
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt- and K-promoted supported gallia as a highly stable alternative catalyst for isobutane dehydrogenation†
Anna N.
Matveyeva
 a,
Nadezhda A.
Zaitseva
b,
Nikolai A.
Pakhomov
c and
Dmitry Yu.
Murzin
*d
a,
Nadezhda A.
Zaitseva
b,
Nikolai A.
Pakhomov
c and
Dmitry Yu.
Murzin
*d
aLaboratory of Materials and Processes for Hydrogen Energy, Ioffe Institute, Politekhnicheskaya ul. 26, St. Petersburg 194021, Russia
bBoreskov Institute of Catalysis SB RAS, Prospekt Akademika Lavrentieva 5, Novosibirsk 630090, Russia
cLaboratory of Catalytic Technologies, St. Petersburg State Institute of Technology (Technical University), Moskovsky pr. 26, St. Petersburg 190013, Russia
dLaboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University, Biskopsgatan 8, Turku/Åbo 20500, Finland. E-mail: dmurzin@abo.fi
First published on 14th September 2020
Abstract
Addition of Pt and K to Ga2O3/Al2O3 was shown to be beneficial for isobutane dehydrogenation. This catalytic system can be successfully realized in both fluidized and fixed-bed reactors. In contrast to supported catalysts based on CrOx and Pt–Sn, the catalyst performance of the supported Ga–Pt–K catalyst only marginally changed after 3 hours of dehydrogenation.
Isobutene is an important chemical building block used in a variety of applications ranging from fuel additives and polymers to pharmaceutical and agricultural industries. The primary driver for the isobutene market is the growing aerospace market as well as the growing demand for rubber in the automotive market.1
In industry, isobutene is obtained by separating it from the butane–butene fraction in the production of gasoline or ethene by catalytic or steam cracking, and pyrolysis of liquid petroleum products and oil gases.2 The yield of the C4 fraction in these processes is small and, as a result, cannot meet the demand for isobutene growing every year. Therefore, there is a need for catalytic alkane dehydrogenation processes, which would selectively produce alkenes.
Commercial catalysts typically used for dehydrogenation contain either CrOx or Pt as active components. Catalysts of these types give different by-products and require different regeneration treatments.3 Their activity decreases with time on stream (TOS) due to blockage of the active sites by coke. As a consequence, these catalysts must be oxidatively regenerated. Although substantial improvements have been made for both types of catalytic materials, several economic, environmental, and technological challenges have still to be solved. Some of them are mentioned below.
Spent CrOx/Al2O3 dehydrogenation catalysts contain highly toxic species of Cr(VI) up to 1.5 wt%, which limits their use as secondary raw materials. Therefore, such catalysts, as a rule, are located in underground bunkers, which unfortunately, leads to the leaching of hexavalent Cr into ground water and soil. In general, dehydrogenation over CrOx/Al2O3 is carried out by alternating the stages of dehydrogenation and regeneration for 5–8 min.
The Oleflex process operates with a Pt–Sn/Al2O3 catalyst,4 which is characterized by a high cost and operation difficulties, namely, stringent requirements for the catalyst support such as high mechanical strength and a very specific size of spherical particles of 1.6 mm,5 relatively fast deactivation and cumbersome catalyst regeneration using a chlorine–air mixture.6
With the purpose of overcoming the above drawbacks, various attempts were reported in the literature to improve available commercial catalysts or develop feasible alternatives.
Among various oxides, gallium-containing materials are considered as very promising for dehydrogenation.2 For example, our previous studies showed that the moderate activity of gallia dispersed on Al2O3 in isobutane dehydrogenation7,8 requires further improvement. Several studies on the promotion of gallia have shown that addition of Pt to Ga is beneficial for dehydrogenation of propane,9–13 isobutane9,12 and ethylbenzene.14 It is also known that commercial dehydrogenation catalysts contain a small amount of alkali metals to suppress coke.2
Using this fundamental knowledge, alumina-supported gallia was promoted with tiny amounts of Pt (0.01, 0.05 and 0.1 wt%) and K (0.25 and 1.25 wt%). Such amounts of Pt supported on alumina are not active in dehydrogenation per se. Note that in commercial catalysts, the Pt content is ca. 0.3 wt%.2 In previous studies reporting the promotion of supported Ga2O3 with Pt, the loading was 0.1 wt% (ref. 10–12) or higher.13,14 In contrast to the patent9 with 0.002 wt% Pt, where catalyst stability was not specifically addressed, the current work explicitly addressed the performance of Pt- and K-promoted gallia/alumina catalysts with TOS. Thus, the novel and important contribution of the current work is in generating and elucidating the experimental data on long-term stability of this system with TOS, which has not been reported previously.
Another feature of this study is that the catalyst support is produced using a waste-free and reagentless technology by thermal activation of gibbsite.15 This technology results in the particle size of alumina in the range of 40–100 μm allowing thus the dehydrogenation to be carried out using a fluidized-bed reactor. The thermal activation method is based on the transformation of nonporous crystalline gibbsite (Al(OH)3) into an amorphous product via pulsed heating of gibbsite on a metallic preheated surface (or in a gas flow) to the temperature of dehydration, which is followed by a rapid cooling step. Incomplete decomposition of the hydroxide gives a gibbsite pseudomorph, which, in contrast to gibbsite, has a developed pore structure, a large specific area, and a high chemical reactivity. The support precursor for this work was obtained from gibbsite under the following conditions: heating surface temperature = 620 °C; gibbsite mass flow = 175 kg h−1. According to DTA and X-ray diffraction, the activated gibbsite was composed of boehmite (ca. 17 wt%), an amorphous phase, and χ- and γ-Al2O3 (Fig. S1 and S2†). After calcination at 750 °C, the support had the following properties, which were determined using N2 physisorption: specific surface area = 143 m2 g−1, total pore volume = 0.26 cm3 g−1, average pore diameter = 5.7 nm and bulk density = 1.1 g cm−3 (Table S1†).
The promoted Ga2O3/Al2O3 catalysts were prepared using capillary impregnation of the alumina precursor by adding aqueous solutions of Ga(NO3)3·9H2O, H2[PtCl6]·6H2O and KOH. For convenience, the obtained catalysts were designated as in the following example, i.e. in xGa-700, x stands for the gallium content, and 700 – calcination temperature in °C.
According to SEM and EDX, some of the Ga2O3 is present on the surface of the support particles in the form of a coarse dispersed precipitate, while the rest is in the pores (Fig. S3 and Table S2†). The fact that Ga2O3 fills the pores of the support also follows from the pore size distribution (Fig. S4†), because the height of the maximum changes with a small displacement. The proportion of coarse particles of Ga2O3 on the support surface is small, therefore, the X-ray diffraction result displays only reflections characteristic of alumina. Supported Ga2O3 is in the β-modification, as shown in the XRD result of the sample with a high content of the active component (15 wt% Ga).8
A list of the prepared catalysts and their experimental conditions is summarized in Table S3.† The experiments were carried out in a cyclic mode for at least sixteen or more successive dehydrogenation–regeneration cycles in fluidized-bed and fixed-bed reactors. A standard duration of dehydrogenation was 10 min at 580 °C, and the regeneration in air was performed no longer than 30 min at 630 °C.
It is characteristic that the introduction of even small K additives to Ga2O3/Al2O3 leads to a sharp decrease in i-C4H10 conversion with an increase in selectivity to i-C4H8. The conversion of i-C4H10 and i-C4H8 selectivity after the 16th cycle for 6Ga-700 were 46% and 69%, respectively. In the case of adding 1.25 wt% K to the same amount of gallium, the isobutane conversion decreased by about half (up to 25%), while the selectivity increased to 86%. Such behaviour typical of consecutive reactions was observed in the literature.16 Interestingly in ref. 10, such significant effects upon addition of K were not observed.
It was considered that the promotion of supported gallia with alkali metals reduces coke deposition by poisoning the Brønsted acid sites (BASs).10 To assess how the Brønsted acidity depends on doping Ga2O3/Al2O3 with potassium, three different methods were used: TPD of NH3, FTIR using pyridine and selective adsorption of a series of acid–base indicators with different intrinsic pKa values in the range from −4.4 to 14.2.
Undoubtedly, potassium decreases the acidity of the samples according to NH3-TPD shown in Fig. 1. However, the BASs were not detected by FTIR of adsorbed pyridine. Dehydrogenation is known to occur on the Lewis acid sites (LASs),17 which are present in small amounts as weak sites on the supported gallia (Table S1†), according to FTIR of adsorbed pyridine.
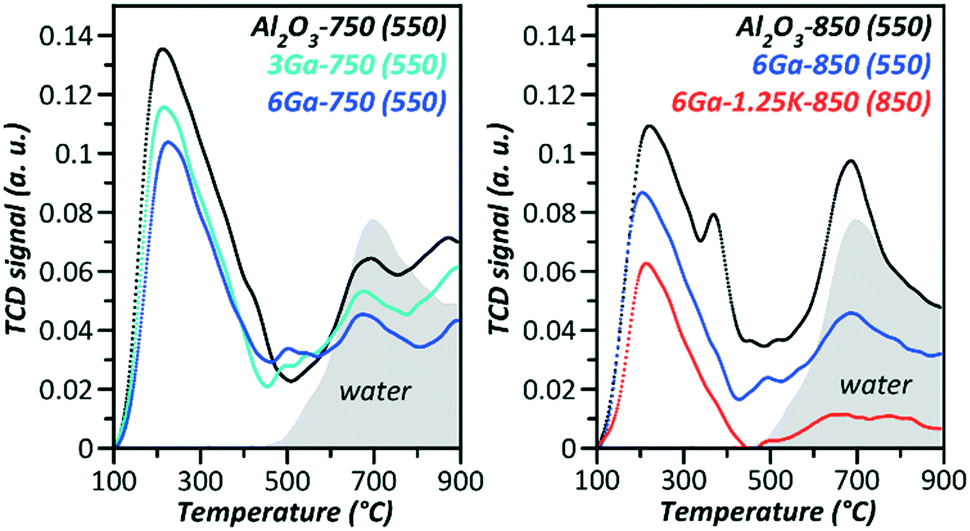 | ||
| Fig. 1 NH3-TPD spectra (in brackets are the designated pretreatment temperatures of a particular sample). | ||
In contrast to TPD and FTIR of adsorbed molecules, the indicator method turned out to be a more efficient tool for surface functionality characterization. When an indicator interacts with the solid surface, the changes in the color intensity because of adsorption on certain active sites can be measured spectrophotometrically and subsequently quantified.
Fig. 2 presents the distribution of the acid–base adsorption sites on the support, pure Ga2O3/Al2O3 and that promoted with potassium. The introduction of a relatively small amount of Ga2O3 (3% Ga) leads to a decrease of LASs with pKa = 14.2, which are pronounced in the support. After the introduction of potassium, there is an even larger decrease in LASs, while the Brønsted acidity (pKa = 6.4), in contrast, increases to the same extent as the decrease of LASs. Such apparently unexpected results on the influence of potassium on the Brønsted acidity can be explained by assuming the scheme of BAS formation upon interactions of KOH with LASs as presented in Fig. 2.
 | ||
| Fig. 2 Distribution of sites according to the acid–base strength, where Q(pKa) is the surface concentration of the adsorbed indicator; scheme of the BAS formation upon interactions of KOH with LASs. | ||
It is also important to note that in the case of simultaneous deposition of Pt and K without Ga, the catalyst exhibits a significantly lower isobutane conversion and isobutene selectivity than a sample containing only Ga and K. Moreover, an increase of conversion from the 1st to the 16th cycle was obtained for 3Ga-0.25K-750, while, in contrast, an activity decline could be seen for 0.1Pt-0.25K-750. It should be mentioned that the reducibility of supported Ga2O3 is controversial, as such a hypothesis is for example not supported by H2-TPR in ref. 8. This intriguing question requires a dedicated study, being, however, outside of the scope of the present work, which is focused on the synergy of Pt and K.
Promotion of Ga2O3/Al2O3 with Pt micro-additives makes it possible to increase both conversion and selectivity. One of the possible explanations is related to the reduction of Ga2O3, which is facilitated in the presence of easily reduced metals.18 However, in another work based on the data obtained by XPS and H2-TPR, it was concluded that the supported Ga2O3 is too stable to be reduced even in the presence of Pt.10 Similarly, it was shown in ref. 12 that Ga3+ with Pt was not reduced during the reaction.
According to FTIR of pyridine and N2-physisorption data, no significant changes in the specific surface area and acidity were observed upon doping Ga2O3/Al2O3 with Pt (Table S1 and Fig. S5†). Thus, the experimental dataset generated in the present work does not allow elucidation of any potential links between acidity changes and the catalytic performance.
The authors of ref. 10 postulated that Pt assists in the recombination of the H atoms on the catalyst, making the active sites available for the following dehydrogenation cycle.
Obviously, there is some optimal ratio of Ga and Pt, because there is no difference in the i-C4H10 conversion and selectivity to i-C4H8 between 3Ga-0.1Pt-750 and 1.5Ga-0.05Pt-750, contrary to 3Ga-0.01Pt-750.
The largest effect can be achieved upon promoting Ga2O3 with both Pt and K. Addition of 0.25% K to the Ga–Pt/Al2O3 catalyst increases the isobutene selectivity by 10% keeping the same i-C4H10 conversion. This effect is fundamentally different from the effect of K on the catalysts containing only Ga or Pt, i.e. no drop in activity. With a decrease in the Pt content to 0.01 wt%, the selectivity of the K-promoted Ga2O3/Al2O3 catalyst increases.
In ref. 10, it was shown that propane conversion and propene selectivity dropped after the 8th cycle for all supported Ga–Pt–K catalysts. In the current study, the obtained Ga–Pt–K catalysts are very stable, even after more than 300 cycles, and isobutane conversion and isobutene selectivity remain unchanged, which proves that the catalysts did not deactivate. Explanation for such behaviour is challenging as the properties of the support were not specified in ref. 10. Moreover, the results indirectly indicate good adhesion of the active components to the catalyst surface despite abrasion in a fluidized-bed reactor.
The equilibrium conversion for isobutane dehydrogenation is ∼66% when the selectivity is 100%. In the current case, considering the obtained selectivity, the equilibrium conversion was achieved for all the catalysts except for 3Ga-0.25K-750 and 0.1Pt-0.25K-750. Similar conversion values of 60% for 3Ga-0.1Pt-0.25K-750 obtained in the fixed and fluidized bed reactors, with GHSV ca. 5 times higher in the former case, can be also attributed to the attainment of equilibrium.
Fig. 3 shows the changes in the isobutene selectivity vs. isobutane conversion. Essentially, the catalytic behavior should not depend on the reactor type if the reaction occurs under the same conditions in the kinetic regime as in the current case. The absence of mass and heat transfer limitations was confirmed by calculating the Weisz–Prater and Anderson criteria,19 which were at least two orders of magnitude below the threshold. The dependence of selectivity on the conversion for the Ga–Pt catalysts is typical for parallel or parallel–consecutive reactions. Namely, there are regions in which selectivity remains unchanged with a change in conversion, as expected for parallel reactions (i.e. dehydrogenation of isobutene and its cracking to propene and methane). At higher concentrations, a decrease in selectivity is visible in Fig. 3 associated with secondary transformations of a consecutive nature (e.g. further reactions of formed olefins). A clearly different dependence was observed for the Ga–K and Pt–K catalysts in a fixed-bed reactor displaying an increase of selectivity with conversion. For the Pt–K catalyst, this is most likely due to sintering of the Pt-containing catalyst, with a concomitant loss of active sites resulting in catalyst deactivation.2
 | ||
| Fig. 3 Dependence of isobutene selectivity on isobutane conversion for the samples calcined at 750 °C and tested at 580 °C in the presence of H2; in brackets are cycles and point numbers. | ||
An increase in selectivity and conversion was not previously detected for Ga2O3/Al2O3;7 therefore, the reason for the catalytic behavior observed in the current work is the promotion of the Ga2O3/Al2O3 catalyst with K. The effect of potassium addition can be due to either a direct action on Ga2O3 or to an indirect influence on the support, thereby altering the active phase–support interactions.
For the sake of comparison, the dehydrogenation performance of an industrially relevant reference material based on CrOx/Al2O3 was evaluated using the same set-up (Fig. 4 and Table 1).
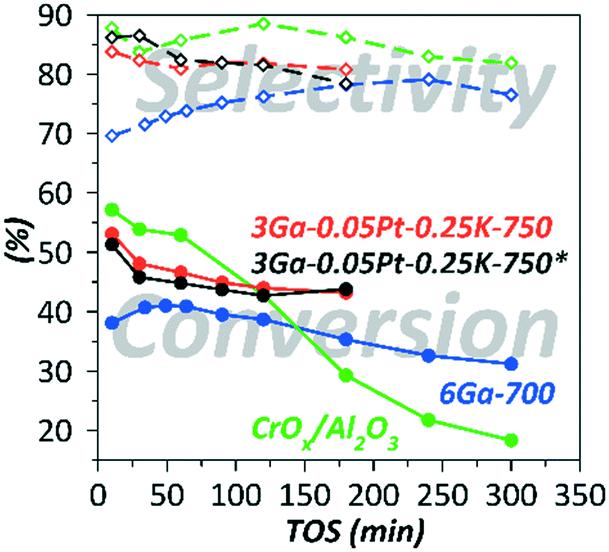 | ||
| Fig. 4 Changes in the catalytic behavior with TOS in a fluidized-bed reactor at 580 °C and GHSV = 400 (i-C4H10); *in the presence of H2. | ||
| Catalyst | Temperature (°C)/time (min)/GHSV of feed (h−1) during dehydrogenation | Bed/dilution | End cycle | X (%) | r(i-C4H10) (mmol h−1 gcat−1) | Y (wt%) | S (%) | C (wt%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 10.95Cr-0.74Zr-1.25K (KDM-16) | 580/10/400 | Fluidized/− | 16 | 65 | — | 56 | 89 | 1.8 | 7 |
| 3Ga-0.05Pt-0.25K | 580/10/400 | Fluidized/− | 199 | 63 | 2.9 | 52 | 83 | 0.6 | This work |
| 3Ga-0.01Pt-0.25K | 580/10/2100 | Fixed/H2 | 128 | 54 | 11.9 | 51 | 94 | 0.1 | This work |
| 0.2Pt-0.7Sn | 580/15/400 | Fluidized/− | 155 | 40 | — | 25 | 64 | — | 9 |
| 0.9Ga-0.002Pt-0.15K | 580/15/400 | Fluidized/− | 155 | 44 | — | 37 | 86 | — | 9 |
| 0.7Pt-0.3Sn-0.8K | 550/60/7.1 | Fixed/He | 20 | ∼7 | — | ∼6 | ∼93 | — | 12 |
| 3Ga-0.1Pt-1Ce | 550/60/7.1 | Fixed/He | 20 | 51 | — | ∼49 | ∼96 | — | 12 |
The selectivity of the Ga–Pt–K-containing catalyst did not exceed the values obtained for CrOx/Al2O3. However, from Fig. 4, where the results of experiments performed for 3 hours of TOS are presented, it can be seen that CrOx/Al2O3 exhibited the highest initial isobutane conversion into a more reactive isobutene; therefore the coke formation accelerated with conversion resulting in an increase of the deactivation rate with TOS. In contrast to the Cr-based catalyst, the Ga-containing catalysts exhibited a typical exponential type of activity decay.
Moreover, the Ga–Pt–K catalyst demonstrated a performance superior to Ga and Pt-containing catalysts obtained in the previous studies on isobutane dehydrogenation in terms of isobutane conversion and selectivity to isobutene (Table 1).
It can also be noted that the doping of gallia/alumina catalysts with Pt and K microadditives does not affect the catalyst stability, which is ascribed to the behavior of gallium oxide.
Dilution of the feed with H2, which previously was not considered for Ga-containing catalysts, prevents the formation of coke. Despite the small differences in the catalyst performance with and without H2, there was a clear effect on the coke formation.
The coke content after dehydrogenation of pure i-C4H10 on 3Ga-0.1Pt-0.25K-750 is 1.6 wt%. The coke content could be diminished two-fold (to 0.8 wt%) when diluting the feed with H2. Even more spectacular results were obtained for the catalyst denoted as 3Ga-0.05Pt-0.25K-750, where the coke content decreased from 1.4% to 0.3%. Data on the distribution of products also indicate suppression of cracking reactions with the formation of C1–C3 in the presence of H2 (Table S4 and Fig. S6†).
In summary, an alumina supported gallia catalyst promoted with tiny amounts of Pt and K showed a significantly increased activity and selectivity for isobutane dehydrogenation both in fixed-bed and fluidized-bed reactors. The catalyst has a remarkably enhanced long-term stability in the isobutane stream in comparison with commercial catalysts. This catalyst is also environmentally friendly, not requiring utilization of any wash water during preparation of the support. In terms of the cost, it is competitive with commercial platinum catalysts; moreover gallium, similar to platinum, can be recovered and reused, avoiding disposal costs.
Conflicts of interest
There are no conflicts to declare.Notes and references
- https://www.transparencymarketresearch.com/isobutene-market.html.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS.
- K. J. Caspary, H. Gehrke, M. Heinritz-Adrian and M. Schwefer, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knozinger and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 3206–3229 Search PubMed.
- https://www.uop.com/processing-solutions/petrochemicals/olefins/; https://pet-oil.blogspot.com/2012/10/uop-oleflex-process-for-light-olefin.html.
- https://www.honeywell-uop.cn/wp-content/uploads/2011/02/UOP-DeH16-Catalyst-Data-Sheet.pdf.
- T. Imai and C.-W. Hung, US Pat., US4438288, 1984 Search PubMed.
- A. N. Matveyeva, N. A. Zaitseva, P. Mäki-Arvela, A. Aho, A. K. Bachina, S. P. Fedorov, D. Yu. Murzin and N. A. Pakhomov, Ind. Eng. Chem. Res., 2018, 57, 927–938 CrossRef CAS.
- A. N. Matveyeva, Sh. O. Omarov, D. A. Sladkovskiy and D. Yu. Murzin, Chem. Eng. J., 2019, 372, 1194–1204 CrossRef CAS; A. N. Matveyeva, J. Wärnå, N. A. Pakhomov and D. Yu. Murzin, Chem. Eng. J., 2020, 381, 122741 CrossRef.
- R. Iezzi, A. Bartolini and F. Buonomo, US Pat., US7235706B2, 2007 Search PubMed.
- J. J. H. B. Sattler, I. D. González-Jiménez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, M. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2014, 53, 9251–9256 CrossRef CAS.
- Q. Yu, T. Yu, H. Chen, G. Fang, X. Pan and X. Bao, J. Energy Chem., 2020, 41, 93–99 CrossRef.
- J. Im and M. Choi, ACS Catal., 2016, 6, 2819–2826 CrossRef CAS.
- T. Bauer, S. Maisel, D. Blaumeiser, J. Vecchietti, N. Taccardi, P. Wasserscheid, A. Bonivardi, A. Görling and J. Libuda, ACS Catal., 2019, 9, 2842–2853 CrossRef CAS; T. Wang, F. Jiang, G. Liu, L. Zeng, Z.-J. Zhao and J. Gong, AIChE J., 2016, 62, 4365–4376 CrossRef; E. L. Jablonski, A. A. Castro, O. A. Scelza and S. R. de Miguel, Appl. Catal., A, 1999, 183, 189–198 CrossRef; P. Sun, G. Siddiqi, M. Chi and A. T. Bell, J. Catal., 2010, 274, 192–199 CrossRef; G. Siddiqi, P. Sun, V. Galvita and A. T. Bell, J. Catal., 2010, 274, 200–206 CrossRef.
- R. A. Pierce, L. Luo, M. M. Olken, S. Domke and H. W. Clark, US Pat., US8653317B2, 2014 Search PubMed.
- A. N. Matveyeva, N. A. Pakhomov and D. Yu. Murzin, Ind. Eng. Chem. Res., 2016, 55, 910–9108 CrossRef.
- P. Michorczyk and J. Ogonowski, Appl. Catal., A, 2003, 251, 425–433 CrossRef CAS.
- M. Chen, J. Xu, F. Su, Y. Liu, Y. Cao, H. He and K. Fan, J. Catal., 2008, 256, 293–300 CrossRef CAS.
- L. Rodriguez, D. Romero, D. Rodriguez, J. Sanchez, F. Dominguez and G. Arteaga, Appl. Catal., A, 2010, 373, 66–70 CrossRef CAS.
- D. Yu. Murzin, Engineering Catalysis, De Gruyter, 2020, p. 540 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01366h |
| This journal is © The Royal Society of Chemistry 2020 |