
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triarylborane catalysed N-alkylation of amines with aryl esters†‡
Valeria
Nori
 ab,
Ayan
Dasgupta
*a,
Rasool
Babaahmadi
c,
Armando
Carlone
b,
Alireza
Ariafard
c and
Rebecca L.
Melen
*a
ab,
Ayan
Dasgupta
*a,
Rasool
Babaahmadi
c,
Armando
Carlone
b,
Alireza
Ariafard
c and
Rebecca L.
Melen
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, Cymru/Wales, UK. E-mail: MelenR@cardiff.ac.uk
bDepartment of Physical and Chemical Sciences, Università degli Studi dell'Aquila, via Vetoio, 67100 L'Aquila, Italy
cSchool of Natural Sciences-Chemistry, University of Tasmania Private Bag 75, Hobart, Tasmania 7001, Australia
First published on 16th September 2020
Abstract
The ability of halogenated triarylboranes to accept a lone pair of electrons from donor substrates renders them excellent Lewis acids which can be exploited as a powerful tool in organic synthesis. Tris(pentafluorophenyl)borane has successfully demonstrated its ability to act as a metal-free catalyst for an ever-increasing range of organic transformations. Herein we report the N-alkylation reactions of a wide variety of amine substrates including diarylamines, N-methylphenyl amines, and carbazoles with aryl esters using catalytic amounts of B(C6F5)3. This mild reaction protocol gives access to N-alkylated products (35 examples) in good to excellent yields (up to 95%). The construction of a C–N bond at the propargylic position has also been demonstrated to yield synthetically useful propargyl amines. On the other hand, unsubstituted 1H-indoles and 1H-pyrroles at the C3/C2 positions afforded exclusively C–C coupled products. Extensive DFT studies have been employed to understand the mechanism for this transformation.
Introduction
Over the last decade there has been a surge of interest in the applications of triarylboranes as metal-free catalysts for a range of transformations. This research originates from the seminal work of Piers who, in 1996, demonstrated the use of tris(pentafluorophenyl)borane [B(C6F5)3] as an effective catalyst for the hydrosilylation of carbonyl compounds.1 A decade later a derivative of this borane, containing a p-PMes2 functionality on one of the aryl rings, was reported to be capable of reversibly activating H2 in the absence of a transition metal.2 This system, comprising unquenched Lewis acidic and basic sites, was subsequently termed a frustrated Lewis pair (FLP).3 These two key findings by Piers and Stephan, as described above, have altered the way chemists think about the reactivity of triarylborane Lewis acids prompting the discovery of new catalytic reactivity of these boron compounds.4,5 Many researchers have investigated the breadth of reactions B(C6F5)3 can catalyze, whereas others have focused on catalyst modification through subtle alterations of the aryl rings to influence the accessibility and energy of the empty p-orbital at boron.5 One of the most studied reactions in FLP chemistry has been the application of B(C6F5)3 (and derivatives) for the FLP-catalyzed hydrogenation of imines to generate amines.6–8 Owing to the importance of nitrogen-containing compounds in nature, new synthetic methods to develop C–N bonds in a mild and efficient manner are continuously being sought.9 One common approach to synthesize C–N bonds is through reductive amination, a reaction that has been commonly mediated using transition metal catalysts.10,11 Several groups (including ourselves) have employed a metal-free FLP-catalyzed strategy for reductive amination using the Lewis acidic boranes B(C6F5)3, B(2,6-Cl2C6H3)(p-HC6F4)2, and B(2,6-Cl2C6H3)2(2-Cl-6-FC6H3) (Scheme 1). In these reactions, the primary or secondary amine is initially condensed with an aldehyde or ketone and, in the second step, the borane catalyzes the reduction of the imine using H2 (ref. 12) or a silane.13 Recent studies indicate that B(C6F5)3 can also be used as effective catalyst towards the N-alkylation of various amines. Chan et al. reported the solvent dependent chemoselective N-alkylation of aryl amines with alcohols using B(C6F5)3 as a catalyst.14 Furthermore, N-alkylation of primary and secondary aromatic amines and amides with primary, secondary, tertiary benzylic alcohols has been demonstrated by Maji et al. Activation of the dibenzyl ether intermediate by triarylfluoroboranes was identified as the rate determining step.15 Here we report an alternative borane catalyzed approach to Csp3–N bond formation through the N-alkylation of an amine with an ester catalyzed by B(C6F5)3. Unlike the FLP reductive amination approach, these reactions occur at lower temperatures and do not require the use of hydrogen gas or silane reductants. A mild reaction protocol for synthesis of propargyl amines has also been highlighted.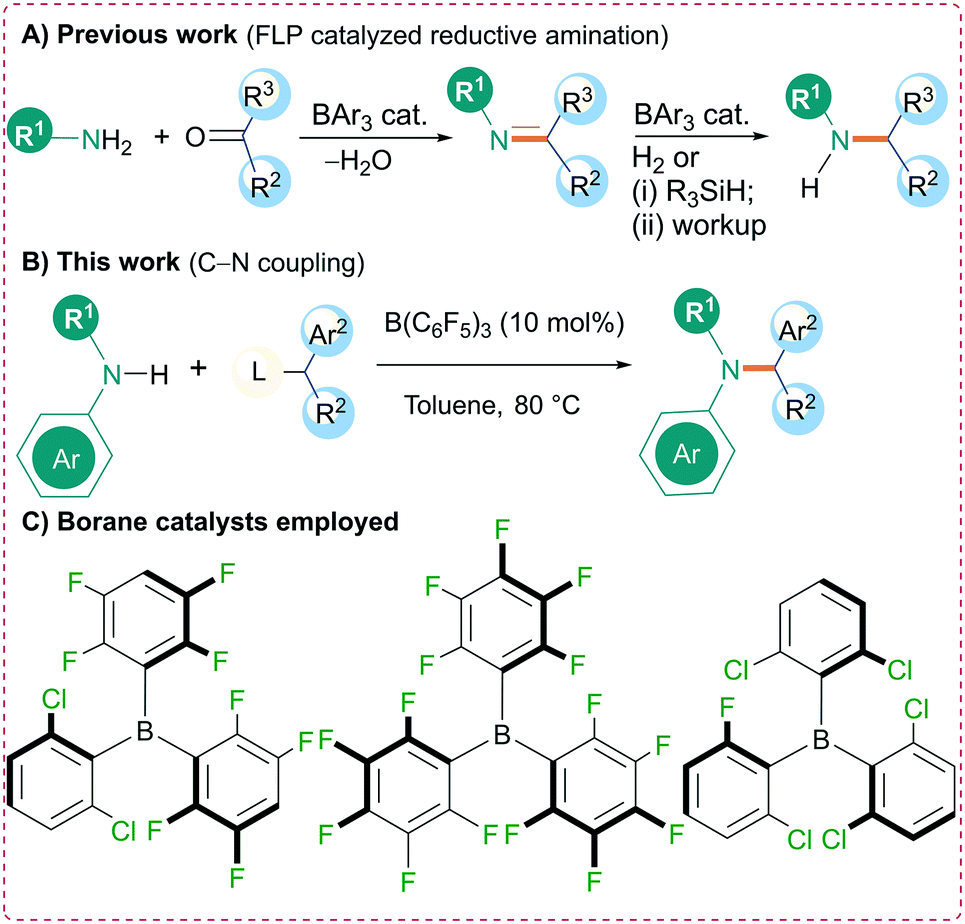 | ||
| Scheme 1 Amine synthesis catalyzed by triarylboranes. A) Previous work on the use of triarylboranes in reductive amination. B) This work on borane catalyzed N-alkylation. C) Borane catalysts employed in current and previous studies. | ||
Initially, we investigated the N-alkylation reaction of diphenyl amine (1a) with p-fluorobenzoate diarylester 2a as a model system (Table 1). In the absence of a borane catalyst no reaction occurs in toluene. However, addition of 10 mol% B(C6F5)3 led to C–N bond formation with elimination of p-fluorobenzoic acid to generate the N-alkylated product 3a in excellent isolated yields of 92% after 16 h at 80 °C. The p-fluorobenzoate esters were selected due to the ease of reaction monitoring by 19F NMR spectroscopy.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temperature (°C) | Time (h) | Yield (%) |
| Yields reported are isolated yields. All reactions were carried out in 0.1 mol% scale using 10 mol% catalyst. NR indicates no reaction. a 5 mol% catalyst employed. | |||||
| 1 | None | Toluene | 80 | 24 | NR |
| 2 | B(C6F5)3 | Toluene | 80 | 16 | 92 |
| 3 | BCl3 | Toluene | 80 | 24 | NR |
| 4 | BPh3 | Toluene | 80 | 24 | 5 |
| 5 | CF3SO3H | Toluene | 80 | 22 | 39 |
| 6 | p-FC6H4CO2H | Toluene | 80 | 22 | NR |
| 7 | B(C6F5)3 | Toluene | 110 | 16 | 52 |
| 8 | B(C6F5)3 | Toluene | RT | 42 | NR |
| 9 | B(C6F5)3 | CH2Cl2 | 45 | 24 | 56 |
| 10 | B(C6F5)3 | THF | 60 | 24 | 45 |
| 11 | B(C6F5)3 | TFT | 80 | 24 | 51 |
| 12 | B(C6F5)3 | Hexane | 65 | 24 | 64 |
| 13 | B(C6F5)3 | Acetonitrile | 80 | 24 | 25 |
| 14 | B(C6F5)3 | Et2O | 40 | 24 | 36 |
| 15 | B(C6F5)3a | Toluene | 80 | 24 | 30 |
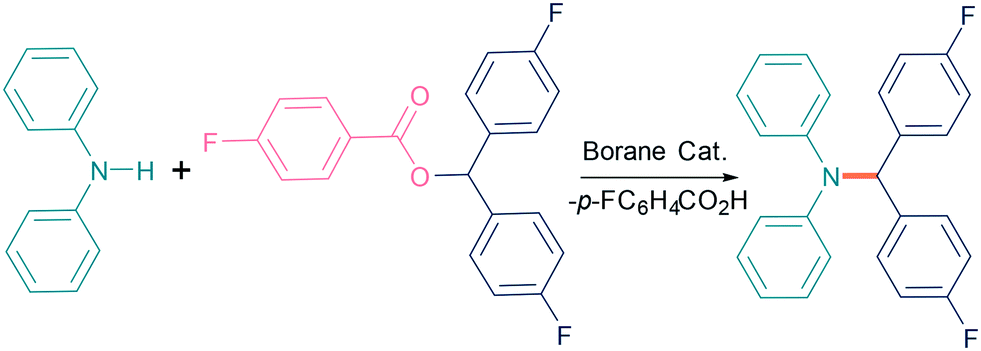
Other boranes, including the more Lewis acidic BCl3 and less Lewis acidic BPh3, gave little or no reaction under identical conditions. Likewise, Brønsted acids such as triflic or p-fluorobenzoic acid gave low or no conversion. Reaction at higher temperature (110 °C) using 10 mol% B(C6F5)3 led to the formation of many other small impurities and lower isolated yields (52%) than the reaction at 80 °C. On the other hand, the room temperature reaction showed no conversion toluene was found to be the best solvent for the reaction with CH2Cl2 (56%), α,α,α-trifluorotoluene (TFT) (51%), and hexane (64%) showing only moderate yields. In addition, the more coordinating solvents THF (45%), Et2O (36%), and acetonitrile (25%) showed a further reduction in yields. Reducing the catalytic loading to 5 mol% also drastically reduced the yield to just 30% after 24 h.
With the optimized conditions in hand, the substrate scope for the reaction was investigated (Scheme 2). Initially, secondary diaryl amines 1a–c were reacted with a range of symmetrical diaryl esters 2a–d (see ESI† for details of starting material structures and their synthesis) to generate the N-alkylated products 3a–j in good to excellent yields (63–92%). The reactions were found to be tolerant to electron withdrawing (p-F) and electron donating (p-OMe) groups on the ester but were generally limited to more electron rich amines. Unsymmetrical diaryl esters 2e–f also worked well giving products 3k–n in good to excellent yields (70–89%). Several alkynyl/aryl substituted esters 2g–i also performed well showing N-alkylation at the propargylic position to generate propargyl amines 3o–q in 55–75% isolated yields. Indeed, the synthesis of propargylic amines is often metal catalyzed16 and new routes to these compounds are important owing to their prevalence in biologically active compounds, as well as their applications in medicinal and synthetic chemistry. Aryl/alkyl esters 2j–l could also be employed; however, with the tert-butyl group, the reaction provided the N-alkylated product 3s in only 21% yield. Fortunately, the efficiency of the reaction could be restored by employing cyclohexyl and methyl substituents yielding 3r, t–v in 65–72% isolated yields. Likewise, changing the amine to have an aliphatic group was also successful and reactions of N-methyl aniline (1d) with esters gave 3w–y in 71–82% yield. Indoline (1e), on the other hand gave poorer yield for the alkylated product 3z of 32% as calculated from crude NMR. Limitations of this reaction included the aliphatic amines diisopropylamine, morpholine, and piperidine, which showed no reaction even when elevating the temperature to 120 °C. The higher basicity of these aliphatic amines, and the potential to react with B(C6F5)3 through hydride abstraction,17 compared with aromatic amines makes them incompatible with the Lewis acidic boranes to afford the desired N-alkylated products. This was also observed in NMR experiments using equimolar mixtures of diisopropyl amine and B(C6F5)3 which showed several reaction products in the in situ11B NMR spectrum.
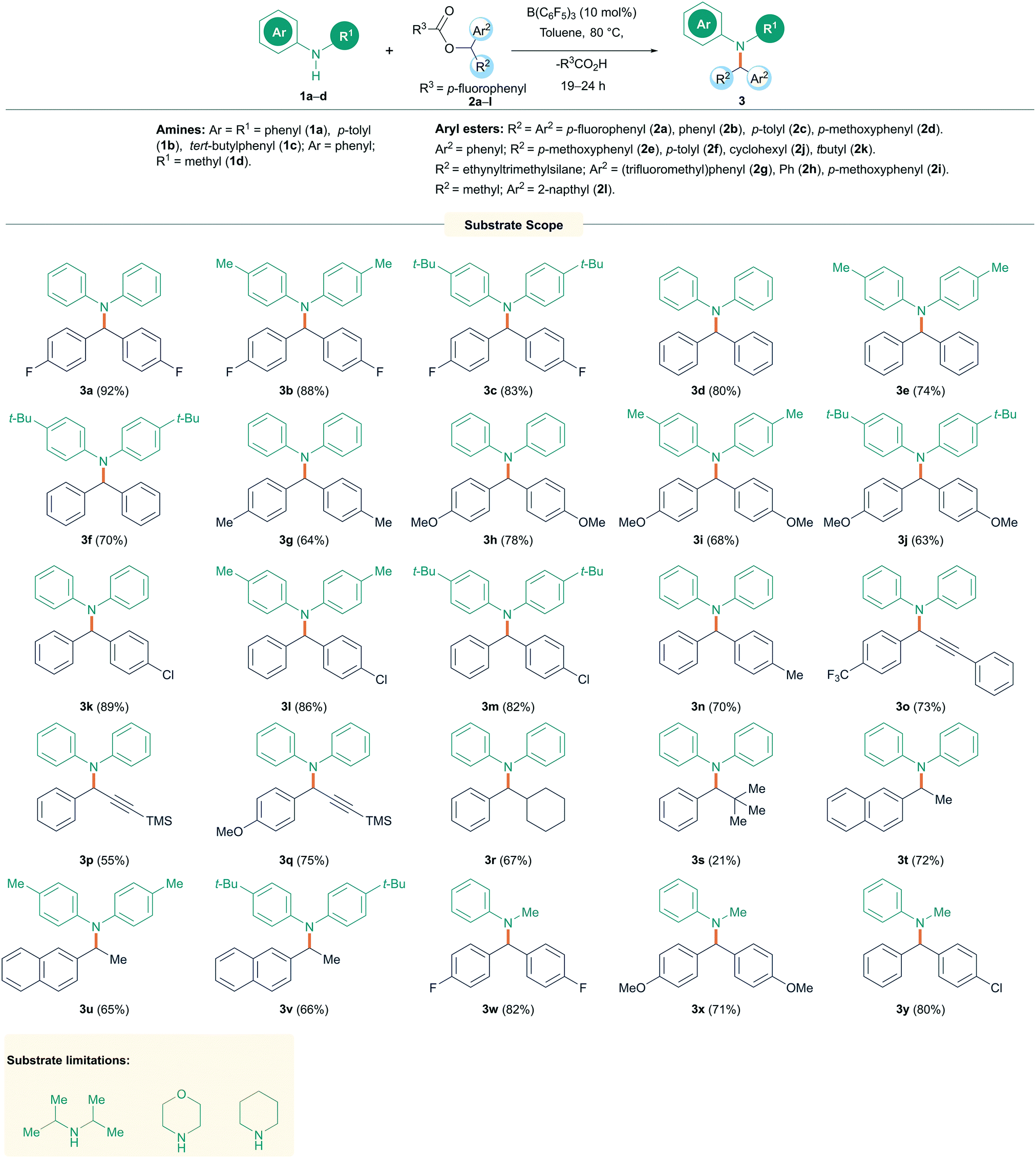 | ||
| Scheme 2 N-Alkylation reactions of amines with diaryl esters. All reactions were carried out on a 0.1 mmol scale. Yields reported are isolated yield. aCrude NMR yield. | ||
The primary amine, aniline, on the other hand gave a complex mixture of products including the secondary amine product from mono alkylation and the tertiary amine product from double alkylation at the nitrogen atom.
Following this, we turned our attention to N-alkylation of aromatic N-heterocycles including carbazoles and indoles with aryl esters (Scheme 3). N-Alkyl carbazoles are an important class of bioactive heteroaromatic compounds.18 Their synthesis often relies on an SN2 reaction, but this is sensitive to more hindered electrophiles, although metal catalyzed processes have recently been reported.19 Other syntheses typically involve heterocycle generation after incorporation of the alkyl group at nitrogen. This includes precious metal catalyzed routes, benzannulation or oxidative cyclization reactions.20 We therefore sought to employ our methodology in the synthesis of N-alkyl heterocycles. 9H-Carbazole (1f) and 3,6-dibromo-9H-carbazole (1g) were reacted with esters 2a, b and d, giving excellent yields (81–95%) of the N-alkylated products (4a–f). As before, the reaction worked well with both electron donating and electron withdrawing groups on the ester. However, when employing electron donating groups on the amine (3,6-dimethoxy-9H-carbazole, 1h), a complicated reaction mixture was obtained, and the presence of unreacted starting material was observed by TLC. The C3 and C2 substituted indole 2,3-dimethyl-1H-indole (1i) could also be employed giving the products 5a–c in good to excellent yields (64–80%) when reacted with esters bearing electron donating or electron withdrawing groups. Suitable crystals of 4c and 5a were obtained from slow evaporation of saturated CH2Cl2 solutions and could be characterized by single-crystal X-ray diffraction (Fig. 1). Surprisingly, when 1H-indole (1j) was treated with 2a, no N-alkylated product was observed and exclusive formation of the C–C cross-coupled product (6a) was isolated in excellent yield (93%). Likewise, 1H-pyrrole (1k) also exhibited similar reactivity and the C–C cross-coupled product (7a) was isolated exclusively in 95% yield. In both cases, addition of further equivalents of the ester 2 still saw no N-alkylation reaction. The generation of the C-alkylated product is likely due to the formation of a Lewis acid–base adduct between the indole or pyrrole and the borane which protects the nitrogen centre from alkylation. We have observed similar selectivity in the reactions of these substrates with diazo esters catalyzed by B(C6F5)3.21 Similarly, blocking all other positions as in 2,3,4,5-tetramethyl pyrrole displayed no product formation. 2-Oxyindole was also tested for the N-alkylation reaction with 2a but complicated mixtures of products resulted along with unreacted 2a.
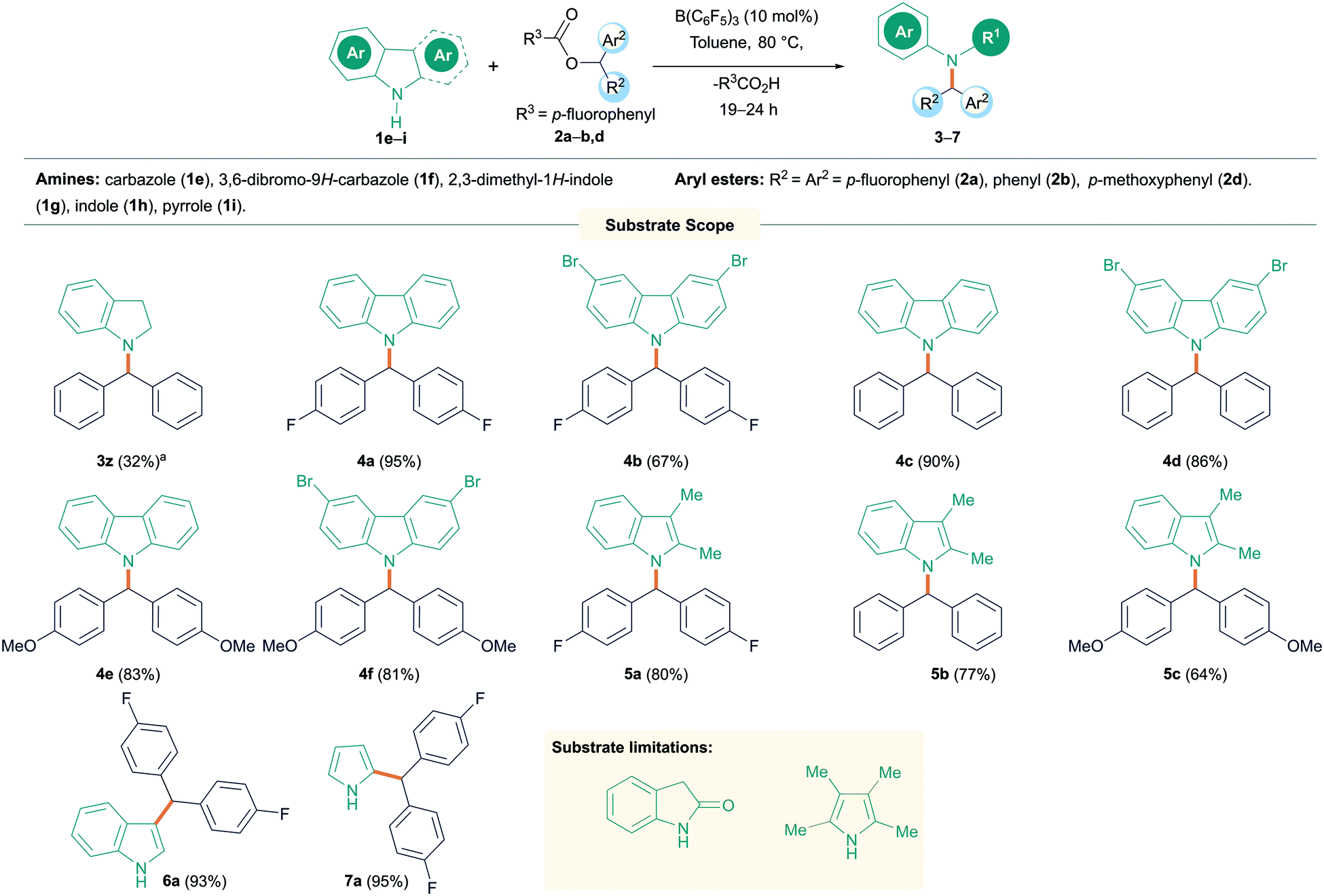 | ||
| Scheme 3 N-Alkylation reactions of amines with diaryl esters. All reactions were carried out on a 0.1 mmol scale. Yields reported are isolated yield. aCrude NMR yield. | ||
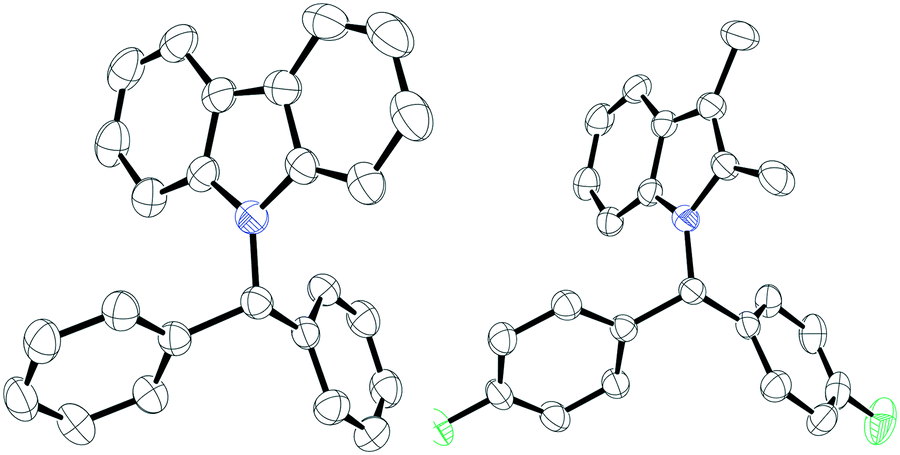 | ||
| Fig. 1 Solid-state structures of compound 4c (left) and 5a (right). Thermal ellipsoids drawn at 50% probability. Carbon: black; nitrogen: blue; fluorine: green. | ||
To investigate the mechanism for the N-alkylation reaction, we undertook a series of DFT calculations. We hypothesized that three mechanisms could be possible for the reaction: a Lewis acid-assisted Brønsted acid-catalyzed reaction from activation of the amine (Scheme 4a) or the carboxylic acid by-product (Scheme 4b), or a Lewis acid-catalyzed reaction (Scheme 4c). In all cases, the Lewis/Brønsted acid activates the ester generating a carbenium species that is subsequently trapped by the amine nucleophile. The generation of the carbenium ion corroborates the observation that esters used in the reaction must bear groups able to stabilize a positive charge. Calculations into the first mechanism (Scheme 4a and ESI,† Fig. S115) at the SMD/M06-2X/def2-TZVP//CPCM/ B3LYP/6-31G(d) level of theory show that the borane initially coordinates to the amine generating an acidic proton. This adduct formation was found to be energetically unfavorable relative to the free Lewis acid and base by 4.4 kcal mol−1 presumably due to steric hindrance. Upon coordination, the acidity of the amine increases, however, the next step of the reaction (protonation of the ester) had a high activation barrier of 40.5 kcal mol−1 (Fig. S115†). An alternative Brønsted acid catalyzed pathway could operate in which the borane activates the 4-fluorobenzoic acid side product, generating an acidic proton (Scheme 4b and Fig. S116†). As in the first case, this protonates the ester 2, yielding carbenium intermediate I2. The activation barrier for the overall reaction was found to be much lower in this case (11.3 to 16.1 kcal mol−1) when calculated at a range of levels of theory (Table 2). However, experimental results (Table 1, entry 6) showed no conversion using p-fluorobenzoic acid as a catalyst.
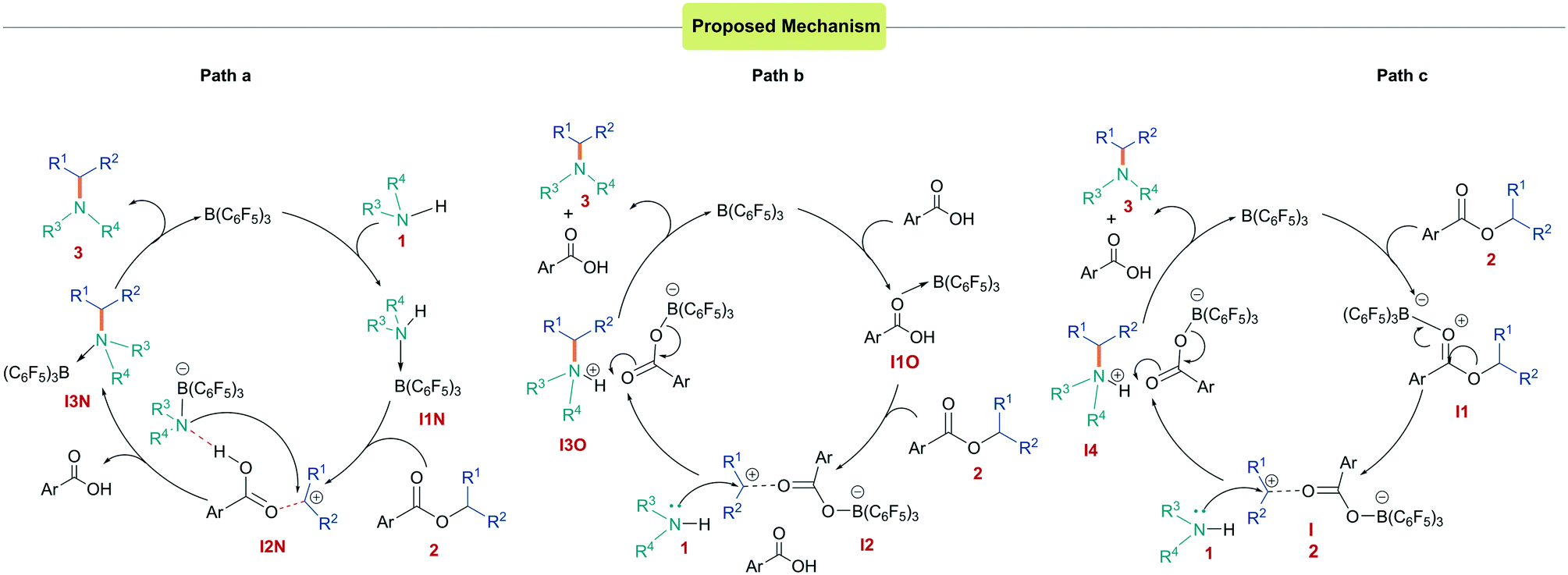 | ||
| Scheme 4 Catalytic cycles investigated for the reaction. (i) Brønsted acid catalyzed from activation of the amine (a) or carboxylic acid (b). (ii) Lewis acid catalyzed (c). | ||
| Level of theory | Lewis acid catalyzed | Brønsted acid catalyzed |
|---|---|---|
| CPCM/ωB97XD/def2-TZVP//CPCM/B3LYP/6-31G(d) | 10.6 kcal mol−1 | 12.3 kcal mol−1 |
| CPCM/M06-2X/def2-TZVP//CPCM/B3LYP/6-31G(d) | 13.5 kcal mol−1 | 14.8 kcal mol−1 |
| SMD/ωB97XD/def2-TZVP//CPCM/B3LYP/6-31G(d) | 11.1 kcal mol−1 | 13.5 kcal mol−1 |
| SMD/M06-2X/def2-TZVP//CPCM/B3LYP/6-31G(d) | 14.1 kcal mol−1 | 16.1 kcal mol−1 |
| SMD/ M06-2X-D3/def2-TZVP//CPCM/B3LYP/6-31G(d) | 10.8 kcal mol−1 | 11.3 kcal mol−1 |
Finally, we turned our attention to the Lewis acid catalyzed pathway in which boron acts as a Lewis acid catalyst to activate the ester (Scheme 4c). DFT studies showed that the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ester:borane adduct was favorable over a 1:2 adduct.22 Calculations into the reaction indicated that this pathway was also favorable under the reaction conditions with an activation barrier of 18.2 kcal mol−1 relative to the starting materials (Fig. 2). It was found that coordination of the borane to the ester 2 was the initial step of the reaction to give the adduct I1via a low energy transition state (TS1) of 7.5 kcal mol−1. I1 was found to have an elongated C–O bond length of 1.510 Å, which results in bond cleavage and the generation of an electrophilic carbenium ion in salt I2 occurring with an activation barrier of 13.6 kcal mol−1 through TS2. I2 exists as a close ion pair and reacts with the amine nucleophile 1viaTS3 (activation barrier 7.6 kcal mol−1) to give I3 then I4. Finally, deprotonation through TS4 with a low activation barrier of 7.0 kcal mol−1 releases the by-product 4-fluorobenzoic acid to generate the N-alkylated product and to regenerate the B(C6F5)3 catalyst. In the 1:1 stoichiometric reaction of the ester with 2a with B(C6F5)3, p-fluorobenzoic acid was isolated supporting this pathway.
:1 ester:borane adduct was favorable over a 1:2 adduct.22 Calculations into the reaction indicated that this pathway was also favorable under the reaction conditions with an activation barrier of 18.2 kcal mol−1 relative to the starting materials (Fig. 2). It was found that coordination of the borane to the ester 2 was the initial step of the reaction to give the adduct I1via a low energy transition state (TS1) of 7.5 kcal mol−1. I1 was found to have an elongated C–O bond length of 1.510 Å, which results in bond cleavage and the generation of an electrophilic carbenium ion in salt I2 occurring with an activation barrier of 13.6 kcal mol−1 through TS2. I2 exists as a close ion pair and reacts with the amine nucleophile 1viaTS3 (activation barrier 7.6 kcal mol−1) to give I3 then I4. Finally, deprotonation through TS4 with a low activation barrier of 7.0 kcal mol−1 releases the by-product 4-fluorobenzoic acid to generate the N-alkylated product and to regenerate the B(C6F5)3 catalyst. In the 1:1 stoichiometric reaction of the ester with 2a with B(C6F5)3, p-fluorobenzoic acid was isolated supporting this pathway.
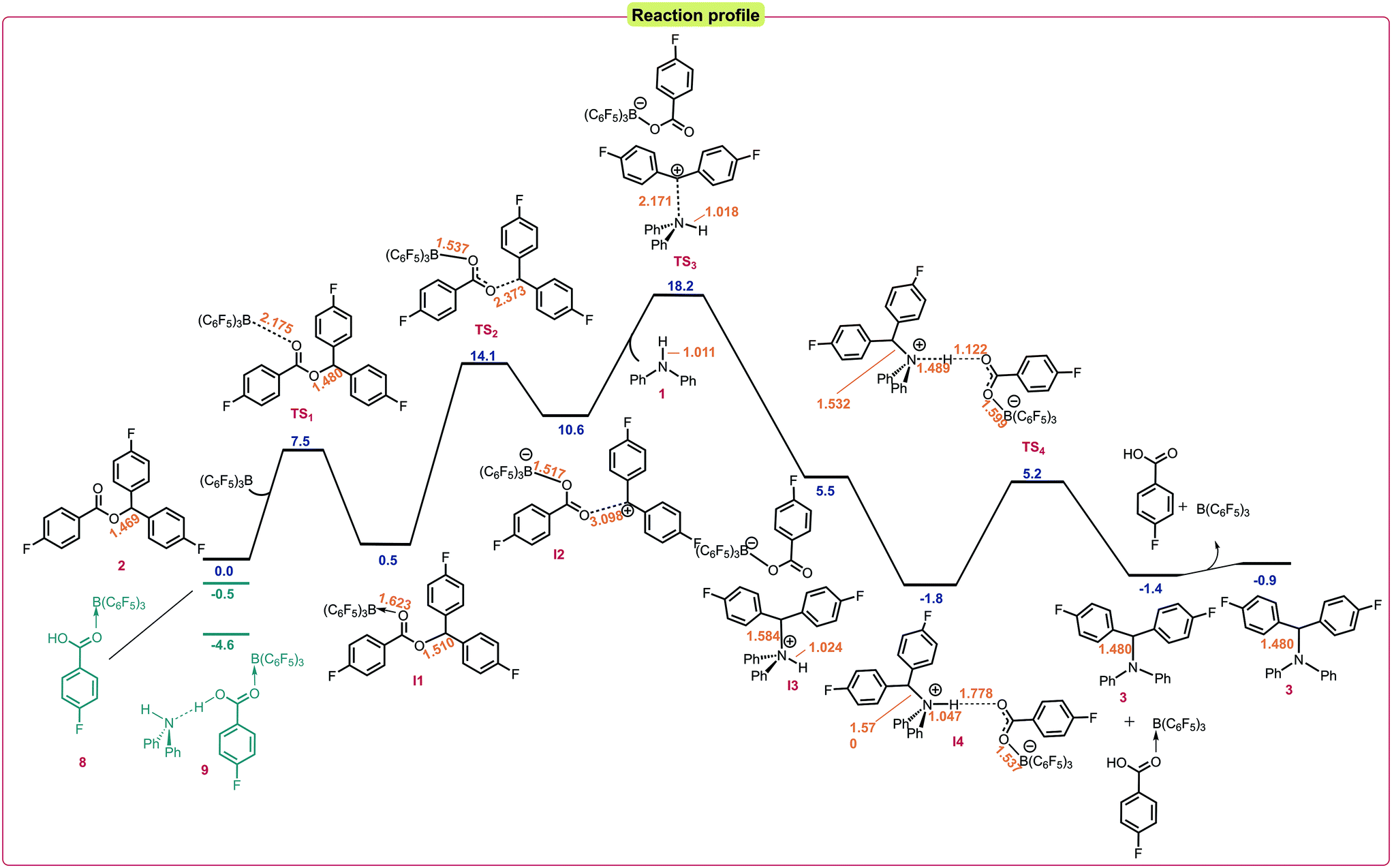 | ||
| Fig. 2 Reaction profile for the DFT calculated mechanism at the SMD/M06-2X/def2-TZVP//CPCM/B3LYP/6-31G(d) level of theory. Bond lengths shown in Å. Relative energies given in kcal mol−1. | ||
Since the overall activation energy for formation of carbenium species for latter two pathways (b and c) were quite similar, we compared the energy barrier for this step at various levels of theory (Table 2). In all cases, the energy barrier was slightly lower by 0.5 to 2.4 kcal mol−1 for the Lewis acid catalyzed pathway over the Brønsted acid catalyzed pathway. Thus, we propose that the Lewis acid catalyzed pathway predominates in this reaction. It should be noted that the reaction of B(C6F5)3 with carboxylic acids can generate RCO2B(C6F5)2 and C6F5H.23 It is therefore possible that an alternative Lewis acid catalyzed pathway could operate with RCO2B(C6F5)2 acting as the active catalyst, however in situ NMR studies showed little correlation between the reaction of stoichiometric B(C6F5)3 and p-fluorobenzoic acid and the catalytic reaction in the 19F NMR spectrum (see ESI† for details).
Lastly, we rationalized why heating is required for the reaction to take place. We hypothesized that several off-cycle catalyst resting states could exist. These include coordination of the borane to the 4-fluorobenzoic acid (8) side product which was found to lie 0.5 kcal mol−1 lower in energy than free B(C6F5)3, and coordination of the borane to the 4-fluorobenzoic acid stabilized by the amine (9) which was calculated to be 4.6 kcal mol−1 lower in energy than free B(C6F5)3 (Fig. 2). This off-cycle intermediate significantly raises the total overall energy barrier for the reaction, which is in accordance with our reaction conditions.
Conclusions
In conclusion, a simple facile metal-free reaction methodology has been demonstrated for the construction of C–N bond through N-alkylation using catalytic amounts of B(C6F5)3. A wide range of substrates was tested including secondary amines, carbazoles and indoles to give good to excellent yields of the N-alkylated products. Interestingly, when 1H-indole and 1H-pyrrole were used for the reaction, exclusive C–C cross-coupled products were obtained. DFT studies have shown that the role of the catalyst is to act as a Lewis acid to generate an electrophilic carbenium ion which is then attacked by the amine nucleophile. Overall, this work represents a mild metal-free approach to construct the C–N bond via N-alkylation reaction and adds to the range of reactions the special Lewis acid B(C6F5)3 can catalyze.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. D. and R. L. M. are grateful to the EPSRC for funding (EP/R026912/1). We acknowledge Dr. Benson Kariuki (Cardiff University) for XRD measurements and for solving and refining data. A. A. and R. B. thank the Australian Research Council (ARC) for project funding (DP180100904) and the Australian National Computational Infrastructure and the University of Tasmania for the generous allocation of computing time.Notes and references
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 Search PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 Search PubMed.
- For selected reviews on FLPs see: (a) A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 Search PubMed; (b) D. W. Stephan, Science, 2016, 354, aaf7229 Search PubMed; (c) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 Search PubMed; (d) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 2–44 Search PubMed; (e) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 Search PubMed.
- For selected reviews see: (a) Y. Hoshimoto and S. Ogoshi, ACS Catal., 2019, 9, 5439–5444 Search PubMed; (b) J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 Search PubMed; (c) G. Erker, Dalton Trans., 2005, 1883–1890 Search PubMed; (d) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 Search PubMed.
- J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 Search PubMed.
- For the first example of B(C6F5)3 in hydrogenation of imines see: P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 Search PubMed.
- For selected reviews see: (a) J. Lam, K. M. Szkop, E. Mosaferia and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 Search PubMed; (b) D. J. Scott, J. M. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689–5700 Search PubMed.
- For selected articles see: (a) S. Tussing, K. Kaupmees and J. Paradies, Chem. – Eur. J., 2016, 22, 7422–7426 Search PubMed; (b) S. Li, G. Li, W. Meng and H. A. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 Search PubMed; (c) N. Gandhamsetty, J. Jeong, J. Park, S. Park and S. Chang, J. Org. Chem., 2015, 80, 7281–7287 Search PubMed.
- For examples see: (a) A. Maity, B. L. Frey, N. D. Hoskinson and D. C. Powers, J. Am. Chem. Soc., 2020, 142, 4990–4995 Search PubMed; (b) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 Search PubMed; (c) A. Ruffoni, F. Juliá, T. D. Svejstrup, A. J. McMillan, J. J. Douglas and D. Leonori, Nat. Chem., 2019, 11, 426–433 Search PubMed; (d) Z. Wu, S. Du, G. Gao, W. Yang, X. Yang, H. Huanga and M. Chang, Chem. Sci., 2019, 10, 4509–4514 Search PubMed; (e) Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559(7712), 83–88 Search PubMed; (f) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 Search PubMed; (g) D. Chusov and B. List, Angew. Chem., Int. Ed., 2014, 53, 5199–5201 Search PubMed.
- For selected examples see: (a) K. Murugesan, Z. Wei, V. G. Chandrashekhar, H. Neumann, A. Spannenberg, H. Jiao, M. Beller and R. V. Jagadeesh, Nat. Commun., 2019, 10, 5443 Search PubMed; (b) T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumann, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Nat. Commun., 2018, 9, 4123 Search PubMed; (c) S. Raoufmoghaddam, Org. Biomol. Chem., 2014, 12, 7179–7193 Search PubMed; (d) A. F. Abdel-Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971–1031 Search PubMed; (e) S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 Search PubMed.
- (a) S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 Search PubMed; (b) J. Zheng, T. Roisnel, C. Darcel and J.-B. Sortais, ChemCatChem, 2013, 5, 2861–2864 Search PubMed; (c) G. B. Giovenzana, D. Imperio, A. Penoni and G. Palmisano, Beilstein J. Org. Chem., 2011, 7, 1095–1099 Search PubMed.
- (a) Y. Hoshimoto, T. Kinoshita, S. Hazra, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2018, 140, 7292–7300 Search PubMed; (b) É. Dorko, M. Szabo, B. Kotai, I. Papai, A. Domjan and T. Soos, Angew. Chem., Int. Ed., 2017, 56, 9512–9516 Search PubMed.
- (a) V. Fasano and M. J. Ingleson, Chem. – Eur. J., 2017, 23, 2217–2224 Search PubMed; (b) L. C. Wilkins, J. L. Howard, S. Burger, L. Frentzel-Beyme, D. L. Browne and R. L. Melen, Adv. Synth. Catal., 2017, 359, 2580–2584 Search PubMed; (c) V. Fasano, J. E. Radcliffe and M. J. Ingleson, ACS Catal., 2016, 6, 1793–1798 Search PubMed; (d) M. R. Tiddens, R. J. M. Klein Gebbink and M. Otte, Org. Lett., 2016, 18, 3714–3717 Search PubMed; (e) J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 Search PubMed.
- S.-S. Meng, X. Tang, X. Luo, R. Wu, J.-L. Zhao and A. S. C. Chan, ACS Catal., 2019, 9, 8397–8403 Search PubMed.
- M. M. Guru, P. R. Thorve and B. Maji, J. Org. Chem., 2020, 85, 806–819 Search PubMed.
- For selected examples see: (a) C. E. Meyet, C. J. Pierce and C. H. Larsen, Org. Lett., 2012, 14, 964–967 Search PubMed; (b) X. Xu and X. Li, Org. Lett., 2009, 11, 1027–1029 Search PubMed; (c) V. K.-Y. Lo, Y. Liu, M.-K. Wong and C.-M. Che, Org. Lett., 2006, 8, 1529–1532 Search PubMed.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskel, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 Search PubMed.
- For selected examples see: (a) M. Blaess, N. Bibak, R. A. Claus, M. Kohl, G. A. Bonaterra, R. Kinscherf, S. Laufer and H.-P. Deigner, Eur. J. Med. Chem., 2018, 153, 73–104 Search PubMed; (b) D. Addla, S.-Q. Wen, W.-W. Gao, S. K. Maddili, L. Zhang and C.-H. Zhou, Med. Chem. Commun., 2016, 7, 1988–1994 Search PubMed; (c) Y. Zhang, V. K. R. Tangadanchu, R. R. Y. Bheemanaboina, Y. Cheng and C.-H. Zhou, Eur. J. Med. Chem., 2018, 155, 579–589 Search PubMed.
- (a) J. M. Ahn, T. S. Ratani, K. I. Hannoun, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 12716–12723 Search PubMed; (b) A. C. Bissember, R. J. Lundgren, S. E. Creutz, J. C. Peters and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 5129–5133 Search PubMed; (c) R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7811 Search PubMed.
- A. Kaga, K. Nogi and H. Yorimitsu, Chem. – Eur. J., 2019, 25, 14780–14784 Search PubMed.
- A. Dasgupta, R. Babaahmadi, B. Slater, B. F. Yates, A. Ariafard and R. L. Melen, Chem, 2020, 6, 2364–2381 Search PubMed.
- S. Mitu and M. C. Baird, Can. J. Chem., 2006, 84, 225–232 Search PubMed.
- S. Mitu and M. C. Baird, Organometallics, 2006, 25, 4888–4896 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary information includes detailed experimental procedures, NMR spectra, DFT data, and X-ray data. Crystallographic data for 4c and 5a have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC: 1996990 and 1996991. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cy01339k |
| ‡ Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2020.0117003506 |
| This journal is © The Royal Society of Chemistry 2020 |