al.15–17 reported e.g. that γ-Al2O3 supported nickel-based catalysts tend to strongly deactivate during methanation of CO2 in the event of fluctuations.
Assuming that CO dissociation is the rate-determining step in CO2 methanation on metallic surfaces, Nørskov, Christensen and co-workers18,19 predicted and verified various alloy surfaces which might provide an enhanced CO and thus also CO2 methanation. Among them, Ni–Fe alloy catalysts appeared very attractive. In fact, recent studies demonstrate that Ni–Fe catalysts show both a high CO2 methanation activity and an improved long-term stability, even under dynamic conditions.20–25 Surprisingly, recent studies reported that nickel remains mainly reduced while iron seems to be oxidized during CO2 methanation, which contradicts other studies and requires modifications in the assumptions made in computational screenings.26–29 Various additional hypotheses are thus reported concerning the role of iron on enhanced CO2 activation, e.g. synergistic effects due to the alloy or the Ni–Fe(Ox) interface such as faster CO dissociation,18–20 enhanced reduction properties of Ni or improved CO2 dissociation.26,27 Further, cascade mechanisms have been considered, i.e. FexC as additional active sites,28 enhanced but limited CO2 dissociation on Fe2+ under irreversible formation of Fe3+,28 or protection of the active Ni0 centers by iron.21,30 Recently, Burger et al. suggested via ex situ studies that segregation of Fe to the particle surface under formation of Fe2+ might provide redox active sites for enhanced CO2 activation.29
In conclusion, the detailed role of iron in bimetallic Ni–Fe catalysts during CO2 methanation remains unclear and requires monitoring of the structure of iron under reaction conditions. This is challenging, as the structure of both iron and nickel are very dynamic. We shed here more light on structural changes of these catalysts in detail by applying advanced synchrotron-based operando tools. Combined X-ray absorption spectroscopy (XAS) and X-ray diffraction (XRD) was used to monitor changes in the amorphous and crystalline bulk phases as they can be applied under realistic reaction conditions.21,31–33 Further, surface sensitivity of these methods was enhanced by coupling modulation excitation spectroscopy with XAS, a technique that has received strong attention in catalysis recently.34–36 With this method, “spectator species”, i.e. species not taking part in the reaction, are filtered out after periodically stimulating active species by alternatingly applied external conditions (e.g., concentration, reactants, temperature, pressure, pH).34–40 To further complement and substantiate the experimental findings, we used density functional theory DFT calculation and developed structural models. With this we were able to get new insight into the role and dynamic surface changes of iron within the bimetallic Ni–Fe CO2 methanation catalyst under realistic reaction conditions.
Experimental
Catalyst preparation
A 17 wt% Ni/γ-Al2O3 and 17 wt% Ni3Fe/γ-Al2O3 catalyst were synthesized via homogeneous precipitation with urea similar as in previous publications.20,21 To ensure an appropriate absorption step during the synchrotron radiation experiments, the catalysts have been diluted with γ-Al2O3 (1/8′′ pellets, Alfa Aesar, crushed to <100 μm, calcined at 600 °C (5 K min−1), 4 h) using a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3.
:1.3.
Operando setup for the combined X-ray based characterization at synchrotron radiation facilities
A schematic drawing of the setup used in the experiments presented in this study is given in Fig. S1.† A micro quartz capillary reactor (1.5 mm diameter, 20 μm wall thickness, 1 cm catalyst bed length, 100–200 μm catalyst sieve fraction) heated by an Oxford GSB-1300 hot air blower was used for all experiments. The gases were dosed with mass flow controllers by Bronkhorst that were calibrated prior to the experiments using a gas flow calibrator by Mesa Labs. For the MES experiments, a pneumatic 4-way valve by Swagelok was used to ensure fast switches (down to 1 s) between the two gas mixtures that were adjusted separately on two different gas lines.Combined operando XAS–XRD (BM31, ESRF)
The combined XAS & XRD experiments were performed at the BM31 beamline41 at the European Synchrotron Radiation Facility (ESRF). XAS spectra were recorded in transmission mode at the Ni K-edge (8333 eV) and Fe K-edge (7112 eV) using a double-crystal Si(111) monochromator. The beam spot size at the sample position was 0.5 mm × 4 mm (v × h). XRD data was collected with a 2D DEXELA detector using a Si(111) channel-cut monochromator, set at a wavelength of 0.4943 Å (beam size 1.0 mm × 1.0 mm). The XAS–XRD measurements were performed quasi-simultaneously in loop: first XRD data were recorded for 3 minutes (5 diffraction patterns), then X-ray absorption spectra. Switching between XAS and XRD measurements took 2 minutes before and after the XRD. Due to the low signal-to-noise quality at the Fe K-edge two X-ray absorption near edge spectra (XANES) were recorded for 5 minutes, followed by four Ni K-edge extended X-ray absorption fine structure (EXAFS) spectra for 4 minutes, respectively. For data analysis, the spectra were averaged resulting in a time resolution of ≈29 minutes.The catalyst was activated by temperature-programmed reduction in 50% H2/N2 (H2-TPR), 2 h at 500 °C, heating rate 10 K min−1 at 6 bar. The total gas flow in the capillary was adjusted to 20 mL min−1. The product feed was additionally diluted after the reactor with a constant flux of 40 mL min−1 N2 in order to ensure an adequate gas flow for the μ-GC sampling. Before and after the H2-TPR, XAS and XRD were recorded at room temperature (RT). The catalyst was heated under TPR conditions to 250 °C at 1 bar, and afterwards the gas feed was changed to 25 vol% H2:CO2 = 4:1 in N2 to start the methanation of CO2. To exclude any temperature effects, the catalyst was cooled down to RT after 60 minutes of reaction and structural changes were monitored at RT by XAS–XRD. This procedure was repeated for the methanation of CO2 at 350 °C and 450 °C, respectively.
For calibration and fingerprinting, XAS spectra of reference compounds NiO, NiCO3, FeO, γ-Fe2O3, as well as Fe and Ni foils were measured. Using the Athena software of the Demeter package (version 0.9.25)42 the XAS spectra at Ni and Fe K-edges were energy calibrated to the respective metal foils, background subtracted and normalized. The normalized XANES spectra were analyzed by the linear combination fitting (LCF) module of Athena in the energy interval from −20 to +50 eV using the spectrum of the fully oxidized sample after calcination at 500 °C in air and the spectrum of a Ni foil as references. For EXAFS analysis, Artemis software was used to fit the models to the experimental data obtained during different stages of catalytic treatment. Model structures of Ni/Fe oxides, Ni/Fe metals and Ni–Fe alloy have been used to fit the EXAFS data in R-space for determining the structural parameters. These parameters include energy shift of the path (ΔE0), change in the half path length (ΔR), amplitude reduction factor (S02), number of identical paths (N) and relative mean-square displacement of the atoms included in path (Debye–Waller factor, σ2). Further details about the EXAFS data analysis are given in the ESI,† section 3.2.1.
The 2D-XRD diffraction patterns were averaged and azimuthally integrated using the software pyFAI (v 0.15.0).43
Powder XRD (PXRD) patterns were refined by the Rietveld method using the FullProf software package.44 An instrumental resolution file was first obtained by profile fitting of a LaB6 NIST 640b standard to correct instrumental line broadening. The wavelength used for refinements is 0.049434 nm. The refinement was conducted using a Thompson-Cox-Hastings pseudo-Voigt description of the profile. The background was defined by linear interpolation between background points with refinable heights. A polynomial convolution was initially tested but could not describe the background well. A stack of the full range of all five refined diffraction patterns is shown in ESI† in section 3.2.2. Due to the broad feature at about 7° caused by the capillary the refinement was performed for a range from 10 to 30°. Initial structural models for γ-Al2O3 by Zhou et al.45 and Ni by Rouquette et al.46 were used from literature data. Further information on XRD evaluation and calculations based on Vegard's law is provided in the ESI.†
Modulation excitation spectroscopy during QEXAFS experiments (SuperXAS, SLS)
MES was performed in quick scanning EXAFS (QEXAFS)47,48 mode at the Super-XAS beamline49,50 at the Swiss Light Source Synchrotron (SLS), Paul Scherrer Institute (PSI). The 17 wt% Ni/Al2O3 and 17 wt% Ni–Fe/γ-Al2O3 catalysts were activated by H2-TPR in 20 mL min−1 50% H2/N2, 2 h at 500 °C, 10 K min−1 heating rate at 1 bar. The MES experiments were performed at 250 °C, 350 °C and 450 °C for each catalyst. After the activation, the catalysts were heated to the desired reaction temperature under reducing conditions. Subsequently, the reaction was started by switching to methanation conditions (50 mL min−1 25 vol% H2:CO2 = 4:1 in N2). The catalytic performance was monitored for 1 h under steady state conditions. Afterwards, modulations were applied by periodically switching between 50 mL min−1 25 vol% H2:CO2 = 4:1 (60 s) and 20 vol% H2 in N2 (60 s) using a pneumatic 4-way valve while simultaneously recording the corresponding XAS data and writing the valve position in the files (details see ESI,† section 2.1). For collimation of the X-ray beam, prior to monochromatisation, a Si coated mirror at 2.9 mrad was used. The QEXAFS monochromator was equipped with the Si(111) channel-cut crystal scanning at 4 Hz. The subsequent monochromatic beam was focused onto the sample position using a toroidal Rh coated mirror. XAS spectra were collected in transmission geometry using a simultaneously measured Ni foil for energy calibration. One period including both conditions took 120 s in which 480 EXAFS spectra were recorded. 30 periods were applied and recorded at each temperature step. The energy range of the recorded transmission mode XAS spectra was adjusted to cover both the Fe and Ni K-edge in one scan.
After the MES experiment, methanation conditions were applied for 1 h to follow changes in the catalytic activity under steady state conditions after the modulations. Subsequently, the MES experiment was repeated at 50 °C as blind test.
The recorded spectra were energy calibrated, normalized and exported as normalized μ(E) files using the “ProXAS-GUI” software (version 2.9).51 As in case of the bimetallic catalyst the Ni and Fe K-edges were recorded in one scan, the corresponding data files were cut in two after calibration and normalization. One period was divided into 24 sections (resolution 5 s). Each section contained 20 spectra representing a step of 0.25 s, respectively. These 20 spectra per section were averaged to obtain one “time-resolved spectrum” per section. Subsequently, to enhance the signal-to-noise ratio, the time-resolved spectra were averaged over all 30 periods. Phase-resolved spectra were obtained by correlation to periodic modulations using eqn (2).34,35
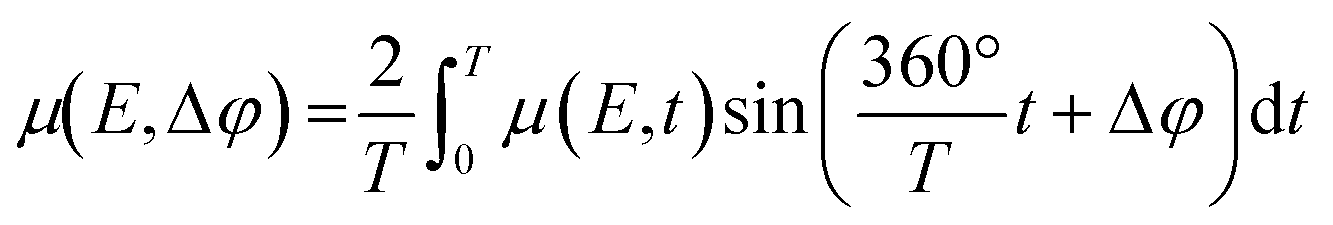 | (2) |
Product analysis
All experiments presented in this study were performed operando. For this purpose, the gaseous products were analyzed on-line using a ThermoStar GSD 320 mass spectrometer by Pfeiffer Vacuum and an Agilent 490 μ-GC (channel 1: 10 m PoraPLOT Q, 0.25 mm diameter, carrier gas helium; channel 2: 10 m mole sieve column with 5 Å, 0.25 mm diameter, carrier gas argon). N2 was used as internal standard during all experiments. The micro-GC measured with a time-resolution of 4 minutes per scan (1 minute sampling, 3 minutes analyzing). The conversion X(CO2) and selectivity S(CH4) were calculated using eqn (3) and (4):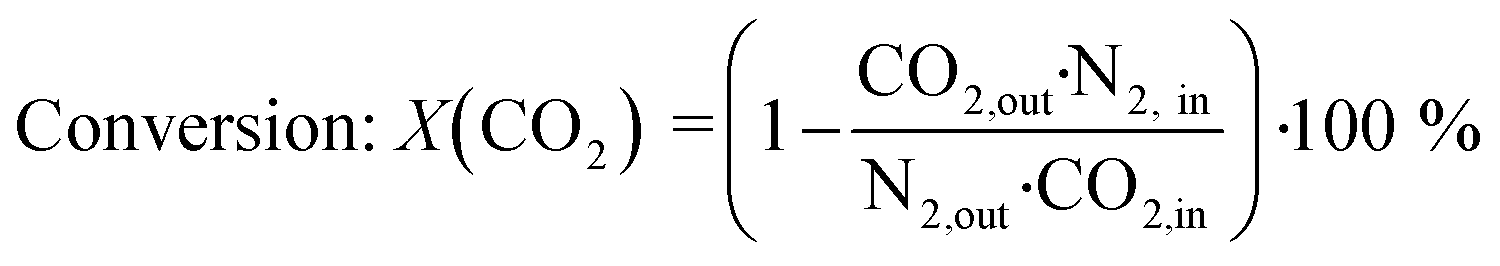 | (3) |
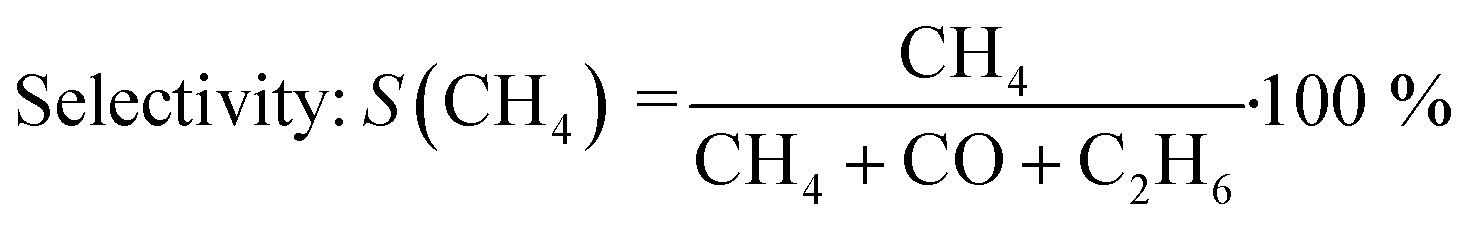 | (4) |
DFT calculations on the stability of NiFe and FeOx on Ni and NiFe
Periodic DFT calculations were performed with the projector-augmented-wave (PAW) method using the Vienna ab initio simulation package (VASP) in version 5.4.1 as well as the standard VASP-PAWs53,54 in connection with the Atomic Simulation Environment (ASE).55 The Bayesian error estimation functional with van-der-Waals correlations (BEEF-vdW)56 together with a plane-wave cutoff energy of 450 eV was used. The GGA+U method57,58 was used in order to improve the description of delocalized Fe d states. Using the reduction energy of FeO we obtained a U of 2.7 eV (See ESI†). Lattice constants for Ni, Ni3Fe and FeO were calculated with a 9 × 9 × 9 Monkhorst–Pack k-point grid.59 The optimized lattice constants are 3.538, 3.599 and 4.363 Å for Ni, Ni3Fe and FeO, respectively. Infinite slab models with a thickness of four layers and separated by more than 15 Å of vacuum were used to represent Ni3Fe(111). The slabs were of the size 2 × 2 in x and y-direction and sampled with a Monkhorst–Pack k-point grid of 6 × 6 × 1. A slab with 5 × 2 atoms in x and y-direction with a k-point sampling of 2 × 6 × 1 was used to model the Ni4Fe(111) surface containing 2 Fe and 8 Ni atoms in each layer of the slab. (FeO)4/Ni4Fe(111) and (FeO)6/Ni4Fe(111) contain FeO periodically repeated in x direction with four and six FeO units per unit cell, respectively. In all calculations, the bottom two layers were kept fixed at the bulk positions whereas all other atoms were allowed to relax during geometry optimization. Convergence criteria of 10−7 eV and 0.01 eV Å−1 were applied to SCF cycles and geometry optimization, respectively. Spin polarization was considered in all calculations. Vibrational analyses were carried out in the harmonic approximation using a finite difference with a magnitude of displacements of 0.01 Å. Ab initio XANES calculations were performed using the DFT optimized model structures employing the FEFF9 code.52 More details can be found in the ESI.†Results and discussion
Catalyst activation and Ni–Fe alloy formation
To follow a possible alloy formation, the activation of the Ni–Fe/γ-Al2O3 catalyst was monitored by combined XAS–XRD during temperature-programmed reduction in H2. Further, the results of the Ni–Fe/γ-Al2O3 catalyst were compared to a monometallic Ni/γ-Al2O3 catalyst to elucidate an influence of iron on the reduction properties of nickel (Fig. 1).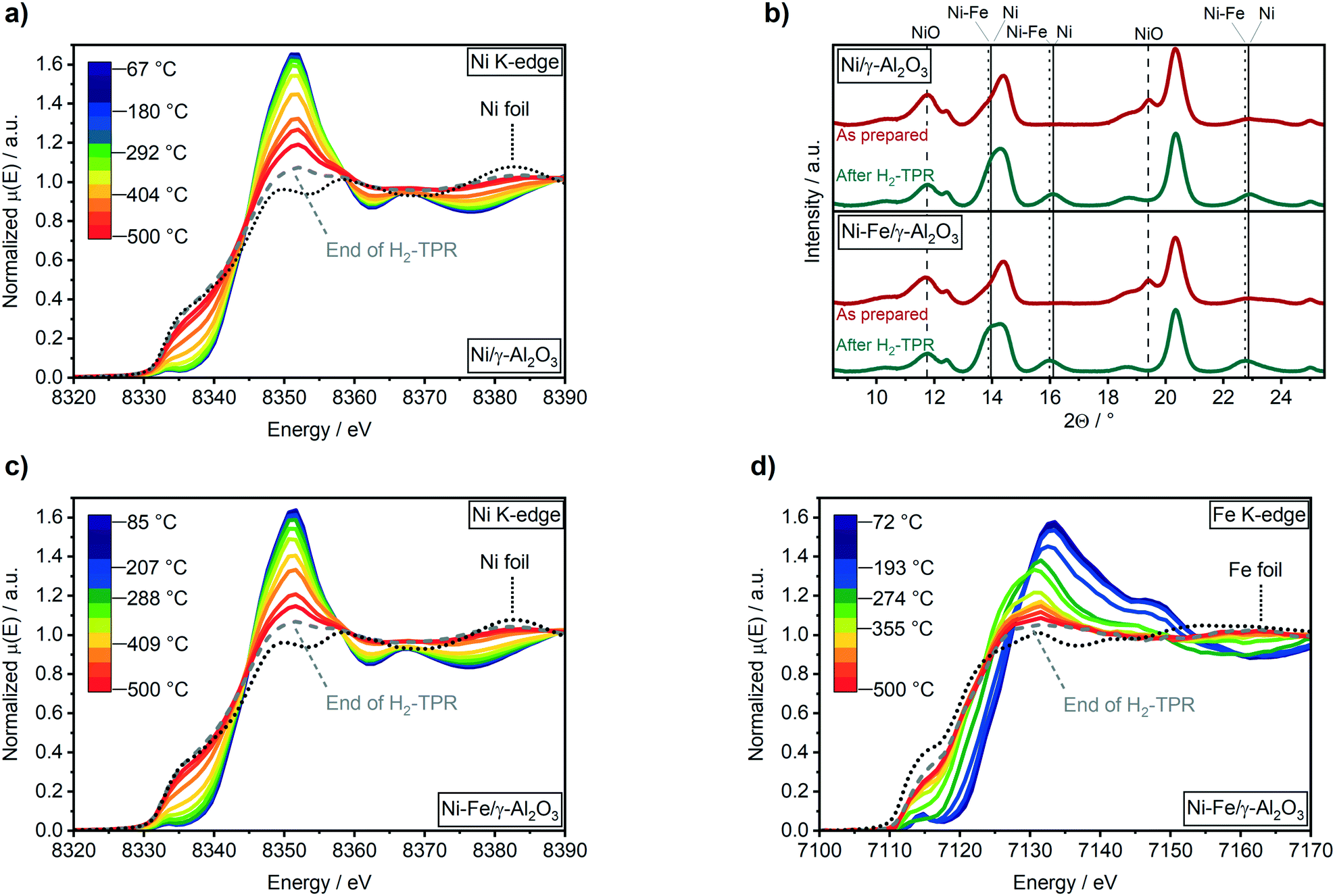 | ||
| Fig. 1 H2-TPR recorded in transmission mode at the Ni K-edge of the 17 wt% Ni/γ-Al2O3 catalyst (a) and at the Ni (c) and Fe (d) K-edge of the 17 wt% Ni–Fe/γ-Al2O3 catalyst using 50% H2 in N2 with a total flow of 20 mL min−1 from RT to 500 °C (10 K min−1). (b) XRD patterns (λ = 0.4943 Å) of the 17 wt% Ni/γ-Al2O3 catalyst (top) and 17 wt% Ni–Fe/γ-Al2O3 catalyst (bottom) as prepared (red) and after (green). | ||
The reduction of the Ni/γ-Al2O3 catalyst was initiated between 290 °C and 330 °C, as depicted by the decrease in the white line intensity at 8352 eV and the shift to lower energies of the pre-edge feature in the Ni K-edge XANES spectra (Fig. 1a). Contribution of about 86% of Ni0 after the reduction step was estimated by linear combination analysis (LCA) after the H2-TPR. Ni reflections at 2Θ = 13.9° and 16.1° were observed in the XRD patterns (Fig. 1b) at temperatures above 390 °C (Fig. S2a, ESI†). Simultaneously, NiO reflections at 2Θ = 11.7° and 19.5° declined. The formation of Ni0 in the Ni–Fe/γ-Al2O3 catalyst was also observed around 290 °C to 330 °C (Fig. 1c), while bulk formation was found above 380 °C in the XRD data at 2Θ = 13.8° and 16.0° (Fig. S2b† and 1b). About 96% of reduced nickel were obtained after 2 h reduction at 500 °C on the bimetallic Ni–Fe catalyst (86% Ni0 for Ni/γ-Al2O3).
EXAFS fitting results at Ni K-edge for Ni–Fe catalyst (ESI,† Table S1) also confirms presence of stable Ni metal phase in bimetallic catalyst after H2-TPR and methanation cycles at different temperatures. Hence, the presence of Fe close to Ni(O) atoms improved reduction of nickel during the activation procedure due to a lower activation energy on mixed Ni–Fe-oxides, as proposed by Unmuth et al.60 As there was no shift observed in the reduction temperature of nickel, iron might especially enhance the reduction of the smallest nickel particles that are harder to reduce.
Starting from Fe3+ state in γ-Fe2O3 (Fig. S2c†), the reduction to Fe2+ was visible in XANES from around 85 °C (Fig. 1d). Further reduction of Fe2+ to Fe0 was observed above 315 °C, as depicted by the shift of the pre-edge feature from 7115 eV to lower energies (Fig. 1d). A maximum reduction of up to 75% compared to the metallic state of iron was found after catalyst activation (H2-TPR). No reflections for any iron or other phase were found in the XRD data. However, a shift of the Ni reflections to lower angles was found in presence of Fe (Fig. 1b). Applying Vegard's law61 on lattice parameters obtained by Rietveld refinement of XRD data, this shift evidences the formation of a Ni4.5Fe alloy.
This is in contrast to previously reported ex situ HR-TEM and EDX mappings after catalyst activation under similar conditions by which the formation of a Ni3Fe alloy was reported.20 A Ni–Fe alloy formation could be also observed in fitting of EXAFS data in R space (Fig. 2 and ESI,† S2d) where presence of a Fe–Ni/Fe shell at around 4 Å has been observed.62 Corresponding EXAFS fitting results at Fe K-edge are given in ESI,† Table S2. It is worth mentioning here that the detection of FeO backscattering is difficult in EXAFS region due to weak contribution from Fe–O species as well as lower data quality at elevated temperature. Also, the FeOx clusters are of amorphous nature which, in combination with the smaller particle size, adds to the loss of the backscattering contribution. The crystallite sizes of both catalysts were estimated to 2.7 ± 0.1 nm using the Rietveld refined XRD data after the H2-TPR (see ESI†). These were markedly smaller than the estimated particle size of around 3.9 ± 0.9 nm from a previous study based on scanning transmission electron microscopy (STEM).20
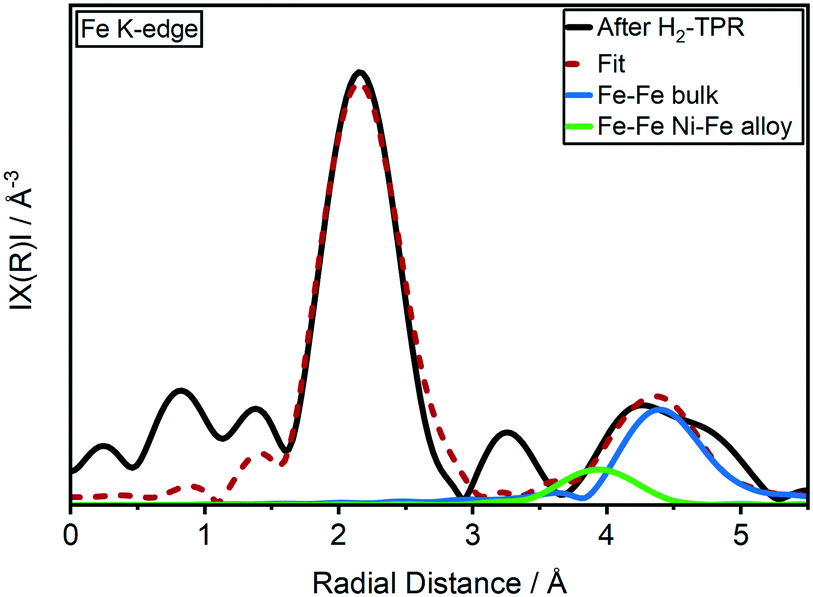 | ||
| Fig. 2 k 2-Weighted Fourier transformed EXAFS (black) and the fitted spectrum (red) at the Fe K-edge of the 17 wt% Ni–Fe/γ-Al2O3 catalyst after H2-TPR in 20 ml min−1 50 vol% H2/N2 at atm (EXAFS fitting results in Table S1†). | ||
Active state of the Ni–Fe catalyst under steady state conditions
A comparison of the catalytic activity of Ni and Ni–Fe catalysts including detailed structural information about monometallic Ni catalysts during CO2 methanation is given in other studies.23,29,30,63,64 Hence, the focus in this study is on gaining information about structural composition of the Ni–Fe catalyst with operando techniques. The Ni–Fe/γ-Al2O3 catalyst was already active at 250 °C, providing a CO2 conversion of 5% (Table 1) and a selectivity to methane of 75%. The only observed byproduct was CO. Based on these results, the presence of Fe led to a higher formation of CO at 250 °C.20 This indicates an enhanced CO2 dissociation on the Ni–Fe catalyst at low temperatures which would be in agreement to the results obtained by Yan et al.27 on a Ni–FeOx catalyst. Compared to the laboratory results,20 the overall activity was lower in this combined operando XAS & XRD synchrotron study. This was due to differences in space velocity caused by catalyst dilution and limitations in catalyst mass that can be loaded in a micro capillary reactor.:CO2 = 4:1 in N2 at atm; mcat = 3.4 mg of 1:1.3 in γ-Al2O3 diluted catalyst
| 250 °C | 350 °C | 450 °C | 350 °C II | |
|---|---|---|---|---|
| X CO2/% | 5 | 61 | 66 | 59 |
| S CH4/% | 75 | 96 | 92 | 96 |
| S CO/% | 25 | 4 | 8 | 4 |
No changes were found during CO2 methanation at 250 °C compared to the oxidation state after H2-TPR in the Ni K-edge XANES spectra (Fig. 3a). However, in the Fe K-edge, the pre-edge feature shifted to higher energy and the intensity of the white line at 7130 eV immediately increased (Fig. 3b). These changes represent an oxidation of Fe0 to Fe2+, e.g. by formation of Fe oxide or carbide. In a recent study it was suggested that iron carbide might be an active species during CO2 methanation, similar as in Fischer–Tropsch synthesis.28 Kureti et al. investigated such an iron carbide formation during CO2 methanation on bulk Fe catalysts and found it only for iron crystallites bigger than 23 nm.65 As the crystallite sizes of the catalysts used in our study were around 2.7 nm, a bulk formation of carbides is unlikely. Further, iron carbide formation was reported mainly for CO methanation, while Fe3O4 (mixture of Fe2+ and Fe3+) was reported to form during CO2 methanation on pre-reduced iron catalysts.66 Finally, a comparison of the Fe K-edge XANES spectra obtained during CO2 methanation with the partially reduced FeOx spectra during H2-TPR provided evidence on FeOx formation (cf. Fig. S3†). As oxygen free gases were used in this study, this clearly demonstrates that the oxygen must originate from CO2 activation.
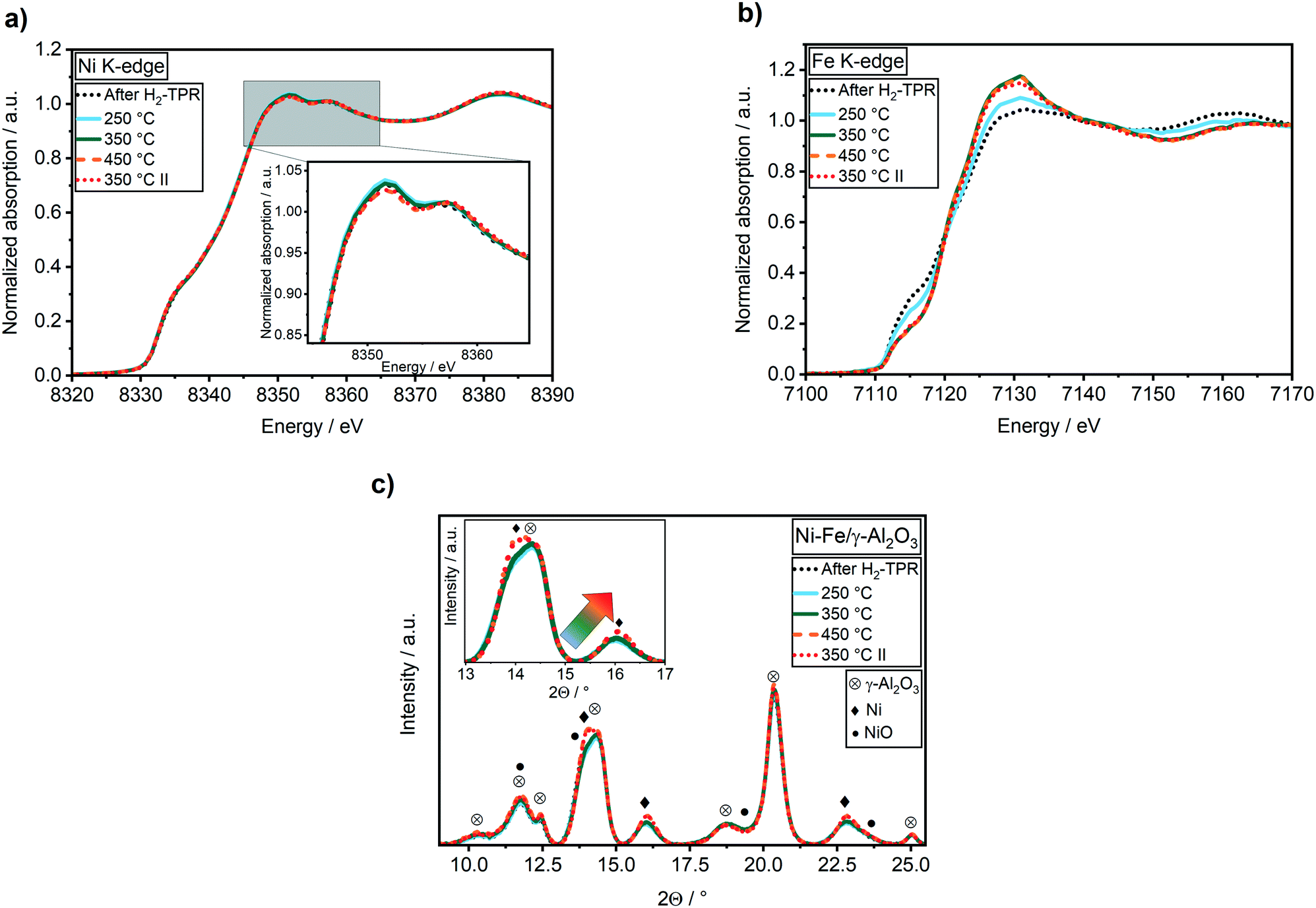 | ||
| Fig. 3 XANES spectra recorded in transmission mode at the Ni (a) and Fe (b) K-edge and (c) XRD patterns (λ = 0.4943 Å) of the 17 wt% Ni–Fe/γ-Al2O3 catalyst at 50 °C after applying methanation conditions of 20 mL min−1 50 vol% H2:CO2 = 4:1 in N2 at atm and various temperatures for 1 h. | ||
By increasing the reaction temperature to 350 °C a CO2 conversion of 61% with a selectivity to CH4 of 96% was achieved (Table 1). While the elevated temperature had no effect on the Ni K-edge XANES spectrum (Fig. 3), iron was further oxidized up to 46% FeO (estimated from LCA) as depicted by a further shift in the feature at 7117 eV and an increase in the intensity at 7130 eV in the Fe K-edge spectrum (Fig. 3b). In contrast to the temperature step at 250 °C, a slight shift of the Ni(200) reflection to a slightly higher diffraction angle at 2θ = 16.10° was observed in the XRD patterns (Fig. 3c). According to analysis of lattice parameters from Rietveld refinement (Table 2) using Vegard's law, this shift represents an enrichment of Ni in the alloy from 4.8 to about 6.0. Hence, the increased temperature and CO2 conversion led to a segregation of iron to the surface including formation of a higher fraction of FeO. Such a structural change was also reported by Yan et al.27 based on ex situ ADF-STEM and EELS elemental maps after methanation of CO2. However, in contrast to their results, the higher fraction of FeO did not lead to a higher CO selectivity in the present study (Table 1). A further increase in temperature to 450 °C resulted in a CO2 conversion of 66% (Table 1). The selectivity to methane declined from 96% to 92% due to the thermodynamically favored water–gas-shift reaction. Interestingly, no further significant increase in FeO was observed (Fig. 3b). This provides evidence, that the effect of FeO formation correlates rather to catalytic activity than temperature. Concurrently, an enrichment of Ni in the alloy from 6.0 to 7.3 was found, while the crystallite size increased from 2.7 nm to 3.2 nm (Table 2). Hence, alloy stability and sintering depend on reaction temperature.
:CO2 = 4:1 in N2 at atm and various temperatures for 1 h
| 250 °C | 350 °C | 450 °C | 350 °C II | |
|---|---|---|---|---|
| Lattice parameter/pm | 354.3 ± 0.3 | 353.9 ± 0.2 | 353.6 ± 0.2 | 353.6 ± 0.2 |
| Crystallite size/nm | 2.5 ± 0.1 | 2.7 ± 0.1 | 3.2 ± 0.1 | 3.3 ± 0.1 |
| Bulk: ratio Ni:Fe/− |
4.8 ± 0.8 | 6.0 ± 0.7 | 7.3 ± 0.9 | 7.5 ± 1.0 |
| Bulk: oxidized Fe/% | 19.5 ± 0.8 | 45.8 ± 1.2 | 51.0 ± 1.4 | 49.5 ± 1.4 |
To investigate the influence of dealloying and sintering on the catalyst performance, a second methanation step was performed for comparison at 350 °C. According to the Ni and Fe K-edge XANES spectra (Fig. 3a and b) and the results obtained by XRD data (Table 2) the changes were not reversible. A slightly lower CO2 conversion of 59% compared to 61% was observed (Table 1). This might be caused either by the higher Ni:Fe ratio of 7.5 compared to 6.0 or by the increase in crystallite sizes from 2.7 nm to 3.3 nm.
Surface changes and FeOx cluster formation
The segregation of iron to the particle surface might lead to formation of, e.g. a core–shell like structure or to formation of clusters on the surface. To obtain further information about the structural composition at the catalyst surface, we calculated the number and composition of the surface atoms. For this purpose, we assumed that nickel and iron were both in fcc structure (premise for alloy formation). A particle size of 3.9 ± 0.9 nm was determined for this catalyst via STEM in a previous study.20For calculation of volume vp (eqn (5)) and surface area as (eqn (6)) of one particle, we assumed hemispherical particles on the support and a linear correlation of changes in crystallite (operando XRD data) with particle size.
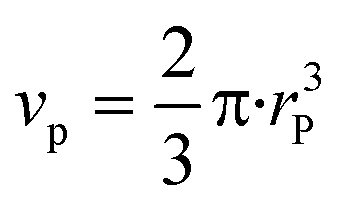 | (5) |
| as = 2π·r2P | (6) |
| X(Fe) = 1 − X(Ni) | (7) |
| vm(Ni–Fe) = X(Ni)·vm(Ni) + X(Fe)·vm(Fe) | (8) |
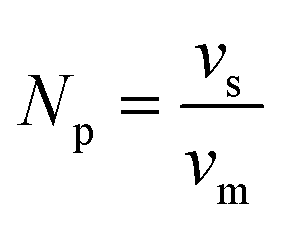 | (9) |
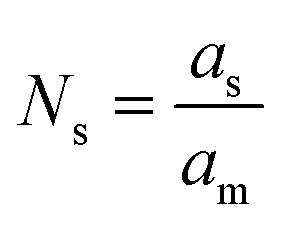 | (10) |
Assuming the number of atoms is constant at each temperature step “T”, iron that formally leaves the Ni–Fe alloy must agglomerate at the surface. The amount of segregated iron Xseg(Fe) out of total number of iron atoms after H2-TPR (“TPR”) was calculated by eqn (11):
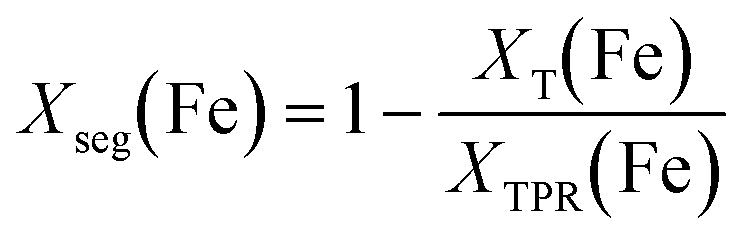 | (11) |
| NT(Fe) = XTPR(Fe)·Ns + Xseg(Fe)·Np(Fe) | (12) |
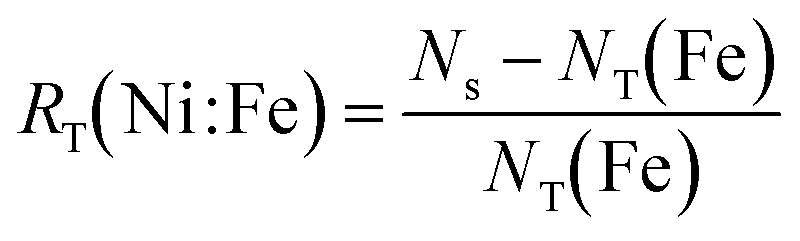 | (13) |
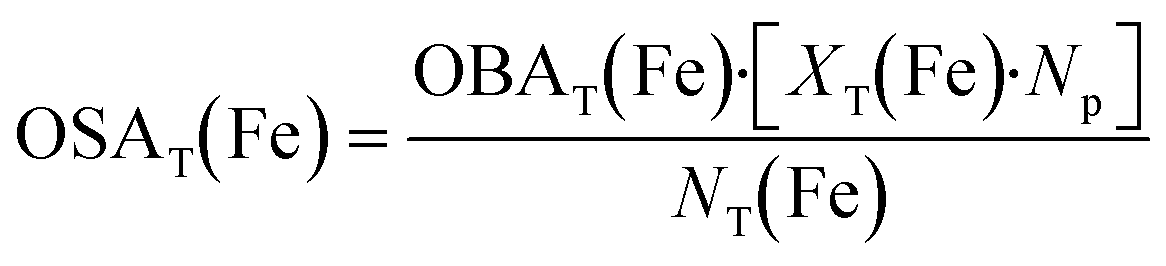 | (14) |
| ATPR | 250 °C | 350 °C | 450 °C | 350 °C II | |
|---|---|---|---|---|---|
| Iron atoms segregated/% | — | 4.4 ± 1.4 | 20.8 ± 1.8 | 32.8 ± 1.8 | 35.0 ± 1.0 |
| Surface: ratio Ni:Fe/− |
4.5 ± 0.8 | 3.7 ± 0.8 | 2.3 ± 0.9 | 1.7 ± 0.8 | 1.6 ± 0.8 |
| Surface: oxidized Fe/% | — | 59.5 ± 14.5 | 86.6 ± 15.2 | 78.4 ± 11.2 | 74.2 ± 9.7 |
Nevertheless, assuming that the model particle is 3.9 nm without any variation, the values given in Table 3 can be considered as relevant to observe and discuss trends. In future, this may be substantiated by in situ XPS under selected model conditions.
We observed that up to 35% of the total iron moved from bulk (cf.Table 2) to the particle surface. This resulted in changes in the Ni:Fe surface ratio from 4.9 up to 1.9 at 450 °C. An amount of ≈60% oxidized surface iron atoms were determined at 250 °C, ≈87% at 350 °C and ≈78% at 450 °C. Hence, we can conclude that the iron surface species were in a mainly oxidized state. The segregation and enrichment of iron on the surface under formation of oxide species might either lead to a “core–shell”-like structure of an FeOx layer or to formation of FeOx clusters on top of the Ni–Fe alloy particles. We simulated several structures for these cases with DFT and calculated the respective XANES spectra using the FEFF9-code52 to compare them to our experimental data (Fig. 4). The spectra were simulated by averaging the core-holes in Ni/Fe atoms at all possible positions on the surface to get the final spectrum. Details about the model generation and simulation procedure with parameters used in FEFF9 are given in ESI,† section 3.2.1.
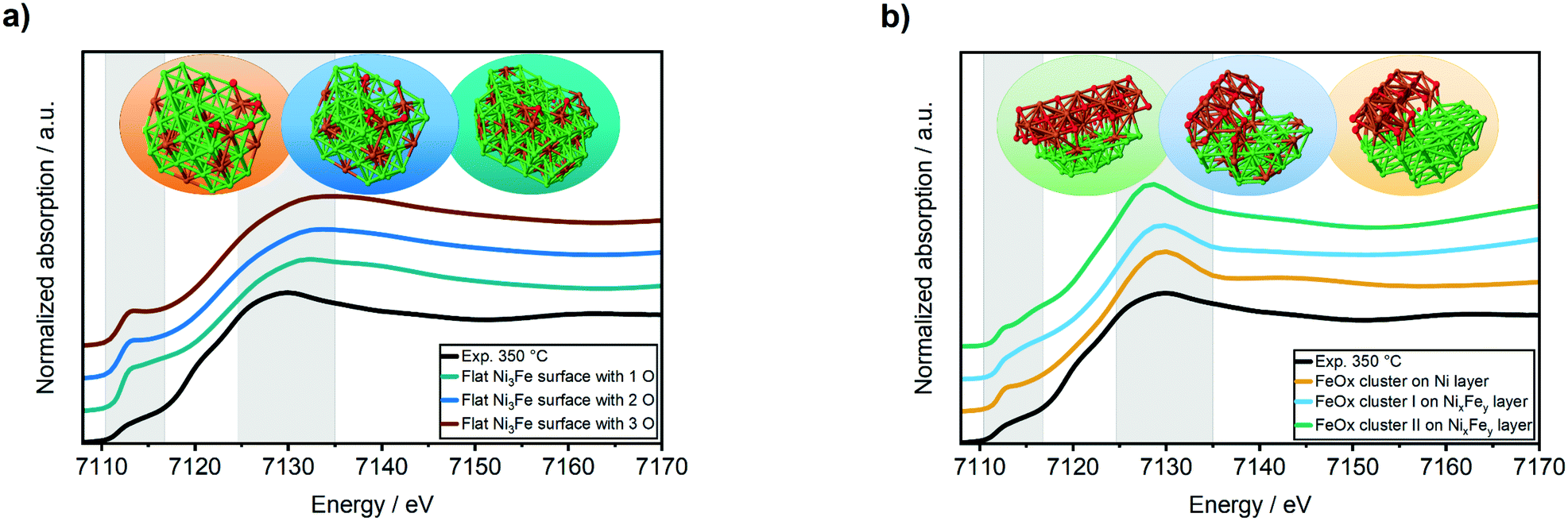 | ||
| Fig. 4 Fe K-edge XANES spectra of the experimental data obtained from the bimetallic Ni–Fe/Al2O3 catalysts after methanation of CO2 at 350 °C compared to FEFF calculated XANES spectra of iron-rich surface with increasing amount of oxygen (a) and to FeOx clusters-like structures (b). | ||
The simulated XANES spectra of the oxidized “core–shell”-like flat Ni3Fe surface (Fig. 4a) were not in a good agreement to the experimentally obtained Fe K-edge XANES spectrum during methanation of CO2 at 350 °C (Fig. 4a, black curve): the white line feature was shifted to 7133 eV instead of 7130 eV and the pre-edge feature at ≈7112 eV was too sharp. In contrast, a good match was found for FeOx clusters on both, a Ni3Fe and a Ni surface (Fig. 4b): the white line feature appeared at the same energy of 7130 eV as the experimental data and the pre-edge feature at ≈7112 eV provided a similar shape. Concerning the latter, the best match was found for the FeOx cluster I on a NixFey layer.
Dynamic changes of the active centres at the surface (MES)
To investigate the influence of these FeOx clusters and to understand their role on CO2 activation at the particle surface, we coupled the bulk technique XAS with modulation excitation spectroscopy (MES)35 to enhance its surface sensitivity. For this purpose, periodic switches (modulation) between H2/N2 (0–60 s) and H2/CO2/N2 (60–120 s) in the gas feed were conducted at various temperatures while monitoring the dynamic responses at the Ni and Fe K-edges during these CO2 modulations with QEXAFS. Subsequently, “spectator” species (those which do not respond to the atmosphere) were filtered out.34 In our case, we decided to use a sinusoidal function as approximation for the stimuli we applied, similar to other catalytic studies.36,69 Note, that external stimuli in a real experiment do not perfectly match a mathematical sinusoidal wave. Hence, in some cases, the signal processing can be enhanced by using square wave modulation and/or corrections terms for amplitude.34,70 These were not applied in our study, as the focus was laid on relative phase changes rather than kinetics or quantitative analysis. Prior to the modulation experiments, the catalyst was activated by H2-TPR and treated under methanation conditions until a steady state was reached. As these structural changes were discussed in detail in the previous section and are the same as during the XAS-XRD experiments, the data are only shown for completeness in the ESI.† In addition, we focus here on the role of iron, hence only a short summary on the MES results at the Ni K-edge will be given.At 250 °C, the shape of the demodulated Ni K-edge spectrum was similar for both catalysts (cf. Fig. S18 and S19†). This demonstrates that the origin in the difference in catalytic activity at 250 °C (Table 1) can be traced back to reactions on iron sites. None of the difference spectra obtained from measured and simulated references matched precisely to experimental demodulated MES data obtained at the Ni K-edge. Hence, surface changes at Ni atoms are very complex. Nevertheless, we can conclude with certainty that we have no indication of Ni–O species formed during CO2 modulation, and that the same surface species were formed on Ni on both catalysts. For further details and discussion see ESI.†Fig. 5a (top) shows time-resolved XANES spectra of the Ni–Fe catalyst at the Fe K-edge during CO2/H2vs. H2 cycling obtained from averaging the complete 30 periods. There are some slight changes observed for the pre-edge feature at ≈7116 eV and for the white line feature at 7129 eV as the zoomed spectra in Fig. 5b and c show, respectively. However, in conventional XAS spectra these changes are too small to predict some phase transitions. In contrast, using demodulation uncovers striking changes. The corresponding demodulated XANES spectra during cycling at various phase angles are shown in Fig. 5a (bottom) where noticeable changes were both observed at the energy positions mentioned for pre-edge and white line region in the Fe K-edge spectra. An asymmetry can be observed in these demodulated spectra, especially at 7140 eV. This demonstrates that complex changes occurred at the Fe K-edge corresponding to changes of more than two species.37 To get further information about these changing species, the demodulated spectrum with the highest amplitude is in a next step compared to experimentally obtained difference spectra from reference spectra of Fe metal, FeO and γ-Fe2O3 (Fig. 6a). A very good correlation to oxidized iron species, such as Fe2O3–FeO and Fe2O3–Fe, was found. This demonstrates that during CO2 cycling Fe provided highly dynamic changes concerning its oxidation state even up to Fe3+. Note, this does not mean that a major Fe2O3 phase is formed during modulation. Fe3+ is known to fit well into the FeO-lattice. In addition, Fe2+ atoms at the interface between FeOx and the Ni–Fe surface may be partially oxidized to Fe3+ due to CO2 activation. As this is the only species that can be periodically correlated to the modulations in the gas phase seen in MES, these changes can be clearly correlated to CO2 activation.
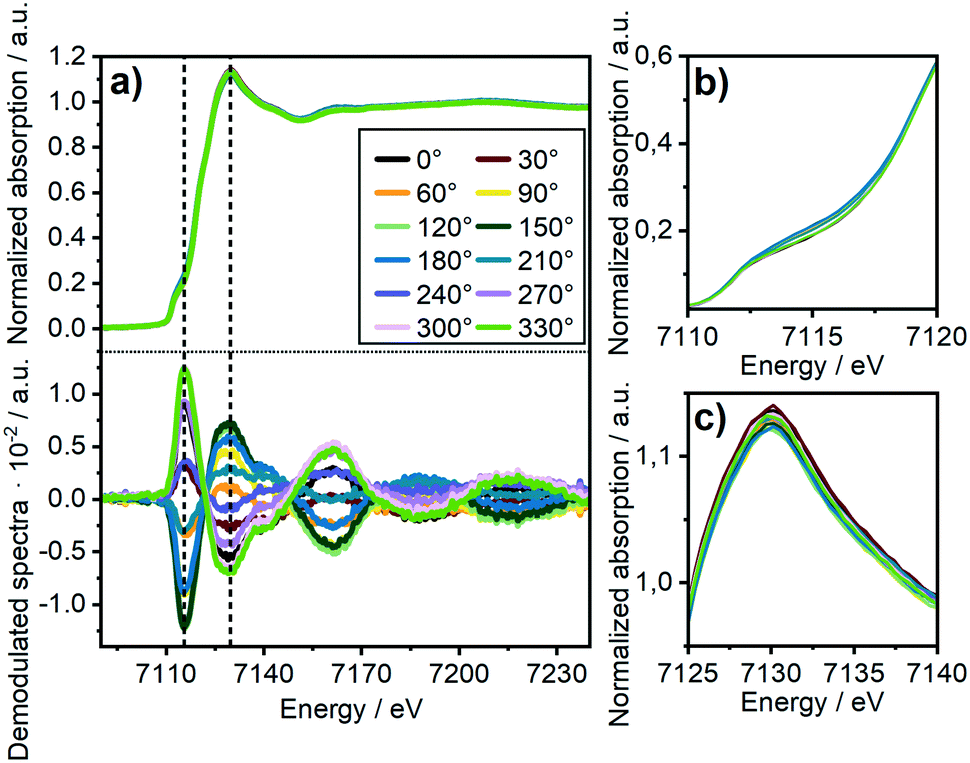 | ||
| Fig. 5 (a) Normalized time-resolved Fe K-edge XANES consisting of a total of 24 spectra covering 5 seconds each for the 17 wt% Ni3Fe/γ-Al2O3 catalyst during CO2/H2/N2 (0–60 s) vs. H2/N2 (60–120 s) cycling and 30 periods average (top) and corresponding demodulated spectra (bottom) at selected values of phase angle (Δϕ). (b) Zoomed edge region, (c) zoomed white line region. | ||
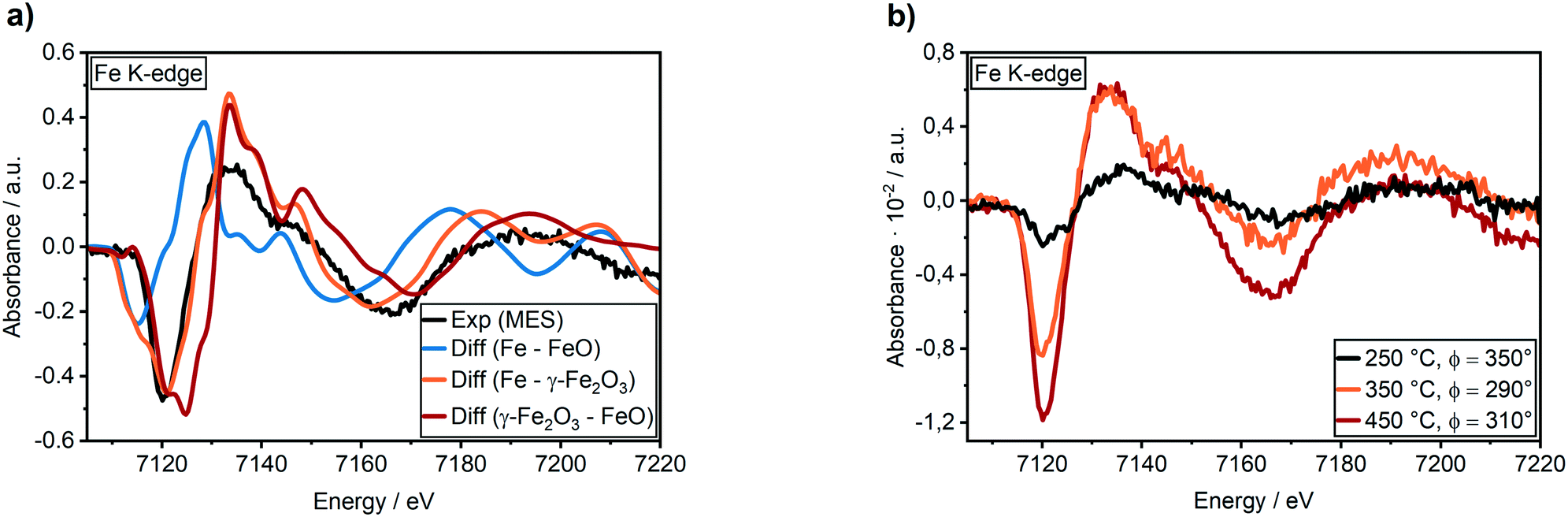 | ||
| Fig. 6 (a) Demodulated Fe K-edge spectra of the 17 wt% Ni–Fe/γ-Al2O3 catalyst at 450 °C (black) compared to difference spectra obtained from experimental Fe reference spectra and (b) demodulated Fe K-edge spectra at 250 °C (black), 350 °C (orange) and 450 °C (red) during CO2/H2/N2 (0–60 s) vs. H2/N2 (60–120 s) cycling with 30 periods average. | ||
The temperature dependency is given in Fig. 6b. At 250 °C a very low intensity in the features of the demodulated spectrum was observed. As we observed a higher selectivity to CO for the bimetallic Ni–Fe sample at 250 °C (Table S9†), we can assume that CO2 is only slowly activated at the Fe0 centers under formation of CO and FeO. In this temperature range, Ni provides a comparatively high affinity to CO, as observed in the MES at the Ni K-edge (cf. ESI† section 3.4.7). DFT calculations demonstrated that the presence of Fe does not influence the CO adsorption energy on Ni (cf. section 3.5.3). Hence, the Ni surface is covered by CO probably hampering the hydrogen activation that led to the high fraction of CO formation. An increase in temperature from 250 °C to 350 °C and 450 °C showed large increase in intensity of the demodulated spectrum at Fe K-edge as shown in fig. 6b. This provides evidence of strong dynamic responses of Fe to the CO2 modulations at elevated temperature which can be correlated well to the determined catalytic activity (see Table 1).
However, there are various iron positions where these dynamic changes might occur, e.g. on top of the cluster or at the Ni–FeOx interface (cf.Fig. 4b). We observed that the amplitude of the response signal obtained from stimulated Ni (see ESI,† Fig. S20) and Fe atoms was in the same range for each temperature step and that it increased with temperature. As this correlates well with the degree of CO2 conversion (Table 1), this indicates that stimulated atoms represent the most active centers and that the “redox”-like behavior they provide is important for CO2 activation and the overall catalyst performance.
Stability of Ni–Fe alloy and formation of FeOx clusters
In order to shed light on the driving forces of the iron oxide cluster formation and on the stability of the Ni–Fe alloy during reaction, DFT calculations of this process have been performed. The calculated heat of formation of the Ni3Fe alloy is −0.60 eV. We tested the surface segregation of Fe using a Ni(111) slab. Under vacuum, this energy is 0.16 eV, such that Fe is more stable in the subsurface. When CO2 and H2O are present in the reaction atmosphere, there is a change of the oxygen chemical potential. We calculated the oxidation of a Ni–Fe alloy with Ni4Fe stoichiometry. Here, we considered the segregation of Fe on top of the Ni4Fe(111) surface where it forms FeO (this is calculated by a corresponding decrease of the iron content in the alloy, see ESI† for further details). The result is depicted as a phase diagram in Fig. 7 for two different FeO structures the stability of whose is calculated as a function of the H2O/H2 pressure and temperature (see Fig. S24 in the ESI† for the corresponding diagram with CO2/CO pressure). As can be seen from Fig. 7, Fe is according to the calculation predicted to segregate on top of the Ni4Fe(111) surface where it is oxidized to small FeO clusters, as also observed experimentally. Note that these structures were used to calculate the XANES spectra in Fig. 4, which showed a reasonable agreement. We considered oxidation of (FeO)6/Ni4Fe(111) such that there is now an extra oxygen atom at the interface between FeO and the Ni4Fe(111) surface (see Fig. 7). This oxygen, however, is slightly unstable under the applied conditions and we therefore speculate that CO2 methanation could take place via a redox-mechanism at the FeO–alloy interface, that is, CO2 splits at the interface into CO (adsorbed on the Ni4Fe(111) facet) and O* located at the interface. We calculated the energy to remove this O* via hydrogenation with H2 to be downhill by −0.61 eV in energy. Removal of stoichiometric oxygen of the FeO with H2 is about thermoneutral (−0.02 eV), again pointing towards a possible redox mechanism at the FeO–alloy interface.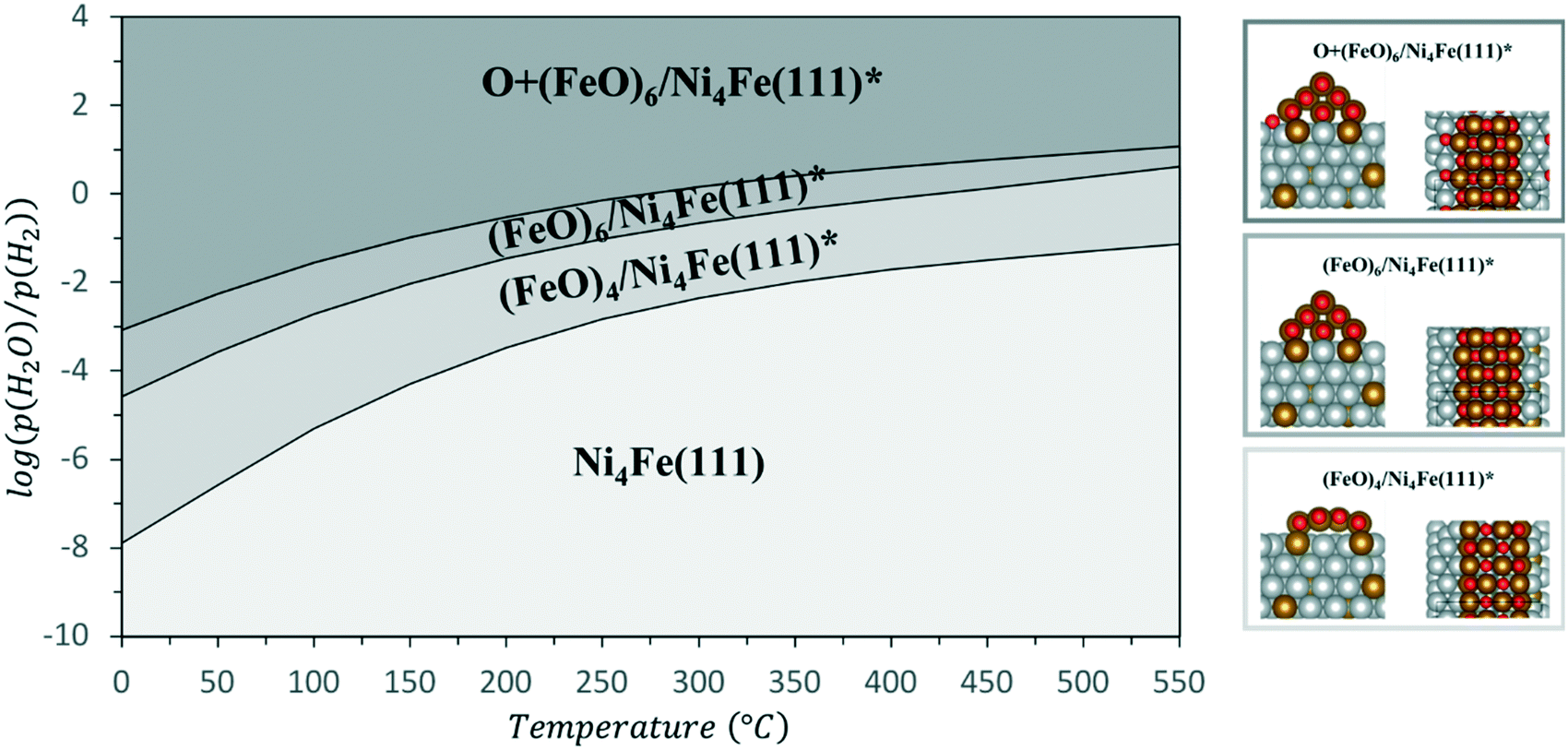 | ||
| Fig. 7 Phase diagram of Ni4Fe(111) and (FeO)x/Ni4Fe(111) system as a function of the pH2O/pH2 ratio and for temperature range between 0 and 550 °C. see ESI† for a phase diagram using pCO2/CO and for further details. | ||
Mechanism of CO2 activation on Ni–Fe alloy catalysts
In our operando studies on the bimetallic Ni–Fe alloy catalyst, we observed partial segregation of iron under formation of FeOx at the particle surface during CO2 methanation. The increasing amount of FeOx at the surface led to formation of FeOx cluster on top of the Ni–Fe particles, which is schematically shown in Fig. 7. These clusters offer new actives sites for CO2 activation and provide, as we demonstrated by MES, a redox cycle between Fe0 ⇌ Fe2+ ⇌ Fe3+. Such a redox cycle was suggested e.g. on bulk Fe3O4 catalysts during CO2 activation71–73 under distinct formation of carbonaceous surface species which led to a superior CH4 yield compared to iron carbide based catalysts.71,74 However, we were not able to detect any surface carbon species on the bimetallic Ni–Fe catalyst during our operando studies, probably due to the high CO2-pressure. Previous operando Raman studies on a Ni–Fe based catalyst from our group16 did not provide any hints for formation of carbonaceous surface species. Besides CH4, CO remains the main product on FeOx catalysts in the gas-phase during CO2 methanation.75 The formation of CO can be attributed to the RWGS activity of FeOx and is, according to our experimental observations and calculations, another key step in enhanced CO2 activation of Ni–FeOx catalysts (cf.Fig. 8). On monometallic Ni catalysts, CO2 dissociation proceeds fast, but CO is strongly bound on reduced Ni0 centers and therefore adsorption sites for CO2 are blocked.76–79 These steps can proceed separately on the Ni–FeOx system: CO2 can be activated and hydrogenated either directly to CH4 or dissociated under formation of CO at the interface between the FeOx clusters and the Ni–Fe surface (Fig. 8). The strong adsorption of CO on Ni0 might subsequently attract it to the Ni centers where it can be further hydrogenated via the CO methanation route. This RWGS activity of the FeOx clusters on the Ni–FeOx catalyst further explains the high CO selectivity at low temperatures: while the FeOx clusters are already active in RWGS activity, the subsequent hydrogenation step at the Ni0 centers does not proceed at low temperature. These two suggested mechanisms are in line with the MES data obtained at the Ni K-edges of both catalysts which demonstrated the formation of identic intermediates on the Ni and the Ni–Fe catalyst (ESI,† Fig. S22).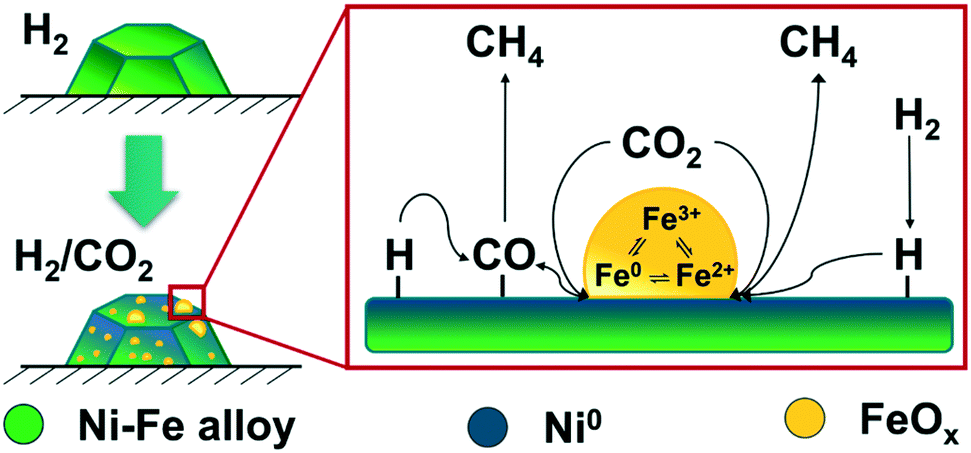 | ||
| Fig. 8 Scheme of the CO2 activation mechanism on Ni–Fe alloy-based catalysts during methanation reaction under realistic conditions. | ||
To summarize, the Fe0 ⇌ Fe2+ ⇌ Fe3+ redox cycle provided by the Fe(Ox) entities at the interface between the clusters and the metal surface improved both, dissociative and associative CO2 activation. Beneficial in the case of the bimetallic Ni–Fe catalyst is that different active sites are available on the reduced Ni surface, the interface and the iron oxide clusters for the adsorption of the respective reactants and, thus, always free sites are available for both CO2 activation and hydrogenation in contrast to monometallic Ni catalysts.
Conclusions
In this study, we investigated in detail the role of iron in a highly active Ni–Fe/γ-Al2O3 catalysts during CO2 methanation. For this purpose, advanced synchrotron-based tools, such as XAS–XRD and XAS coupled MES have been applied and the findings further substantiated by DFT calculations.We noticed a synergistic effect of iron on nickel which led already during catalyst activation by H2-TPR to a higher fraction of active Ni0 species. First, surface atoms or amorphous species were reduced followed by the bulk phase up to formation of nanocrystalline nickel species at elevated temperature. The reduction of Fe2+ to Fe0 occurred simultaneously to the reduction of Ni2+ to Ni0 which led to the formation of the desired Ni–Fe alloy. Notably, no reflections were found in the XRD data for any iron phases during the experiments, which, supported by the changes in the lattice parameters, evidences an incorporation into an alloy.
During stoichiometric CO2 methanation in the temperature range from 250 °C to 450 °C, a good comparability of the operando synchrotron studies to laboratory studies could be achieved concerning CO2 conversion and selectivity to CH4. Under all applied CO2 methanation conditions nickel remained in its reduced metal state as obtained after H2-TPR. In contrast, an oxidation of Fe0 to Fe2+ was immediately observed upon addition of CO2. The amount of Fe2+ thereby correlated to CO2 conversion, either due to CO2 dissociation or from other intermediates/products, such as H2O. Increasing the reaction temperature from 250 °C up to 450 °C resulted in sintering, and agglomeration of Fe at the particle surface under formation of FeOx clusters, as demonstrated by XAS–XRD and DFT calculations.
By enhancing the sensitivity of XAS with MES, it was shown that iron exhibits a highly dynamic behavior during CO2 activation. A Fe0 ⇌ Fe2+ ⇌ Fe3+ redox cycle, highly likely located at the interface between the FeOx clusters and the surface of the metal particles, promotes CO2 dissociation during the methanation reaction. Combination of these findings with DFT calculation conclude that FeOx clusters on reduced Ni particles are at the origin of the high catalytic activity of Fe–Ni catalysts and, by this, CO2 activation and the following hydrogenation are improved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The combined XAS–XRD experiments were performed on beamline BM31 (SNBL) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to Dr. Hermann Emerich (ESRF) and Dr. Dragoş Stoian (ESRF) for assistance and technical support during the beamtime. The X10DA (SuperXAS) beamline, Dr. Maarten Nachtegaal and Dr. Olga Safonova (PSI) at the Swiss Light Source (SLS) in Villigen (Switzerland) are thanked for providing the QEXAFS beam time and support during the experiments. Further, we would like to thank Dr. Henning Lichtenberg, Florian Maurer and Matthias Stehle for beamtime support, as well as Dr. Alexey Boubnov for discussion on MES coupled XAS. The German Federal Ministry of Education and Research (BMBF) is gratefully acknowledged for financial support within the Kopernikus Project P2X and we thank the SPP2080 (DFG-priority program, GR 3987/13-1 and 14-1) both for financial support and a discussion forum for “Catalysts and reactors under dynamic conditions for energy storage and conversion”. Jelena Jelic and Felix Studt acknowledge support by the state of Baden-Württemberg through bwHPC (bwUnicluster and JUSTUS, RV bw17D01).References
- R. Schlögl, Angew. Chem., Int. Ed., 2019, 58, 343–348 CrossRef.
- G. Centi and S. Perathoner, Greenhouse Gases: Sci. Technol., 2011, 1, 21–35 CrossRef CAS.
- M. Sterner, Erneuerbare Energien und Energieeffizienz - Renewable Energies and Energy Efficiency, Kassel University Press GmbH, Kassel, 2009 Search PubMed.
- M. Robinius, A. Otto, P. Heuser, L. Welder, K. Syranidis, D. S. Ryberg, T. Grube, P. Markewitz, R. Peters and D. Stolten, Energies, 2017, 10, 956 CrossRef.
- P. Sabatier and J.-B. Senderens, C. R. Hebd. Seances Acad. Sci., 1902, 134, 514–516 CAS.
- M. Jentsch, T. Trost and M. Sterner, Energy Procedia, 2014, 46, 254–261 CrossRef.
- I. Stadler, in Energiespeicher-Bedarf, Technologien, Integration, Springer, 2017, pp. 327–493 Search PubMed.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Gläser, U. Krewer, K. Reuter and J.-D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS.
- S. Rahmani, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 1346–1352 CrossRef CAS.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef CAS.
- G. Garbarino, P. Riani, L. Magistri and G. Busca, Int. J. Hydrogen Energy, 2014, 39, 11557–11565 CrossRef CAS.
- J. Ashok, M. L. Ang and S. Kawi, Catal. Today, 2017, 281, 304–311 CrossRef CAS.
- F. Ocampo, B. Louis, L. Kiwi-Minsker and A.-C. Roger, Appl. Catal., A, 2011, 392, 36–44 CrossRef CAS.
- B. Mutz, H. W. P. Carvalho, S. Mangold, W. Kleist and J.-D. Grunwaldt, J. Catal., 2015, 327, 48–53 CrossRef CAS.
- B. Mutz, A. Gänzler, M. Nachtegaal, O. Müller, R. Frahm, W. Kleist and J.-D. Grunwaldt, Catalysts, 2017, 7, 279 CrossRef.
- B. Mutz, P. Sprenger, W. Wang, D. Wang, W. Kleist and J.-D. Grunwaldt, Appl. Catal., A, 2018, 556, 160–171 CrossRef CAS.
- B. Mutz, H. W. P. Carvalho, W. Kleist and J.-D. Grunwaldt, J. Phys.: Conf. Ser., 2016, 712, 012050 CrossRef.
- J. Sehested, K. E. Larsen, A. L. Kustov, A. M. Frey, T. Johannessen, T. Bligaard, M. P. Andersson, J. K. Nørskov and C. H. Christensen, Top. Catal., 2007, 45, 9–13 CrossRef CAS.
- M. P. Andersson, T. Bligaard, A. Kustov, K. E. Larsen, J. Greeley, T. Johannessen, C. H. Christensen and J. K. Nørskov, J. Catal., 2006, 239, 501–506 CrossRef CAS.
- B. Mutz, M. Belimov, W. Wang, P. Sprenger, M.-A. Serrer, D. Wang, P. Pfeifer, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2017, 7, 6802–6814 CrossRef CAS.
- M.-A. Serrer, K. F. Kalz, E. Saraçi, H. Lichtenberg and J.-D. Grunwaldt, ChemCatChem, 2019, 11, 5018–5021 CrossRef CAS.
- T. Burger, F. Koschany, O. Thomys, K. Köhler and O. Hinrichsen, Appl. Catal., A, 2018, 558, 44–54 CrossRef CAS.
- C. Mebrahtu, S. Abate, S. Chen, A. F. Sierra Salazar, S. Perathoner, F. Krebs, R. Palkovits and G. Centi, Energy Technol., 2018, 6, 1196–1207 CrossRef CAS.
- D. Pandey and G. Deo, J. Mol. Catal. A: Chem., 2014, 382, 23–30 CrossRef CAS.
- D. Pandey, K. Ray, R. Bhardwaj, S. Bojja, K. V. R. Chary and G. Deo, Int. J. Hydrogen Energy, 2018, 43, 4987–5000 CrossRef CAS.
- L. R. Winter, E. Gomez, B. Yan, S. Yao and J. G. Chen, Appl. Catal., B, 2018, 224, 442–450 CrossRef CAS.
- B. Yan, B. Zhao, S. Kattel, Q. Wu, S. Yao, D. Su and J. G. Chen, J. Catal., 2019, 374, 60–71 CrossRef CAS.
- G. Giorgianni, C. Mebrahtu, M. E. Schuster, A. I. Large, G. Held, P. Ferrer, F. Venturini, D. Grinter, R. Palkovits, S. Perathoner, G. Centi, S. Abate and R. Arrigo, Phys. Chem. Chem. Phys., 2020, 22, 18788–18797 RSC.
- T. Burger, S. Ewald, A. Niederdränk, F. E. Wagner, K. Köhler and O. Hinrichsen, Appl. Catal., A, 2020, 604, 117778 CrossRef CAS.
- C. Mebrahtu, S. Perathoner, G. Giorgianni, S. Chen, G. Centi, F. Krebs, R. Palkovits and S. Abate, Catal. Sci. Technol., 2019, 9, 4023–4035 RSC.
- S. A. Theofanidis, V. V. Galvita, H. Poelman and G. B. Marin, ACS Catal., 2015, 5, 3028–3039 CrossRef CAS.
- J.-D. Grunwaldt and B. S. Clausen, Top. Catal., 2002, 18, 37–43 CrossRef CAS.
- M. Rønning, N. E. Tsakoumis, A. Voronov, R. E. Johnsen, P. Norby, W. van Beek, Ø. Borg, E. Rytter and A. Holmen, Catal. Today, 2010, 155, 289–295 CrossRef.
- A. Urakawa, T. Bürgi and A. Baiker, Chem. Eng. Sci., 2008, 63, 4902–4909 CrossRef CAS.
- A. Gaur, T. M. Hartmann Dabros, M. Høj, A. Boubnov, T. Prüssmann, J. Jelic, F. Studt, A. D. Jensen and J.-D. Grunwaldt, ACS Catal., 2019, 9, 2568–2579 CrossRef CAS.
- C. F. J. König, J. A. van Bokhoven, T. J. Schildhauer and M. Nachtegaal, J. Phys. Chem. C, 2012, 116, 19857–19866 CrossRef.
- G. L. Chiarello and D. Ferri, Phys. Chem. Chem. Phys., 2015, 17, 10579–10591 RSC.
- D. Ferri, M. A. Newton and M. Nachtegaal, Top. Catal., 2011, 54, 1070 CrossRef CAS.
- P. Müller and I. Hermans, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef.
- A. Urakawa, W. Van Beek, M. Monrabal-Capilla, J. R. Galán-Mascarós, L. Palin and M. Milanesio, J. Phys. Chem. C, 2011, 115, 1323–1329 CrossRef CAS.
- W. van Beek, O. V. Safonova, G. Wiker and H. Emerich, Phase Transitions, 2011, 84, 726–732 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- R.-S. Zhou and R. L. Snyder, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 617–630 CrossRef.
- J. Rouquette, J. Haines, G. Fraysse, A. Al-Zein, V. Bornand, M. Pintard, P. Papet, S. Hull and F. A. Gorelli, Inorg. Chem., 2008, 47, 9898–9904 CrossRef CAS.
- R. Frahm, Nucl. Instrum. Methods Phys. Res., Sect. A, 1988, 270, 578–581 CrossRef.
- R. Frahm, Rev. Sci. Instrum., 1989, 60, 2515–2518 CrossRef CAS.
- R. Frahm, M. Nachtegaal, J. Stötzel, M. Harfouche, J. A. van Bokhoven and J.-D. Grunwaldt, AIP Conf. Proc., 2010, 1234, 251–255 CrossRef CAS.
- O. Müller, M. Nachtegaal, J. Just, D. Lützenkirchen-Hecht and R. Frahm, J. Synchrotron Radiat., 2016, 23, 260–266 CrossRef.
- A. H. Clark, J. Imbao, R. Frahm and M. Nachtegaal, J. Synchrotron Radiat., 2020, 27, 551–557 CrossRef.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- E. E. Unmuth, L. H. Schwartz and J. B. Butt, J. Catal., 1980, 61, 242–255 CrossRef CAS.
- L. Vegard, Z. Angew. Phys., 1921, 5, 17–26 CAS.
- S. M. Kim, P. M. Abdala, T. Margossian, D. Hosseini, L. Foppa, A. Armutlulu, W. van Beek, A. Comas-Vives, C. Copéret and C. Müller, J. Am. Chem. Soc., 2017, 139, 1937–1949 CrossRef CAS.
- D. Pandey and G. Deo, J. Ind. Eng. Chem., 2016, 33, 99–107 CrossRef CAS.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- J. Kirchner, J. K. Anolleck, H. Lösch and S. Kureti, Appl. Catal., B, 2018, 223, 47–59 CrossRef CAS.
- H. Ando, Y. Matsumura and Y. Souma, J. Mol. Catal. A: Chem., 2000, 154, 23–29 CrossRef CAS.
- G. Bergeret and P. Gallezot, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2008, pp. 738–765, DOI:10.1002/9783527610044.hetcat0038.
- C. N. Singman, J. Chem. Educ., 1984, 61, 137 CrossRef CAS.
- D. Ferri, M. S. Kumar, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC.
- A. Urakawa, T. Bürgi and A. Baiker, Chem. Phys., 2006, 324, 653–658 CrossRef CAS.
- T. Yoshida, K. Nishizawa, M. Tabata, H. Abe, T. Kodama, M. Tsuji and Y. Tamaura, J. Mater. Sci., 1993, 28, 1220–1226 CrossRef CAS.
- Y. Tamaura and K. I. Nishizawa, Energy Convers. Manage., 1992, 33, 573–577 CrossRef CAS.
- H. Kato, T. Sano, Y. Wada, Y. Tamaura, M. Tsuji, T. Tsuji and S. Miyazaki, J. Mater. Sci., 1995, 30, 6350–6354 CrossRef CAS.
- T. Kodama, Y. Kitayama, M. Tsuji and Y. Tamaura, Energy, 1997, 22, 183–187 CrossRef CAS.
- G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1984, 87, 352–362 CrossRef CAS.
- T. Van Herwijnen, H. Van Doesburg and W. A. De Jong, J. Catal., 1973, 28, 391–402 CrossRef CAS.
- M. B. I. Choudhury, S. Ahmed, M. A. Shalabi and T. Inui, Appl. Catal., A, 2006, 314, 47–53 CrossRef CAS.
- D. C. D. Da Silva, S. Letichevsky, L. E. P. Borges and L. G. Appel, Int. J. Hydrogen Energy, 2012, 37, 8923–8928 CrossRef CAS.
- D. E. Peebles, D. W. Goodman and J. M. White, J. Phys. Chem., 1983, 87, 4378–4387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01396j |
| This journal is © The Royal Society of Chemistry 2020 |